

Abstract
Several 2-imido substituted furans were found to undergo a rapid intramolecular [4+2]-cycloaddition to deliver oxabicyclo adducts in good to excellent yields. By using a Rh(I)-catalyzed ring opening of the resulting oxabicyclic adduct, it was possible to prepare several highly functionalized tetrahydro-1H-indol-2(3H)-one derivatives which were then used to prepare several erythrina alkaloids. By taking advantage of the Rh(I)-catalyzed reaction, it was possible to convert tert-butyl 3-oxo-5-carbomethoxy-10-oxa-2-azatricyclo[5.2.1.01,5]dec-8-ene-2-carboxylate into the ring opened boronate by reaction with phenylboronic acid. Treatment of the boronate with pinacol/acetic acid afforded the corresponding diol which was used in a successful synthesis of racemic 3-demethoxyerythratidinone. During the course of these studies, several novel rearrangement reactions were encountered while attempting to induce an acid-initiated Pictet Spengler cyclization of a key lactam intermediate. The IMDAF/Rh(I)-catalyzed ring opening cascade sequence was also applied to the total synthesis of (±)-erysotramidine as well as the lycorine type alkaloid (±)-epi-zephyranthine.
Introduction
The tetrahydroindoline nucleus is a structurally characteristic component found in a wide variety of alkaloids,1 including the amaryllidaceae,2 aspidosperma,3 strychnos4 and erythrina5 families. The 1-azaspirocyclic structure of the erythrina family has long been of interest in the development of synthetic strategies for its efficient formation.6 Many members of this family possess curare-like and hypnotic activity, and a variety of pharmacological effects are associated with the erythrinane skeleton, including sedative, hypotensive, neuromuscular blocking and CNS activity.7 As a result, significant effort has been devoted to the assembly of the tetrahydroindoline framework associated with such alkaloids as well as to the synthesis of the natural products themselves.8-15 Erythrina alkaloids are generally classified into two groups according to their structural features;16 those whose D-rings are aromatic (e.g. 3-demethoxyerythratidinone (1) and erysotramidine (2)) and the others whose Drings possess an unsaturated lactone (e.g. cocculolidine (3)) (Figure 1).17 Taking the final step of bond formation into consideration, the methods for building up the erythrinan ring system have been loosely classified into seven different reaction types.18-25
Figure 1.

Some Representative Erythrina Alkaloids
Our approach toward the synthesis of a typical Erythrina alkaloid such as 3-demethoxyerythratidinone (1) derives from a program underway in our laboratory designed to exploit the facile intramolecular Diels-Alder reaction (IMDAF) of 2-imidofurans for the purpose of natural product synthesis.26 3-Demethoxyerythratidinone (1) was first isolated in 1973 by Barton and his collaborators from Erythrina lithosperma.27 Even though several syntheses have been reported,20,28 we felt that this compound could serve to illustrate our methodology and also provide a basis for a general cycloaddition approach toward Erythrina alkaloids. In particular, we considered the possibility that the erythrina framework of both 1 and erysotramidine (2) could be assembled from a tetrahydroindoline of type 5 by making use of the IMDAF cycloaddition of 2-imidofuran 4. It was expected that appropriate precursors (i.e. 6 and 7) to these two alkaloids would be derived from a Rh(I)-catalyzed ring opening reaction (vide infra) of the initially formed oxabicyclic adduct 5 with various nucleophilic reagents (Scheme 1). We now detail the successful implementation of this strategy. In addition, by using related chemistry, we were able to synthesize (±)-epi-zephyranthine (9), an azapolycycle that possesses the challenging tetracyclic galanthan skeleton found in the Amaryllidaceae family of alkaloids.
Scheme 1.

Some Key Disconnections Towards Several Alkaloids
Results and Discussion
In an earlier report, we showed that 2-imidofuran 4a rapidly reacted at room temperature to deliver the Diels-Alder cycloadduct 5a in 77% yield.29 The isolation of the somewhat labile (acid, heat) oxabicyclo adduct 5a was attributed to the low reaction temperature employed as well as the presence of the carbonyl group, which diminished the basicity of the nitrogen atom thereby retarding the ring cleavage/rearrangement reaction generally encountered with related furanyl carbamates.30 The facility of the cycloaddition was also shown to be due to the placement of the carbonyl center within the dienophile tether31 as well as the presence of the carbomethoxy group which lowers the LUMO energy of the π-bond, thereby facilitating the cycloaddition.
In recent years, the Rh(I)-catalyzed addition of arylboronic acids to olefins has become an active research area in organic synthesis.32 Conjugate addition generally occurs with electron-deficient olefins such as enones,33 alkenyl-phosphonates34 and nitroalkenes.35 The facile addition of boronic acids to oxabenzonorbornenes has also been achieved using a catalytic amount of a rhodium(I) complex.36 A common step in these reactions is the carborhodation of the carbon-carbon double bond followed by hydrolysis of the organorhodium intermediate. Lautens and co-workers have shown that the Rh(I)-catalyzed ring-opening reaction of unsymmetrical oxabicyclic compounds is a highly regioselective process, giving rise to products derived from the attack of the nucleophile distal to the bridgehead substituent.37 By taking advantage of this Rh(I)-catalyzed reaction, we were able to convert 5a into the ring-opened boronate 6 (97%), which was then converted to the corresponding diol 12 by treatment with pinacol/acetic acid (Scheme 2).38 It was also possible to prepare the same diol 12 by first treating 5a with catalytic amounts of SnCl2 in acetone39 to give dioxolane 11 followed by a subsequent hydrolysis reaction. The conversion of 5a to the corresponding acetonide 11 most likely proceeds by an initial ring opening of 5a by the mild Lewis acid SnCl2, followed by addition to acetone to give intermediate 10. Cyclization of this transient species onto the neighboring π-bond ultimately generates dioxolane 11 in 95% yield.
Scheme 2.
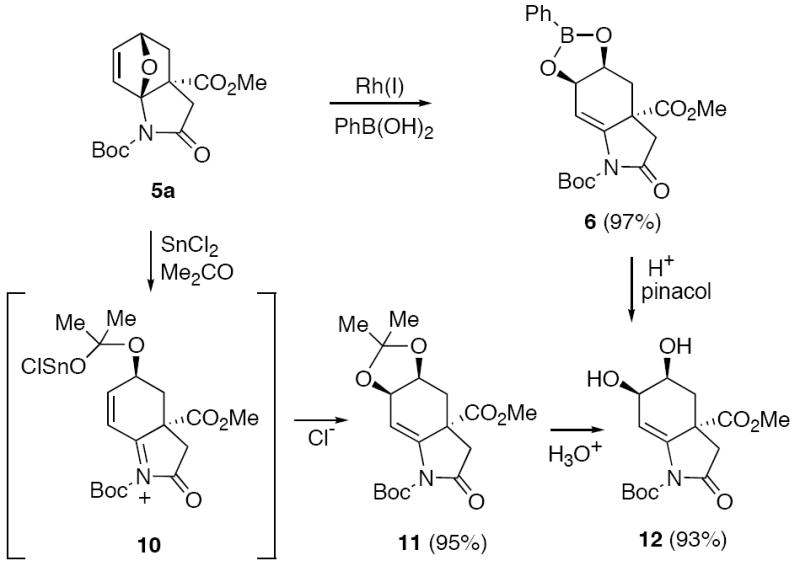
Rh(I) and SnCl2 Catalyzed Reactions
Acid Catalyzed Rearrangements
Our initial plan to synthesize 3-demethoxyerythratidinone (1) involved the cyclization of a N-acyliminium ion derived from a suitable aryl enamide precursor40,41 emanating from diol 12. Toward this end, oxidation of the allylic alcohol group of 12 was carried out with MnO2 and this was followed by protection of the secondary OH group with TBSCl. Removal of the Boc group, and subsequent N-alkylation with 4-(2-bromoethyl)-1,2-dimethoxybenzene afforded enamido lactam 13 in 65% yield for the three-step sequence (Scheme 3). Several acids were examined in our attempt to promote the planned acidinitiated Pictet-Spengler cyclization of lactam 13. During the course of these studies, we encountered several novel rearrangement reactions. For example, when 13 was treated with polyphosphoric acid (PPA) in refluxing CH2Cl2, the rearranged benzo[4,5]azepino lactam 16 was isolated in 80% yield and its structure was unequivocally established by X-ray crystallography. This unusual reorganization can be rationalized by the pathway proposed in Scheme 4. We assume that the first step involves generation of the tetracyclic erythrina intermediate 14 by the traditional Pictet-Spengler type cyclization.40 Intermediate 14 then undergoes a nitrogen-assisted 1,2-bond migration with simultaneous expulsion of water (or TBSOH) to produce the ring-expanded N-acyliminium ion 15. Loss of a proton and subsequent tautomerization perfectly accounts for the formation of the observed product 16.41c
Scheme 3.
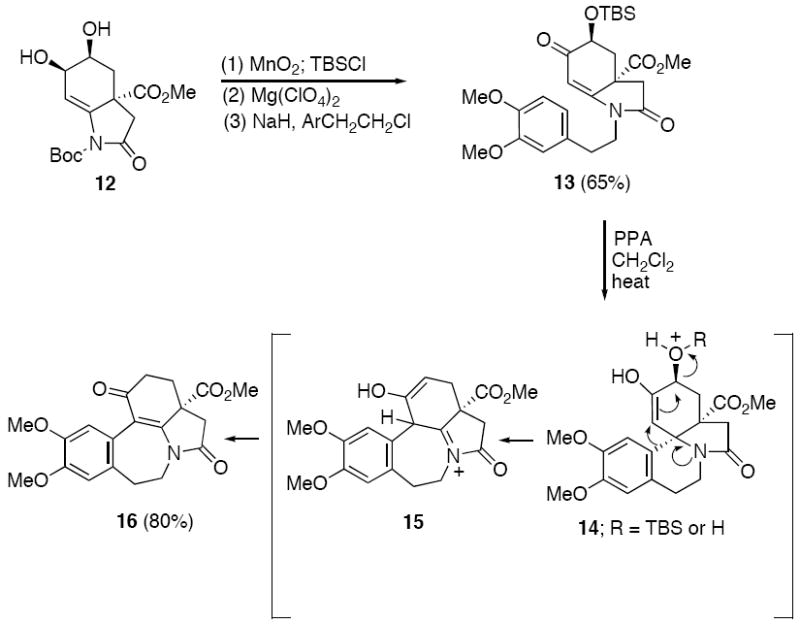
PPA Induced Rearrangement
Scheme 4.

Triflic Acid Induced Rearrangements
In contrast to the rearrangement observed using PPA, heating a sample of 13 in CH2Cl2 with trifluoromethanesulfonic acid (TfOH) followed by base workup afforded phenol 17 in 76% yield.42 Analysis of the rearrangement by 1H-NMR spectroscopy revealed that the reaction proceeded via the intermediacy of lactone 18, which could be isolated in 80% yield by terminating the thermolysis after 1 h. Additional heating of 18 in the presence of TfOH afforded phenol 17 in 95% yield. Furthermore, when p-TsOH was employed as the acid promoter, a new, interesting intermediate (i.e., 19) was obtained in 95% yield. The isolation of 19 under these milder acidic conditions suggests that the initial step in the conversion of 13 to 17 involves formation of the γ-lactone ring. Exposure of 19 to TfOH in refluxing CH2Cl2 (1 h) resulted in the preferential cyclization of the activated aromatic ring onto the amido carbonyl group, producing 18 in 90% yield (Scheme 4).
Free Radical Induced Cyclizations
Considering the difficulty encountered with the traditional Pictet-Spengler reaction of enamido lactam 13, we modified our approach toward 3-demethoxyerythratidinone (1). Acetonide 20 was prepared by removal of the Boc group of 11 followed by N-alkylation with 4-(2-bromoethyl)-1,2-dimethoxybenzene. In earlier work, we had found that NBS in acetonitrile could be used to promote an electrophilic substitution reaction of a tetrahydroindolinone to assemble the tetracyclic core of the erythrinone skeleton.41d Unfortunately, when acetonide 20 was subjected to an equiv amount of NBS in acetonitrile, bromoenamide 21 was obtained as the exclusive product in 97% yield and its formation can be attributed to a faster deprotonation (vs. cyclization) of the presumed N-acyliminium ion intermediate. If more than 1 equiv of NBS was used in this reaction, varying amounts of the dibrominated hydroindolinone 22 were isolated. When a sample of 21 was exposed to an additional equivalent of NBS in acetonitrile, dibromide 22 was isolated in 93% yield.
Radical cyclizations of highly reactive aryl and vinyl radicals onto double and triple bonds have proven to be very useful for construction of both carbocycles and heterocycles.14,43 To investigate whether these bromosubstituted tetrahydroindolinones could be used as precursors for radical cyclization chemistry, we explored the reaction of 21 with n-Bu3SnH under the standard radical-forming conditions. Exposure of 21 to n-Bu3SnH/AIBN at 80 °C provided the azepinoindole derivative 23 in 61% yield. In this case, selective 7-endo-cyclization took place by addition of the vinyl radical onto the aromatic ring (Scheme 5). Interestingly, the closely related dibromide 22 also afforded 23 in 55% yield when it was allowed to react under the radical conditions. Even though it is not clear which particular bromine atom is attacked by the stannyl radical, 7-ring cyclization followed by bromine atom ejection represents the major reaction pathway that occurs with tetrahydroindolinone 22.
Scheme 5.
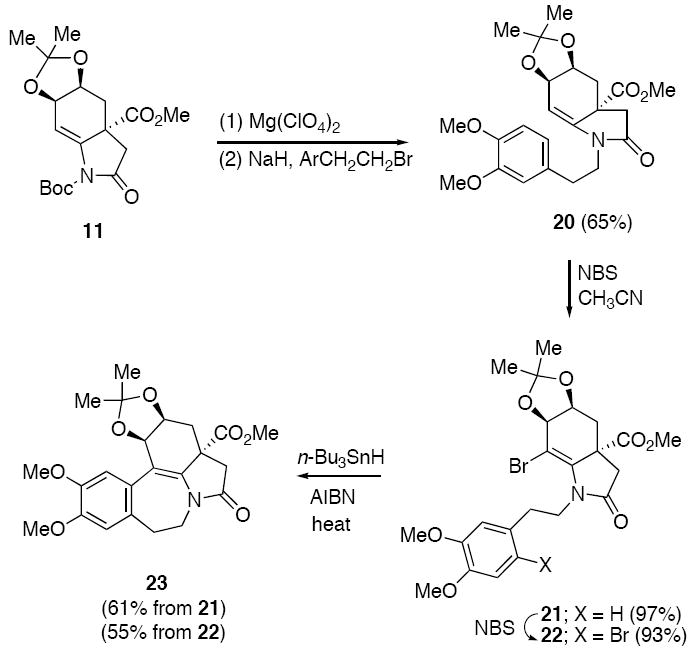
Free Radical Cyclization to form the Azepinoindole Ring
The radical cyclization approach was also extended to tetrahydroindolinone 24 which was readily prepared by the reaction of acetonide 11 with NaH and 1-bromo-2-(2-bromoethyl)-4,5-dimethoxybenzene. In this case, both 6-exo-trig and 7-endo-trig modes of cyclization are possible. The regiochemical preferences in cyclization of vinyl and aryl radicals generally parallel the alkyl analogues.44 When compound 24 was heated at 80 °C with n-Bu3SnH/ AIBN, the only product that could be isolated (72%) was that derived from a 7-endo-trig cyclization pathway (Scheme 6). Apparently, steric hindrance of the substituents sufficiently retards the 6-exo-trig cyclization so that the 7-endo closure becomes the predominant pathway.
Scheme 6.

7-endo-trig Cyclization of Tetrahydroindolinone 24
Synthesis of 3-Demethoxyerythratidinone
As a consequence of the above result, we abandoned the radical cyclization approach toward 3-demethoxyerythratidinone (1) and instead decided to reinvestigate the Pictet-Spengler reaction, this time using tetrahydroindolinone 27 as the cyclization substrate. We found that treating acetonide 20 with trifluoroacetic acid (TFA) in CH2Cl2 at 25 °C led to the desired tetrahydroindolinone 27 in 93% yield. As highlighted in Scheme 7, we believe that the reaction of 20 proceeds by an acid-induced loss of acetone to generate N-acyliminium ion 26, which then loses the available allylic proton so as to dissipate the positive charge. Ketonization of the resulting enol produces 27. The Pictet-Spengler reaction of 27 was then carried out uneventfully with PPA to furnish the tetracyclic erythrinane 28 in 90% yield. Base hydrolysis of 28 gave carboxylic acid 29, which was then subjected to Barton decarboxylation conditions45 using BrCCl3 as the solvent. Subsequent elimination of HBr from the labile tertiary bromide afforded the known 5H-indolo[7a, 1a]isoquinolinedione 30.28a This compound was converted to 3-demethoxyerythratidinone (1) following the reductive method of Tsuda and co-workers.28a
Scheme 7.
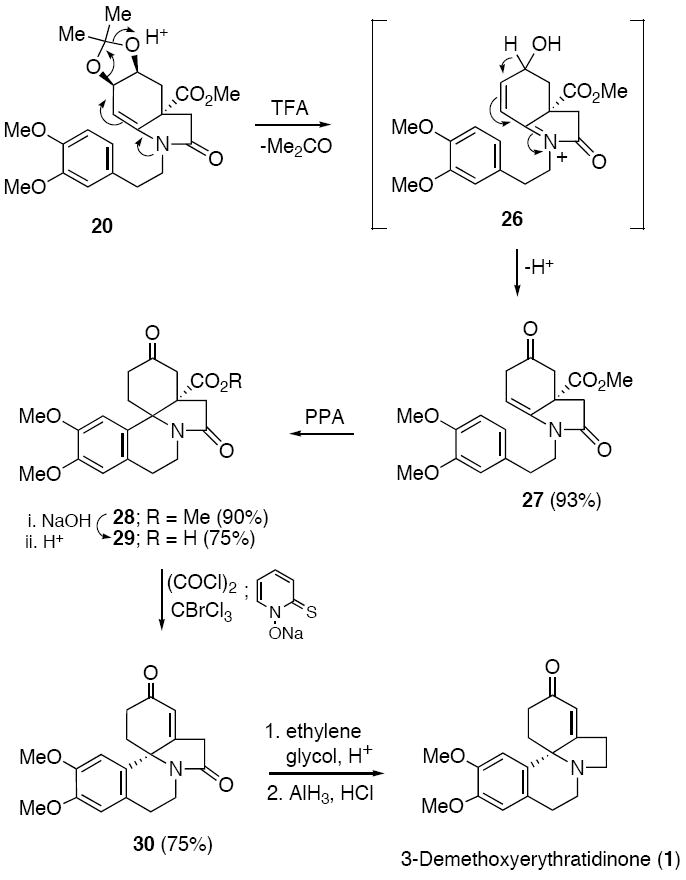
Pictet-Spengler Route toward 3-Demethoxyerythratidinone (1)
Synthesis of Erysotramidine
As a further demonstration of the efficiency of the IMDAF/Rh(I)-catalyzed cascade of 2-imidofurans for the synthesis of erythrina alkaloids, we applied the method towards the synthesis of erysotramidine (2). We had previously demonstrated29 that the reaction of azaoxabicyclohexene 5b with catalytic [Rh(COD)Cl]2 in the presence of various ammonium carboxylates afforded the dienyl alcohol 7 in 80% isolated yield according to the mechanism outlined in Scheme 8. The ready availability of 7 from cycloadduct 5b suggested its use in a synthesis of erysotramidine (2). This approach was bourn out in the following manner. The homoallylic alcohol present in compound 7 was first protected as the TBS ether and the Boc group was then removed with Mg(ClO4)2. Subsequent N-alkylation with NaH and 4-(2-bromoethyl)-1,2-dimethoxybenzene furnished 33 in 60% overall yield for the three-step sequence. Our attempts to induce an acid-promoted cyclization of 33 failed to produce any characterizable products, probably as a consequence of the anti-aromatic nature of the resulting cationic intermediate. We were pleased to discover, however, that bicyclic lactam 33 underwent an extremely smooth cyclization to the desired erythrinan skeleton (i.e., 34) in 89% yield when treated with NBS in acetonitrile. This cyclization reaction was found to be markedly dependent on the nature of the solvent; acetonitrile was the only solvent that was successful. It is not clear to us why the NBS cyclization of 33 proceeded so cleanly and why the related bicyclic lactam 20 only underwent enamido-bromination (i.e., 20 → 21) under identical conditions. Refluxing a sample of 34 with n-Bu3SnH and AIBN in benzene resulted in reduction of the secondary bromide. This was followed by deprotection of the TBS ether and subsequent dehydration of the alcohol to give 35 in 50% overall yield for the three-step process. Stereoselective allylic oxidation with selenium dioxide in the presence of formic acid gave a 1:1-mixture of formate 36 and alcohol 37 in 60% yield (based on recovered starting material) as single diastereomers. The stereochemical outcome of the oxidation involves attack by the oxidant from the least hindered α-position. Formate 36 was quantitatively transformed into alcohol 37 by treatment with acetyl chloride in ethanol.46 Finally, compound 37 was converted into (±)-erysotramidine (2)25f in 91% yield by O-methylation using KOH/MeI in THF according to Tsuda’s method (Scheme 9).40a-c
Scheme 8.
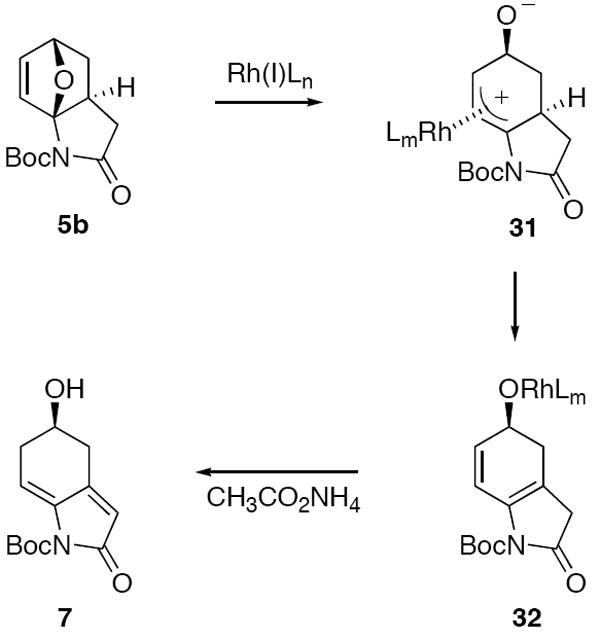
Rh(I)-Catalyzed Ring-Opening Reaction
Scheme 9.

Total Synthesis of (±)-Erysotramidine (2)
Synthesis of (±)-epi-Zephyranthine
To further demonstrate the wide potential of this methodology for alkaloid natural product synthesis, we opted to use dioxolane 8, which had been previously prepared by the IMDAF/Rh(I)-catalyzed route,29 for the synthesis of a member of the Amaryllidaceae family of alkaloids.47 In contrast to other members of the lycorine (38) family such as dihydrolycorine (39), lycoricidine (40), pancratistatin (41), only a limited number of syntheses of zephranthine48 (42) have been carried out and there are no reports dealing with the synthesis of the stereoisomeric epi-zephyrantine (9). Dioxolane 8 was readily available from the oxabicyclic adduct 5b via an acid induced cleavage of boronate 43 to first produce the corresponding diol followed by a subsequent reaction with 2,2-dimethoxypropane. We were now in a position to apply the experience gained from our erythrina syntheses to the preparation of epi-zephyranthine (9). To this end, dioxolane 8 was easily converted to the corresponding benzamide 44 in 81% yield by first removing the t-Boc group using Mg(ClO4)2 in acetonitrile followed by benzoylation with the acid chloride derived from 6-iodobenzo[1,3]-dioxazole-5-carboxylic acid (Scheme 10). Exposure of 44 to n-Bu3SnH in benzene at reflux in the presence of AIBN afforded the anticipated tetracyclic product 45 in 55% yield. The trans-B,C- and cis-C,D-ring fusion of the cyclized product was unambiguously assigned by an X-ray crystal structure of compound 45. SYBYL force field calculations suggest that this stereochemistry corresponds to the most stable isomer, and is also in agreement with the earlier reports of Rigby49 and Schultz.50 Reduction of 45 with BH3·THF followed by hydrolysis of the 1,3-dioxolane furnished (65%) epi-zephyranthine (9) in 14.5% overall yield for the 7-step sequence starting from the 2-imidofuran 4b.
Scheme 10.
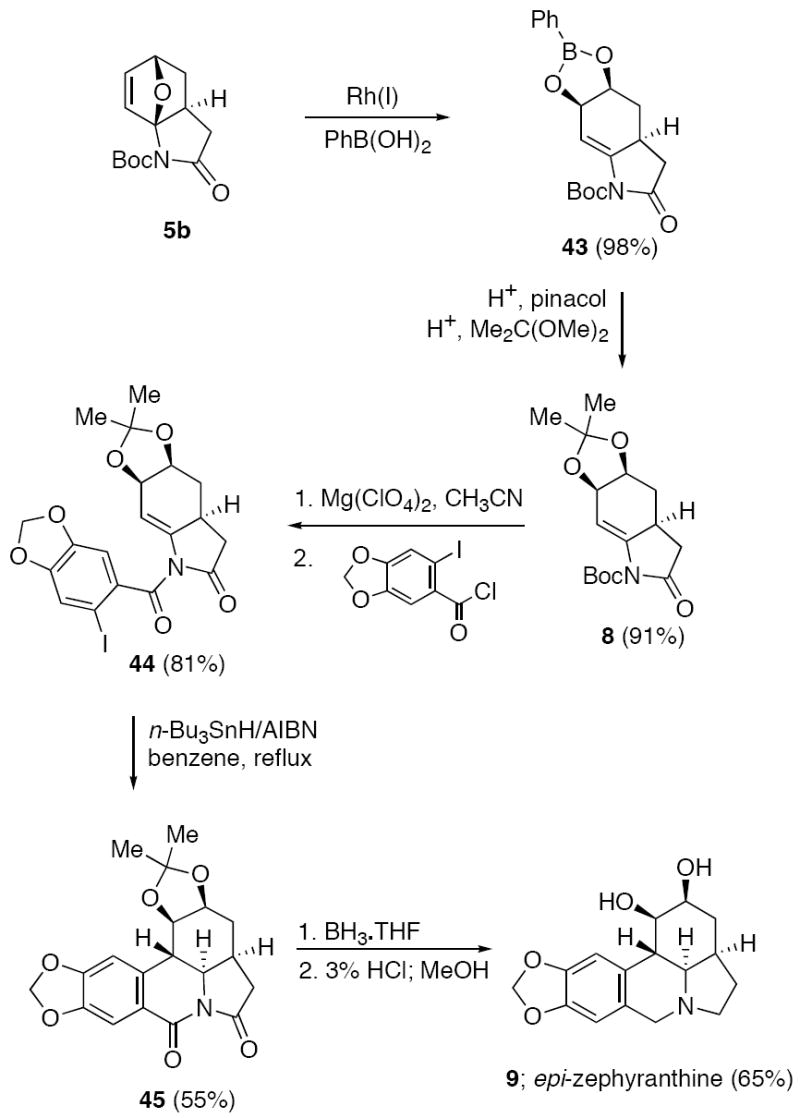
Total Synthesis of epi-Zephyranthine (9)
In summary, a new strategy for the synthesis of the tetrahydroindolinone core found in the erythrina and amaryllidaceae alkaloid family has been developed, which is based on an extraordinarily facile intramolecular Diels-Alder reaction of 2-imido-substituted furans. By using a Rh(I)-catalyzed ring opening of the oxabicyclic adduct, it was possible to synthesize the key tetrahydroindolinone necessary for a Pictet-Spengler cyclization. During the course of the synthesis, several novel acid-induced rearrangement reactions were encountered. The Rh(I)-catalyzed reactions proceed in high yield under very mild conditions and occur with excellent diastereoselectivity. The application of this approach to other natural product targets is currently under investigation, the results of which will be disclosed in due course.
Experimental Section
6-Oxo-2-phenyl-6,7,8,8a-tetrahydro-3aH-1,3-dioxa-5-aza-2-bora-s-indacene-5,7a-dicarboxylic Acid 5-tert-Butyl Ester 7a-Methyl Ester (6)
To a solution containing 0.15 g (0.5 mmol) of 3-oxo-5-carbomethoxy-10-oxa-2-azatricyclo[5.2.1.01,5]dec-8-ene-2-carboxylic acid tert-butyl ester (5a)29, 7.0 mg (0.015 mmol) of [Rh(COD)Cl]2, 5.1 μL (0.03 mmol) of P(OEt)3 in 2.5 mL of THF was added 0.12 g (1.0 mmol) of phenylboronic acid. The reaction mixture was heated at 65 °C for 6 h, cooled to room temperature and quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layer was washed with a saturated aqueous NaHCO3 solution and dried over MgSO4. The solvent was removed under reduced pressure and the residue was subjected to flash silica gel chromatography to give 0.2 g (97%) of 6 as a white solid: mp 131-132 °C; IR (neat) 1775, 1736, 1369, 1097 and 701 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 1.53-1.59 (m, 1H), 1.58 (s, 9H), 2.50 (d, 1H, J = 17.2 Hz), 2.88 (dd, 1H, J = 12.8 and 6.0 Hz), 2.95 (d, 1H, J = 17.2 Hz), 3.74 (s, 3H), 5.08 (ddd, 1H, J = 10.0, 8.0 and 6.0 Hz), 5.143 (dd, 1H, J = 8.0 and 3.2 Hz), 6.38 (d, 1H, J = 3.2 Hz), 7.36 (t, 2H, J = 7.6 Hz), 7.47 (tt, 1H, J = 7.6 and 1.6 Hz) and 7.78 (dd, 2H, J = 7.6 and 1.6 Hz); 13C-NMR (CDCl3, 100 MHz) δ 28.1, 36.8, 42.0, 46.2, 53.5, 73.2, 73.4, 85.0, 108.8, 128.0, 131.8, 135.0, 138.6, 148.9, 170.6 and 171.6; Anal. Calcd. for C21H24BNO7: C, 61.04; H, 5.85; N, 3.39. Found: C, 60.90; H, 5.76; N, 3.26.
2,2-Dimethyl-6-oxo-6,7,8,8a-tetrahydro-3aH-[1,3]dioxolo[4,5-f]indole-5,7a-dicarboxylic acid 5-tert-Butyl Ester 7a-Methyl Ester (11)
To a solution containing 0.1 g (0.38 mmol) of oxabicycle 5a in 0.5 mL of acetone and 2 mL of CH2Cl2 was added 5 mg (0.03 mmol) of anhydrous stannous chloride. After stirring at room temperature for 15 min, the solvent was removed under reduced pressure and the residue was subjected to the flash silica gel chromatography to afford 0.11 g (92%) of dioxolane 11 as a white solid: mp 131-133 °C; IR (neat) 1774, 1735, 1297 and 850 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 1.36 (s, 3H), 1.45 (s, 3H), 1.54 (s, 9H), 1.49-1.56 (m,1H), 2.49 (d, 1H, J = 17.2 Hz), 2.62 (dd, 1H, J = 12.8 and 6.0 Hz), 2.86 (d, 1H, J = 17.2 Hz), 3.69 (s, 3H), 4.52 (dt, 1H, J = 10.8 and 6.0 Hz), 4.71 (dd, 1H, J = 6.0 and 3.2 Hz) and 6.21 (d, 1H, J = 3.2 Hz); 13C-NMR (CDCl3, 100 MHz) δ 25.3, 28.0, 28.1, 35.6, 42.4, 46.9, 53.4, 71.2, 71.3, 84.8, 107.6, 109.4, 139.0, 148.8, 170.9 and 171.8; Anal. Calcd. for C18H25NO7: C, 58.84; H, 6.86; N, 3.81. Found: C, 58.77; H, 6.93; N, 3.62.
5,6-Dihydroxy-2-oxo-2,3,5,6-tetrahydro-4H-indole-1,3a-dicarboxylic acid 1-tert-Butyl Ester 3a-Methyl Ester (12)
A 0.1 g (0.24 mmol) sample of boronate 6 in 10 mL of CH2Cl2 to which 0.14 g (1.2 mmol) of pinacol and 2.8 μL (0.05 mmol) of acetic acid had been added was stirred at room temperature for 12 h and then the solvent was removed under reduced pressure. The residue was purified by flash silica gel chromatography to give 0.08 g (95%) of diol 12 as a clear oil: IR (neat) 3447, 2982, 1785, 1734 and 990 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 1.53 (s, 9H), 1.74 (t, 1H, J = 12.4 Hz), 2.44 (dd, 1H, J = 12.4 and 3.6 Hz), 2.54 (d, 1H, J = 17.2 Hz), 2.80 (d, 1H, J = 17.2 Hz), 2.81 (brs, 1H), 2.92 (brs, 1H), 3.69 (s, 3H), 3.88-3.96 (m, 1H), 4.34 (t, 1H, J = 4.4 Hz) and 6.18 (d, 1H, J = 4.4 Hz); 13C-NMR (CDCl3, 100 MHz) δ 28.1, 34.3, 43.1, 49.0, 53.4, 65.0, 66.4, 84.8, 111.4, 139.1, 148.9, 170.7 and 172.5; HRMS Calcd for C15H20NO7: 326.1240. Found: 326.1238.
Diol 12 could also be obtained by treating a sample of dioxolane 11 in 10 mL of methanol which contained 2 drops of aqueous HCl. The mixture was stirred at room temperature for 12 h and then the solvent was removed under reduced pressure. The residue was purified by flash silica gel chromatography to give 0.08 g (93%) of the same diol 12 as was obtained from boronate 6.
5-(tert-Butyl-dimethylsilanyloxy)-1-[2-(3,4-dimethoxyphenyl)ethyl]-2,6-dioxo-1,2,3,4,5,6-hexahydroindole-3a-carboxylic Acid Methyl Ester (13)
To a solution 0.2 g (0.6 mmol) of the diol 12 in 10 mL of CH2Cl2 was added 0.62 g (12 mmol) of MnO2. The mixture was stirred at room temperature for 3 days and filtered through Celite. The solvent was removed under reduced pressure. The black mixture was filtered through a short pad of silica and the filtrate was concentrated under reduced pressure and the crude residue was used for the next step. The mixture was taken up in 5 mL of CH2Cl2 and 0.3 mL (2.7 mmol) of 2,6-lutidine and 0.24 g (0.9 mmol) of TBSOTf was added. The reaction mixture was stirred at room temperature for 20 min. The mixture was quenched by water and the aqueous layer was extracted with EtOAc. The combined organic layer was washed with brine and dried over MgSO4. The solvent was removed under reduced pressure and the crude residue was purified by flash column chromatography to give 0.18 g (80%) of 5-(tert-butyl-dimethylsilanyloxy)-2,6-dioxo-2,3,5,6-tetrahydro-4H-indole-1,3a-dicarboxylic acid 1-tert-butyl ester 3a-methyl ester as a clear oil: IR (neat) 2954, 1804, 1734, 1629, 1299 and 862 cm-1; 1H-NMR (CDCl3, 600 MHz) δ 0.05 (s, 3H), 0.12 (s, 3H), 0.86 (s, 9H), 1.53 (s, 9H), 2.03 (t, 1H, J = 12.0 Hz), 2.61 (d, 1H, J = 16.8 Hz), 2.81 (dd, 1H, J = 12.0 and 5.4 Hz), 2.85 (d, 1H, J = 16.8 Hz), 3.74 (s, 3H), 4.34 (dd, 1H, J = 12.0 and 5.4 Hz) and 6.29 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ -5.4, -4.4, 18.6, 25.9, 28.0, 40.5, 42.1, 48.3, 53.8, 71.0, 86.2, 111.2, 147.6, 154.9, 169.9, 170.8 and 196.5.
To a solution containing 0.52 g (1.2 mmol) of above imide in 10 mL of CH3CN was added 0.05 g (0.24 mmol) of magnesium perchlorate. The mixture was stirred at room temperature for 2 h. The solvent was removed under reduced pressure and the residue was purified by flash silica gel chromatography to give 0.36 g (90%) of 5-(tert-butyl-dimethylsilanyloxy)-2,6-dioxo-1,2,3,4,5,6-hexahydroindole-3a-carboxylic acid methyl ester as a white solid: mp 159-161 °C; IR (neat) 3238, 1737, 1640, 1195, 1125 and 838 cm-1; 1H-NMR (CDCl3, 600 MHz) δ 0.08 (s, 3H), 0.14 (s, 3H), 0.88 (s, 9H), 2.10 (t, 1H, J = 12.0 Hz), 2.61 (d. 1H, J = 16.8 Hz), 2.79 (d, 1H, J = 16.8 Hz), 2.84 (dd, 1H, J = 12.0 and 5.4 Hz), 3.76 (s, 3H), 4.30 (dd, 1H, J = 12.0 and 5.4 Hz), 5.57 (s, 1H) and 9.36 (brs, 1H); 13C-NMR (CDCl3, 150 MHz) δ -5.3, -4.3, 18.6, 25.9, 40.6, 41.5, 50.3, 53.8, 71.4, 104.1, 159.9, 171.2, 175.1 and 196.5; Anal. Calcd. for C16H25NO5Si: C, 56.61; H, 7.42; N, 4.13. Found: C, 56.80; H, 7.59; N, 4.10.
To a solution of 0.15 g (0.44 mmol) of the above amide in 5 mL of DMF was added 22 mg (0.88 mmol) of 95% NaH at 0 °C. After stirring the mixture at 0 °C for 1 h, 0.22 g (0.89 mmol) of 4-(2-bromoethyl)-1,2-dimethoxybenzene was slowly added. The mixture was stirred for 12 h and then quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layer was washed with water, brine, and dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by flash silica gel chromatography to give 0.23 g (89%) of 13 as a white solid: 150-152 °C; IR (neat) 1747, 1633, 1516, 1127 and 838 cm-1; 1H-NMR (CDCl3, 600 MHz) δ 0.10 (s, 3H), 0.16 (s, 3H), 0.90 (s, 9H), 2.04 (t, 1H, J = 12.0 Hz), 2.53 (d. 1H, J = 16.8 Hz), 2.72-2.81 (m, 2H), 2.81 (d, 1H, J = 16.8 Hz), 2.87 (dd, 1H, J = 12.0 and 5.4 Hz), 3.51 (ddd, 1H, J = 14.4, 9.0 and 5.4 Hz), 3.67 (s, 3H), 3.83 (s, 3H), 3.85 (s, 3H), 3.93 (ddd, 1H, J = 14.4, 10.2 and 6.6 Hz), 4.31 (dd, 1H, J = 12.0 and 5.4 Hz), 5.55 (s, 1H), 6.70 (d, 1H, J = 1.8 Hz), 6.71 (dd, 1H, J = 8.4 and 1.8 Hz) and 6.77 (d, 1H, J = 8.4 Hz); 13C-NMR (CDCl3, 150 MHz) δ -5.3, -4.3, 18.6, 25.9, 32.2, 40.5, 40.9, 42.0, 48.7, 53.6, 56.0, 56.1, 71.5, 102.6, 111.5, 112.0, 120.8, 129.8, 148.1, 149.2, 160.6, 171.2, 172.9 and 195.7; Anal Calcd for C24H36NO5Si: C, 64.54; H, 8.13; N, 3.14. Found: C, 64.39; H, 8.24; N, 3.08.
10,11-Dimethoxy-1,5-dioxo-2,3,4,5,7,8-hexahydro-1H-benzo[4,5]azepino[3,2,1hi]-indole-3a-carboxylic Acid Methyl Ester (16)
A mixure of 0.05 g (0.1 mmol) of 13 and 0.5 mL of PPA was heated at 90 °C for 12 h, cooled to room temperature and then quenched with water. The aqueous layer was extracted with CHCl3 and the combined organic layer was dried over anhydrous MgSO4. The solvent was removed under reduced pressure and the residue was purified by flash silica gel chromatography to give 0.03 g (80%) of 16 as a pale yellow solid: IR (neat) 2954, 1734, 1606 and 1272 cm-1; 1H-NMR (CDCl3, 600 MHz) δ 2.12 (dt, 1H, J = 13.2 and 5.4 Hz), 2.62 (ddd, 1H, J = 18.6, 13.2 and 5.4 Hz), 2.64 (d, 1H, J = 17.4 Hz), 2.68 (dd, 1H, J = 13.2 and 5.4 Hz), 2.73 (dd, 1H, J = 18.6 and 5.4 Hz), 2.84 (dd, 1H, J = 15.0 and 5.4 Hz), 2.91 (d, 1H, J = 17.4 Hz), 2.99 (dd, 1H, J = 15.0 and 12.6 Hz), 3.46 (t, 1H, J =12.6 Hz), 3.76 (s, 3H), 3.83 (s, 3H), 3.87 (s, 3H), 4.35 (dd, 1H, J = 12.6 and 5.4 Hz), 6.61 (s, 1H) and 6.88 (s, 1H); 13C-NMR (CDCl3, 150 MHz) δ 29.4, 32.3, 35.5, 41.5, 50.6, 50.9, 53.7, 56.1, 56.2, 111.5, 114.9, 116.9, 122.8, 133.9, 147.2, 148.8, 155.2, 172.1, 173.3 and 196.0; HRMS Calcd. for C20H21NO6 [M+H]: 372.1442. Found: 372.1435.
2,3-Dimethoxy-5,6-dihydroindolo[2,1-a]isoquinolin-9-ol (17)
A solution containing 0.03 g (0.06 mmol) of 13 and 0.1 mL of TfOH in 2 mL of CH2Cl2 was heated at reflux for 4 h, cooled to room temperature and quenched with water. The aqueous layer was extracted with CHCl3 and the combined organic layer was dried over anhydrous MgSO4. The solvent was removed under reduced pressure and the residue was purified by flash silica gel chromatography to give 0.02 g (80%) of lactone 18 as a yellow oil: IR (neat) 1781, 1589, 1510, and 1272 cm-1; 1H-NMR (CDCl3, 600 MHz) δ 2.52 (d, 1H, J = 11.4 Hz), 2.97 (dt, 1H, J = 16.2 and 5.4 Hz), 3.08-3.14 (m, 1H), 3.13 (dd, 1H, J = 11.4 and 5.4 Hz), 3.54 (ddd, 1H, J = 12.0, 9.6 and 5.4 Hz), 3.71 (dt, 1H, J = 12.0 and 5.4 Hz), 3.90 (s, 3H), 3.91 (s, 3H), 4.91 (dd, 1H, J = 5.4 and 1.8 Hz), 5.21 (s, 1H), 5.66 (s, 1H), 6.69 (s, 1H) and 7.04 (s, 1H); 13C-NMR (CDCl3, 150 MHz) δ 28.0, 40.1, 47.8, 56.2, 60.2, 82.9, 91.4, 97.5, 108.0, 110.9, 117.9, 126.7, 146.0, 148.8, 151.1, 166.3, 173.2 and 189.1; HRMS Calcd. for C19H19NO6 [M+H]: 340.1185. Found: 340.1178.
To a sample of lactone 18 in 1 mL of MeOH was added 0.1 mL of Et3N. The reaction mixture was stirred at room temperature for 30 min and concentrated under reduced pressure. The crude NMR spectrum showed only the presence (>95%) of phenol 17: 1H-NMR (C6D6, 600 MHz) δ 2.49 (t, 2H, J = 6.6 Hz), 3.45 (s, 3H), 3.48 (s, 3H), 3.51 (t, 2H, J = 6.6 Hz), 6.41 (s, 1H), 6.54 (dd, 1H, J = 8.4 and 1.2 Hz), 6.55 (d, 1H, J = 1.2 Hz), 6.72 (s, 1H), 7.03 (brs, 2H) and 7.53 (s, 1H); HRMS Calcd. for C18H17NO3 [M+H]: 296.1281. Found: 296.1280.
4-[2-(3,4-Dimethoxyphenyl)ethyl]-9-oxa-4-aza-tricyclo[6.2.1.01,5]undec-5-ene-3,7,10-trione (19)
A solution containing 0.02 g (0.04 mmol) of 13 together with 0.01 g (0.06 mmol) of p-TsOH in 1 mL of toluene was heated at 100 °C for 1 h and then cooled to room temperature. The solvent was removed under reduced pressure and the residue was purified by flash silica gel chromatography to give 0.01 g (95%) of 19 as a clear oil: IR (neat) 1790, 1754, 1673, 1606, and 1264 cm-1; 1H-NMR (CDCl3, 600 MHz) δ 2.59 (d, 1H, J = 18.0 Hz), 2.68 (d, 1H, J = 12.0 Hz), 2.79 (t, 2H, J = 7.2 Hz), 2.87 (dd, 1H, J = 12.0 and 6.0 Hz), 3.18 (d, 1H, J = 18.0 Hz), 3.61 (dt, 1H, J = 13.8 and 7.2 Hz), 3.82-3.87 (m, 1H), 3.83 (s, 3H), 3.86 (s, 3H), 4.79 (dd, 1H, J = 6.0 and 1.8 Hz), 5.25 (d, 1H, J = 1.8 Hz), 6.62 (d, 1H, J = 1.8 Hz), 6.65 (dd, 1H, J = 8.4 and 1.8 Hz) and 6.77 (d, 1H, J = 8.4 Hz); 13C-NMR (CDCl3, 150 MHz) δ 32.3, 32.7, 42.8, 43.0, 47.6, 56.2, 81.0, 96.9, 111.7, 112.1, 121.1, 129.3, 148.4, 149.4, 162.9, 173.0, 173.1 and 189.1; HRMS Calcd. for C19H19NO6 [M+H]: 358.1285. Found: 358.1280.
Heating a sample of 19 with TfOH in CH2Cl2 at reflux for 4 h followed by the standard workup gave 18 in 84% yield.
5-[2-(3,4-Dimethoxyphenyl)ethyl]-2,2-dimethyl-6-oxo-3a,5,6,7,8,8a-hexahydro-[1,3]-dioxolo[4,5-f]indole-7a-carboxylic Acid Methyl Ester (20)
To a solution containing 0.1 g (0.26 mmol) of acetonide 11 in 1 mL of CH3CN was added 15 mg (0.06 mmol) of magnesium perchlorate. The mixture was stirred for 12 h at room temperature. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography to afford 0.06 g (84%) of 2,2-dimethyl-6-oxo-3a,5,6,7,8,8a-hexahydro-[1,3]dioxolo-[4,5-f]indole-7a-carboxylic acid methyl ester as a clear oil: IR (neat) 3198, 2985, 1727, 1699 and 1211 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 1.36 (s, 3H), 1.46 (s, 3H), 1.61 (dd, 1H, J = 12.4 and 10.8 Hz), 2.48 (d, 1H, J = 16.8 Hz), 2.62 (dd, 1H, J = 12.4 and 6.0 Hz), 2.79 (d, 1H, J = 16.8 Hz), 3.71 (s, 3H), 4.49 (dt, 1H, J = 10.8 and 6.0 Hz), 4.66 (dd, 1H, J = 6.0 and 2.8 Hz), 5.21 (d, 1H, J = 2.8 Hz) and 8.42 (brs, 1H); 13C-NMR (CDCl3, 100 MHz) δ 26.3, 28.0, 35.3, 41.4, 49.1, 53.3, 71.4, 72.1, 99.0, 109.5, 141.5, 172.4 and 175.5.
To a solution containing 0.2 g (0.74 mmol) of the above amide in 3 mL DMF was added 0.04 g (0.97 mmol) 60% NaH at 0 °C. After stirring the mixture at 0 °C for 1 h, a 0.33 g (0.1.4 mmol) sample of 4-(2-bromoethyl)-1,2-dimethoxybenzene was added. The reaction mixture was stirred at room temperature overnight and was then quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layer was washed with brine and dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography to give 0.25 g (80%) of 20 as a white solid: mp 128-130 °C; IR (neat) 2983, 1736, 1677, 1027 and 730 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 1.37 (s, 3H), 1.45 (s, 3H), 1.56 (dd, 1H, J =12.4 and 10.4 Hz), 2.42 (d, 1H, J = 16.8 Hz), 2.64 (dd, 1H, J =12.4 and 6.0 Hz), 2.71-2.85 (m, 2H), 2.78 (d, 1H, J = 16.8 Hz), 3.46 (ddd, 1H, J = 14.0, 10.0, and 6.0 Hz), 3.63 (s, 3H), 3.83 (s, 3H), 3.85 (s, 3H), 4.50 (dt, 1H, J = 10.4 and 6.0 Hz), 4.72 (dd, 1H, J = 6.0 and 2.8 Hz), 5.17 (d, 1H, J = 2.8 Hz), 6.72 (s, 1H), 6.75 (d, 1H, J = 8.0 Hz) and 6.77 (d, 1H, J = 8.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 25.2, 27.9, 32.2, 35.2, 40.9, 41.6, 47.5, 53.0, 55.9, 56.0, 71.2, 72.1, 97.2, 109.3, 111.4, 112.0, 120.8, 130.6, 143.6, 147.8, 149.0, 172.5 and 172.7; Anal. Calcd for C23H29NO7: C, 64.02; H, 6.77; N, 3.25. Found: C, 63.83; H, 6.83; N, 3.11.
4-Bromo-5-[2-(3,4-dimethoxyphenyl)ethyl]-2,2-dimethyl-6-oxo-3a,5,6,7,8,8a-hexahydro[1,3]dioxolo[4,5-f]indole-7a-carboxylic Acid Methyl Ester (21)
To a solution containing 0.16 g (0.37 mmol) of above lactam 20 in 6 mL of CH3CN was added 0.07 g (0.37 mmol) of NBS. The reaction mixture was stirred at room temperature for 1 h and was quenched with water. The aqueous layer was extracted with EtOAc and the combined extracts were dried over MgSO4 and concentrated under reduced pressure. Purification of the residue by flash silica gel chromatography afforded 0.18 g (97%) of 21 as a colorless oil: IR (neat) 1732, 1664, 1515, 1213 and 731 cm-1; 1H-NMR (CDCl3, 600 MHz) δ 1.39 (s, 3H), 1.49 (s, 3H), 1.71 (dd, 1H, J = 12.6, 11.4 Hz), 2.50 (d, 1H, J = 16.2 Hz), 2.59 (dd, 1H, J = 12.6 and 6.0 Hz), 2.71 (td, 1H, J = 12.0 and 6.0 Hz), 2.79 (d, 1H, J = 16.2 Hz), 2.90 (td, 1H, J = 12.0 and 4.8 Hz), 3.65 (s, 3H), 3.83 (s, 3H), 3.85, (s, 3H), 4.14 (m, 2H), 4.45 (dt, 1H, J = 11.4 and 6.0 Hz), 4.74 (d, 1H, J = 6.0 Hz) and 6.78 (m, 3H); 13C-NMR (CDCl3, 150 MHz) δ 25.6, 28.1, 34.0, 35.0, 40.9, 72.9, 51.9, 53.3, 56.0, 72.4, 77.3, 94.5, 109.5, 111.4, 112.2, 120.9, 130.6, 140.3, 147.8, 149.0, 171.6 and 173.5.
4-Bromo-5-[2-(2-bromo-4,5-dimethoxyphenyl)ethyl]-2,2-dimethyl-6-oxo-3a,5,6,7,8,8a-hexahydro[1,3]dioxolo[4,5-f]indole-7a-carboxylic Acid Methyl Ester (22)
To a solution containing 0.047 g (0.09 mmol) of above lactam 21 in 6 mL of CH3CN was added 0.016 g (0.09 mmol) of NBS. The reaction mixture was stirred at room temperature for 12 h and was quenched with water. The aqueous layer was extracted with EtOAc and the combined extracts were dried over MgSO4 and concentrated under reduced pressure. Purification of the residue by flash silica gel chromatography afforded 0.05 g (93%) of 22 as a colorless oil: IR (neat) 2950, 1737, 1738, 1506, 912 and 729 cm-1; 1H-NMR (CDCl3, 600 MHz) δ 1.40 (s, 3H), 1.50 (s, 3H), 1.71 (dd, 1H, J = 12.6 and 11.4 Hz), 2.50 (d, 1H, J = 16.8 Hz), 2.60 (dd, 1H, J = 12.6 and 5.4 Hz), 2.80 (d, 1H, J = 16..8 Hz), 2.89 (ddd, 1H, J = 13.8, 10.2 and 6.6 Hz), 2.98 (ddd, 1H, J = 13.8, 10.2 and 4.8 Hz), 3.66 (s, 3H), 3.82 (s, 3H), 3.83, (s, 3H), 4.16 (ddd, 1H, J = 13.8, 10.2 and 4.8 Hz), 4.24 (ddd, 1H, J = 13.8, 10.2 and 6.6 Hz), 4.45 (dt, 1H, J = 11.4 and 5.4Hz), 4.74 (d, 1H, J = 5.4 Hz), 6.84 (s, 1H) and 6.98 (s, 1H); 13C-NMR (CDCl3, 150 MHz) δ 25.7, 28.2, 34.3, 35.2, 41.1, 41.4, 52.1, 53.4, 56.3, 72.5, 94.9, 109.6, 113.4, 114.6, 115.7, 129.7, 140.4, 148.5, 148.6, 171.7 and 173.7.
(7aR,Z)-Methyl 1,2-(4,5-Dimethoxy)benzo-10,10-dimethyl-6-oxo-3,4,6,7,7a,-8,8a,11a-octahydroazepino[3,2,1-hi][1,3]dioxolo[4,5-f]indole-7a-carboxylate (23)
To a solution of 0.13 g (0.25 mmol) of lactam 21 in 25 mL of benzene was added 8.4 mg (0.05 mmol) of AIBN followed by 0.16 mL (0.61 mmol) of Bu3SnH. The mixture was heated at reflux for 10 h, cooled to room temperature and poured into 10 mL of an aqueous KF solution (1M). The aqueous layer was extracted with EtOAc and the combined organic layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography to give 0.06 g (61%) of 23 as a white solid: mp 168-170 °C; IR (neat) 2981, 1727, 1643, 1211, 1050 and 729 cm-1; 1H-NMR (CDCl3, 600 MHz) δ 1.49 (s, 3H), 1.57 (s, 3H), 1.96 (t, 1H, J = 12.0 Hz), 2.59 (d, 1H, J = 16.8 Hz), 2.60 (dd, 1H, J = 12.0 and 4,8 Hz), 2.81 (d, 1H, J = 16.8 Hz), 2.92 (dd, 1H, J = 15.0 and 4.8 Hz), 3.02 (dd, 1H, J = 15.0 and 10.2 Hz), 3.41-3.50 (m, 1H), 3.73 (s, 3H), 3.87 (s, 3H), 3.90 (s, 3H), 4.29 (brs, 1H), 4.36 (dt, 1H, J = 12.0 and 4.8 Hz), 4.78 (d, 1H, J = 4.8 Hz), 6.61 (s, 1H) and 7.66 (s, 1H); 13C-NMR (CDCl3, 150 MHz) δ 26.6, 29.0, 33.7, 34.9, 41.8, 48.4, 51.1, 53.4, 55.8, 56.1, 71.9, 76.2, 108.9, 109.3, 111.5, 112.3, 128.0, 133.9, 141.0, 147.9, 172.0 and 173.3; Anal. Calcd for C23H27NO7: C, 64.32; H, 6.34; N, 3.26. Found: C, 64.06; H, 6.34; N, 3.10.
Treatment of lactam 22 under similar radical conditions also afforded 23 in 55% yield.
5-[2-(2-Bromo-4,5-dimethoxyphenyl)ethyl]-2,2-dimethyl-6-oxo-3a,5,6,7,8,8a-hexahydro[1,3]dioxolo[4,5-f]indole-7a-carboxylic Acid Methyl Ester (24)
To a solution of 0.2 g (0.74 mmol) of acetonide 11 in 3 mL of DMF was added 0.04 g (0.97 mmol) 60% NaH at 0 °C. After the mixture was stirred at 0 °C for 1 h, 0.43 g (1.33 mmol) of 1-bromo-2-(2-bromoethyl)-4,5-dimethoxybenzene was added. The reaction mixture was stirred from 0 °C to room temperature overnight and then quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layer was washed with brine and dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by flash column chromatography to afford 0.2 g (53%) of 24 as a clear oil: 1H-NMR (CDCl3, 600 MHz) δ 1.35 (s, 3H), 1.43 (s, 3H), 1.55 (dd, 1H, J =12.6 and 10.2 Hz), 2.43 (d, 1H, J = 16.2 Hz), 2.63 (dd, 1H, J =12.6 and 6.6 Hz), 2.77 (d, 1H, J = 16.2 Hz), 2.81 (ddd, 1H, J = 13.8, 10.2, and 6.6 Hz), 2.96 (ddd, 1H, J = 13.8, 10.2 and 6.6 Hz), 3.42-3.51 (m, 1H), 3.61 (s, 3H), 3.76-3.84 (m, 1H), 3.78 (s, 3H), 3.80 (s, 3H), 4.48 (dt, 1H, J = 10.2 and 6.6 Hz), 4.70 (dd, 1H, J = 6.6 and 3.0 Hz), 5.29 (d, 1H, J = 3.0 Hz), 6.74 (s, 1H) and 6.96 (s, 1H); 13C-NMR (CDCl3, 150 MHz) δ 25.2, 28.0, 33.0, 35.1, 40.0, 41.0, 47.5, 53.1, 56.2, 60.5, 71.2, 72.1, 97.6, 109.3, 113.6, 114.3, 115.6, 129.4, 143.5, 148.5, 148.6, 172.3 and 172.8.
(7aR,Z)-Methyl 1,2-(4,5-dimethoxy)benzo-10,10-dimethyl-6-oxo-3,4,51,6,7,7a,-8,8a,11a,11b-decahydroazepino[3,2,1-hi][1,3]dioxolo[4,5-f]indole-7a-carboxylate (25)
To a solution of 0.1 g (0.2 mmol) of bromo-lactam 24 in 20 mL of benzene, was added 8.0 mg (0.05 mmol) of AIBN followed by 0.067 mL (0.25 mmol) of Bu3SnH. The mixture was heated to reflux for 10h, cooled to room temperature and poured into 10 mL of an aqueous KF solution (1M). The aqueous layer was extracted with EtOAc and the combined organic layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash silica gel chromatography to give 0.062 g (72%) of 25 as a white solid: mp 138-140 °C; IR (neat) 1732, 1689, 1516, 1214 and 730 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 1.35 (s, 3H), 1.38 (s, 3H), 2.01 (dd, 1H, J = 13.6 and 9.2 Hz), 2.44 (d, 1H, J = 17.6 Hz), 2.52-2.60 (m, 2H), 2.71 (d, 1H, J = 17.6 Hz), 2.71-2.74 (m, 1H), 2.88-2.96 (m, 2H), 3.75 (s, 3H), 3.75-3.81 (m, 1H), 3.84 (s, 3H), 3.88 (s, 3H), 4.42 (dt, 1H, J = 9.6 and 7.2 Hz), 4.51 (ddd, 1H, J = 13.6, 5.2 and 1.6 Hz), 4.57 (dd, 1H, J = 11.6 and 7.2 Hz), 6.67 (s, 1H) and 7.00 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ 25.2, 27.8, 34.2, 35.0, 42.1, 43.0, 44.5, 45.6, 53.2, 56.0, 56.3, 63.6, 71.2, 73.3, 109.2, 111.2, 112.7, 130.8, 133.9, 147.5, 147.6, 171.1, and 175.0; HRMS Calcd. for C23H29NO7: 431.1944. Found: 431.1943.
1-[2-(3,4-Dimethoxyphenyl)ethyl]-2,5-dioxo-1,2,3,4,5,6-hexahydroindole-3a-carboxylic Acid Methyl Ester (27)
To a solution containing 0.38 g (0.88 mmol) of 20 in 5 ml of CH2Cl2 was added 1 mL of TFA. The reaction mixture was stirred at room temperature for 30 min and was then concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give 0.34 g (93%) of 27 as a pale yellow oil: IR (neat) 2954, 1724, 1677, 1516, 1027 and 729 cm-1; 1H-NMR (CDCl3, 100 MHz) δ 2.40 (d, 1H, J = 16.4 Hz), 2.45 (d, 1H, J = 16.4 Hz), 2.80 (t, 2H, J = 8.0 Hz), 2.97 (dd, 1H, J = 22.4 and 6.0 Hz), 3.03 (d, 1H, J = 8.8 Hz), 3.07 (d, 1H, J = 8.8 Hz), 3.12 (dd, 1H, J = 22.4 and 3.6 Hz), 3.54 (dt, 1H, J = 14.8 and 8.0 Hz), 3.68 (s, 3H), 3.86 (s, 3H), 3.88 (s, 3H), 3.89 (dt, 1H, J = 14.8 and 8.0 Hz), 5.07 (dd, 1H, J = 6.0 and 3.6 Hz), 6.73 (d, 1H, J = 8.4 Hz), 6.75 (s, 1H) and 6.79 (d, 1H, J = 8.4 Hz); 13C-NMR (CDCl3, 100 MHz) δ 32.4, 37.0, 40.1, 41.7, 46.5, 47.6, 53.4, 56.1, 56.2, 95.3, 111.4, 112.1, 120.9, 130.6, 140.4, 148.0, 149.2, 171.7, 172.7 and 205.6; HRMS Calcd. for C20H23NO6: 373.1525. Found: 373.1524.
11,12-Dimethoxy-3,6-dioxo-1,2,3,4,5,6,8,9-octahydroindolo[7a,1-a]isoquinoline-4a-carboxylic Acid Methyl Ester (28)
To 0.1 g (0.26 mmol) sample of 27 was added 1.5 mL of PPA. The reaction mixture was heated at 70 °C for 2 h and then cooled to room temperature and quenched with water. The aqueous layer was extracted with CHCl3 and the combined organic layer was dried over MgSO4. Concentration under reduced pressure followed by purification of the residue by silica gel chromatography afforded 0.09 g (90%) of 28 as a clear oil: IR (neat) 1720, 1686, 1260 and 751 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 2.30 (d, 1H, J = 18.0 Hz), 2.34 (s, 2H), 2.29-2.39 (m, 1H), 2.41-2.48 (m, 1H), 2.61 (dd, 1H, J = 15.2 and 5.2 Hz), 2.79 (d, 1H, J = 17.6 Hz), 2.84 (d, 1H, J = 18.0 Hz), 2.87 (dd, 1H, J = 15.2 and 3.2 Hz), 2.97 (dd, 1H, J = 12.8 and 3.2 Hz), 3.07 (s, 3H), 3.28 (d, 1H, J = 17.6 Hz), 3.82 (s, 6H), 4.42 (dd, 1H, J = 12.8 and 5.2 Hz), 6.55 (s, 1H) and 6.56 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ 28.5, 34.6, 35.2, 35.7, 42.0, 45.2, 50.8, 52.2, 56.0, 56.2, 61.1, 108.6, 111.5, 127.3, 129.7, 147.4, 148.2, 170.8, 172.7 and 208.9; HRMS Calcd. for C20H23NO6 : 373.1525. Found: 373.1516.
11,12-Dimethoxy-1,2,8,9-tetrahydro-5H-indolo[7a,1-a]isoquinoline-3,6-dione (30)
To a mixture containing 0.2 g (0.56 mmol) of 28 in 6 mL of MeOH was added 2 mL of 2M NaOH. The mixture was heated at 90 °C for 12 h, cooled to room temperature, and acidified to pH = 3 with HCl. The mixture was extracted with EtOAc and the combined organic layer was dried over MgSO4. The solvent was removed under reduced pressure to give 0.14 g (75%) of 11,12-dimethoxy-3,6-dioxo-1,2,3,4,5,6,8,9-octahydroindolo[7a,1-a]isoquinoline-4a-carboxylic acid (29) as a pale yellow oil: IR (neat) 2935, 1718, 1635 and 1260 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 2.18 (d, 1H, J = 18.0 Hz), 2.24-2.34 (m, 1H), 2.31 (s, 2H), 2.57 (dd, 1H, J = 15.6 and 3.2 Hz), 2.74 (d, 1H, J = 18.0 Hz), 2.78 (d, 1H, J = 17.6 Hz), 2.84 (ddd, 1H, J = 15.6, 12.8 and 5.2 Hz), 2.97 (td, 1H, J = 12.8 and 3.2 Hz), 3.22 (d, 1H, J = 17.6 Hz), 3.77 (s, 3H), 3.79 (s, 3H), 4.33 (dd, 1H, J = 12.8 and 5.2 Hz), 6.51 (s, 1H) and 6.57 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ 28.4, 34.9, 35.2, 35.9, 42.2, 45.0, 50.7, 56.0, 66.3, 108.6, 111.7, 127.4, 129.7, 147.4, 148.3, 171.6, 174.9 and 209.1; HRMS Calcd. for C19H21NO6: 359.1369. Found: 359.1363.
To a solution of 0.04 g (0.1 mmmol) of the above acid in 3 mL of CH2Cl2 was added 10 μL (0.12 mmol) of oxalyl chloride. The reaction mixture was stirred at room temperature for 1 h and then concentrated under reduced pressure. The crude residue was taken up in 2 mL of CBrCl3 and 18 mg (0.12 mmol) of 3-mercapto-pyridine-1-oxide sodium salt and 2 mg of AIBN was added. The mixture was heated at reflux for 2 h, cooled to room temperature and concentrated under reduced pressure. The resulting residue was purified by flash silica gel chromatography to give 0.02 g (75%) of 30 as a pale yellow oil: IR (neat) 2935, 1689, 1684 and 1512 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 2.30-2.37 (m, 1H), 2.45-2.58 (m, 3H), 2.89 (ddd, 1H, J = 16.8, 8.0 and 4.0 Hz), 3.12 (dt, 1H, J = 16.8 and 8.0 Hz), 3.42 (dt, 1H, J = 13.2 and 8.0 Hz), 3.77 (s, 3H), 3.86 (s, 3H), 4.34 (ddd, 1H, J = 13.2, 8.0 and 4.0 Hz), 6.25 (s, 1H), 6.55 (s, 1H) and 6.70 (s, 1H); HRMS Calcd for C18H19NO4: 313.1314. Found: 313.1310.
3-Demethoxyerythratidinone (1)
A 0.02 g sample of 30, 0.5 mL of ethylene glycol and a catalytic amount of p-TsOH in 5 mL of benzene was heated at reflux for 7 h. The cooled mixture was washed with water, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel chromatography and immediately used in the next step. A 0.02 sample of sublimed AlCl3 in 0.4 mL of ether was added to a solution of 0.02 g of LiAlH4 in 5 mL of THF at -15 °C and the mixture was stirred at this temperature for 30 min. The resulting solution was added to a stirred solution of the acetonide derived from 30 in 3 mL of THF. After stirring for 1 h at 25 °C, the mixture was diluted with ether and the excess LiAlH4 was decomposed by the addition of 5% aqueous NH4OH. The ether layer was washed with water, dried over sodium sulfate and concentrated under reduced pressure. The resulting residue was taken up in 1 mL of acetone and 0.5 mL of 5% HCl was added and the mixture was heated at reflux for 1 h. The cooled mixture was washed with 10% NaOH, extracted into CHCl3, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue was crystallized from benzene-hexane to give 0.01 g of 3-demethoxyerythratidinone (1) as a colorless solid, mp 100-101 °C (lit28a 101-102 °C) and whose NMR spectrum matched that reported in the literature.28a
5-(tert-Butyl-dimethylsilanyloxy)-1-[2-(3,4-dimethoxyphenyl)ethyl]-1,4,5,6-tetrahydroindol-2-one (33)
To a solution containing 0.04 g (0.17 mmol) of dienyl alcohol 729 in 1 mL of DMF was added 0.03 g (0.38 mmol) of imidazole at 0 °C followed by 0.03 g (0.19 mmol) of tert-butyldimethylsilyl chloride. The mixture was heated at 50 °C for 2 h and was then diluted with EtOAc. The mixture was washed by water and the aqueous layer was extracted with EtOAc. The combined organic layer was washed with brine and dried over MgSO4. The solvent was removed under reduced pressure and the crude residue was purified by flash column chromatography to give 0.06 g (92%) of 5-(tert-butyl-dimethyl-silanyloxy)-2-oxo-2,4,5,6-tetrahydro-indole-1-carboxylic acid tert-butyl ester as a white solid: mp 118-119 °C; IR (neat) 2960, 1777, 1736, 1322, 1080 and 866 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 0.06 (s, 6H), 0.86 (s, 9H), 1.56 (s, 9H), 2.34 (ddd, 1H, J = 17.5, 8.3 and 3.4 Hz), 2.54 (ddd, 1H, J = 16.2, 10.3 and 1.8 Hz), 2.56-2.61 (m, 1H), 2.86 (dd, 1H, J = 16.2 and 4.4 Hz), 4.00 (ddt, 1H, J = 10.3, 8.3 and 4.4 Hz), 5.73 (s, 1H) and 6.54 (ddd, 1H, J = 6.4, 3.4 and 1.8 Hz); 13C-NMR (CDCl3, 100 MHz) δ -4.6, -4.5, 18.2, 25.9, 28.3, 34.7, 34.8, 67.5, 83.7, 115.8, 116.5, 136.1, 149.6, 150.7 and 168.2; Anal. Calcd for C19H31NO4Si: C, 62.43; H, 8.55; N, 3.83. Found: C, 62.50; H, 8.59; N, 3.97.
To a solution containing 0.05 g (0.13 mmol) of above imide in 1 mL of CH3CN was added 5.6 mg (0.03 mmol) of magnesium perchlorate. The mixture was heated at 50 °C for 2 h and was then cooled to 25 °C. The solvent was removed under reduced pressure and the residue was purified by flash silica gel chromatography to give 0.04 g (87%) of 5-(tert-butyl-dimethylsilanyloxy)-1,4,5,6-tetrahydro-indol-2-one as a white solid: mp 97-99 °C; IR (neat) 3716, 2950, 1678, 1095 and 835 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 0.06 (s, 3H), 0.07 (s, 3H), 0.88 (s, 9H), 2.31 (ddd, J = 17.2, 8.4 and 3.2 Hz), 2.50 (dt, 1H, J = 17.2 and 4.4 Hz), 2.52 (ddd, 1H, J = 16.0, 10.2 and 2.0 Hz), 2.87 (dd, 1H, J = 16.0 and 4.4 Hz), 4.03 (ddt, 1H, J = 10.2, 8.4 and 4.4 Hz), 5.58 (ddt, 1H, J = 4.4, 3.2 and 2.0 Hz), 5.78 (s, 1H) and 8.61 (brs, 1H); 13C-NMR (CDCl3, 100 MHz) δ -4.5, 18.2, 30.0, 34.3, 34.4, 68.7, 109.1, 117.7, 138.3, 148.3 and 173.5; FAB HRMS Calcd for [(C14H23NO2Si)+Li]+: 272.1658. Found: 272.1658.
To a solution containing 0.06 g (0.24 mmol) of the above amide in 3 mL of DMF was added 0.02 g (0.54 mmol) of NaH (60%) at 0 °C. The mixture was stirred at 0 °C for 1.5 h, and then 0.22 g (0.9 mmol) of 4-(2-bromoethyl)-1,2-dimethoxybenzene was added. The reaction mixture was stirred at 25 °C for 12 h and was then quenched by the addition of water. The aqueous layer was extracted with EtOAc and the combined organic layer was washed with brine, dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by flash silica gel chromatography to give 0.07 g (85%) of 33 as a white solid: mp 117-118 °C; IR (neat) 2955, 1687, 1513, 1260 and 861 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 0.06 (s, 3H), 0.07 (s, 3H), 0.87 (s, 9H), 2.24 (ddd, J = 17.2, 8.8 and 3.2 Hz), 2.42-2.52 (m, 2H), 2.78 (t, 2H, J = 7.6 Hz), 2.85 (dd, 1H, J = 16.0 and 4.4 Hz), 3.71 (t, 2H, J = 7.6 Hz), 3.84 (s, 6H), 5.27 (m, 1H), 5.77 (s, 1H), 6.67 (s, 1H), 6.71 (d, 1H, J = 8.0 Hz) and 6.77 (d, 1H, J = 8.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ -4.6, -4.5, 18.2, 25.9, 34.3, 34.5, 34.9, 41.0, 56.0, 56.1, 68.7, 106.7, 111.4, 112.2, 117.1, 120.9, 131.5, 139.7, 146.4, 147.8, 149.1 and 170.5; Anal. Calcd for C24H35NO4Si: C, 67.10; H, 8.21; N, 3.26. Found: C, 67.06; H, 8.16; N, 3.27.
1-Bromo-3-(tert-butyl-dimethylsilanyloxy)-11,12-dimethoxy-1,2,3,4,8,9-hexahydroindolo[7a,1-a]isoquinolin-6-one (34)
To a solution containing 0.05 g (0.12 mmol) of 33 in 5 mL of CH3CN was added 0.02 g (0.12 mmol) of N-bromo-succinimide (NBS). The reaction mixture was stirred at room temperature for 1 h and then quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layer was dried over MgSO4. Concentration under reduced pressure followed by purification by flash silica gel chromatography afforded 0.05 g (89%) of 34 as a white solid: mp 189-190 °C; IR (neat) 2950, 1674, 1508, 1094 and 860 cm-1; 1H-NMR (CDCl3, 600 MHz) δ 0.10 (s, 3H), 0.13 (s, 3H), 0.91 (s, 9H), 2.24-2.33 (m, 2H), 2.86 (ddd, 1H, J = 14.4, 10.2, 1.8 Hz), 2.98 (t, 2H, J = 6.6 Hz), 3.28 (dd, 1H, J = 14.4 and 5.4Hz), 3.94 (dt, 1H, J = 13.2 and 6.6 Hz), 3.88 (s, 3H), 3.89 (s, 3H), 3.91 (dt, 1H, J = 13.2 and 6.6 Hz), 4.39 (tt, 1H, J = 10.2 and 5.4 Hz), 4.56 (t, 1H, J = 3.0 Hz), 6.14 (d, 1H, J = 1.8 Hz), 6.74 (s, 1H) and 7.07 (s, 1H); 13C-NMR (CDCl3, 150 MHz) δ -4.4, -4.5, 18.2, 26.0, 27.7, 36.2, 38.5, 39.5, 55.7, 56.1, 56.5, 67.8, 68.0, 110.2, 112.7, 126.2, 127.3, 128.1, 147.2, 149.1, 156.2 and 169.6; Anal. Calcd for C24H34BrNO4Si: C, 56.69; H, 6.74; N, 2.75. Found: C, 56.65; H, 6.70, N, 2.75.
11,12-Dimethoxy-1,2,8,9-tetrahydroindolo[7a,1-a]isoquinolin-6-one (35)
To a 0.05 g (0.1mmol) sample of bromide 34 in 5 mL of benzene was added 3 mg of AIBN and 43 μL (0.16 mmol) of n-Bu3SnH. The reaction mixture was heated at reflux for 12 h and then cooled to room temperature. Concentration under reduced pressure followed by purification by silica gel chromatography afforded 0.03 g (70%) of 3-(tert-butyl-dimethylsilanyloxy)-11,12-dimethoxy-1,2,3,4,8,9-hexahydroindolo[7a,1a]iso-quinolin-6-one as a colorless oil: IR (neat) 2950, 1687, 1513, 1258, 1092 and 837 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 0.08 (s, 3H), 0.10 (s, 3H), 0.90 (s, 9H), 1.47-1.51 (m, 1H), 1.81-1.87 (m, 2H), 2.31 (dt, 1H, J = 13.0 and 3.5 Hz), 2.83 (dd, 1H, J = 22.6 and 9.6 Hz), 2.87 (dd, 1H, J = 22.6 and 10.5 Hz), 2.96 (dd, 1H, J = 15.6 and 7.6 Hz), 3.15 (dd, 1H, J = 13.7 and 5.7 Hz), 3.38 (ddd, 1H, J = 13.0, 7.9 and 5.7 Hz), 3.76-3.43 (m, 1H), 3.85 (s, 3H), 3.88 (s, 3H), 4.09 (ddd, 1H, J = 13.0, 6.7, 5.4 Hz), 5.91 (d, 1H, J = 1.6 Hz), 6.68 (s, 1H) and 7.11 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ -4.4, 18.2, 26.0, 28.6, 31.1, 36.4, 37.1, 39.7, 56.1, 56.5, 65.7, 72.6, 110.2, 112.6, 122.9, 127.1, 129.6, 146.9, 148.3, 161.9 and 170.1.
To a solution of 0.04 g (0.10 mmol) of the above compound in 1mL of THF was added 0.14 mL (0.14 mmol) of TBAF (1 M). The reaction mixture was stirred at 25 °C for 1 h and was then quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layer was dried over MgSO4 and concentrated under reduced pressure to afford 0.03 g (100%) of 3-hydroxy-11,12-dimethoxy-1,2,3,4,8,9-hexahydroindolo[7a,1-a]isoquinolin-6-one as a colorless oil: IR (neat) 3378, 2936, 1663, 1079 and 730 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 1.53, (td, 1H, J = 13.6 and 4.0 Hz), 1.69 (brs, 1H), 1.83 (tdd, 1H, J = 13.6, 11.2 and 3.6 Hz), 1.96 (dd, 1H, J = 13.6 and 3.6 Hz), 2.36 (dt, 1H, J = 12.8 and 3.6 Hz), 2.45 (d, 1H, J = 3.6 Hz), 2.83-3.00 (m, 3H), 3.29 (ddd, 1H, J = 13.2, 5.6 and 1.6 Hz), 3.42 (dt, 1H, J = 13.2 and 6.4 Hz), 3.85 (s, 3H), 3.88 (s, 3H), 4.05 (dt, 1H, J = 12.8 and 6.4 Hz), 5.96 (d, 1H, J = 1.6 Hz), 6.70 (s, 1H), 7.08 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ 28.5, 30.7, 36.7, 37.2, 39.1, 56.1, 56.6, 65.8, 72.0, 110.0, 112.7, 123.3, 127.2, 129.6, 147.0, 148.4, 161.5 and 170.1; HRMS Calcd. for C18H21NO4: 315.1471. Found: 315.1476.
To a solution containing 0.024 g (0.08 mmol) of the above alcohol in 1.5 mL of CH2Cl2 was added 8.4 μL (0.09 mmol) of POCl3 and 56 μl (0.38 mmol) of DBU. The reaction mixture was stirred at 25 °C for 4 h and was then quenched with a saturated NH4Cl aqueous solution. The aqueous layer was extracted with EtOAc and the organic layer was dried over MgSO4. The solvent was removed under reduced pressure and the residue was purified by flash silica gel chromatography to give 0.016 g (72%) of 35 as a colorless oil: IR (neat) 2929, 1687 and 1513 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 1.84 (dt, 1H, J = 12.0 and 5.6 Hz), 2.14-2.26 (m, 1H), 2.32 (dd, 1H, J = 12.0 and 4.6 Hz), 2.41 (dt, 1H, J = 19.2 and 5.6 Hz), 2.97 (t, 2H, J = 6.8 Hz), 3.55 (dt, 1H, J = 12.8 and 6.8 Hz), 3.76 (s, 3H), 3.84 (s, 3H), 4.03 (dt, 1H, J = 12.8 and 6.8 Hz), 5.88 (s, 1H), 6.28 (ddd, 1H, J = 9.6, 5.6, and 2.0 Hz), 6.69 (s, 1H), 6.81 (dd, 1H, J = 9.6 and 2.8 Hz) and 7.00 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ 24.5, 27.1, 34.9, 37.0, 55.8, 55.9, 64.6, 108.5, 112.0, 118.7, 123.9, 126.2, 128.5, 136.1, 146.8, 148.1 and 158.0; HRMS Calcd. for [(C18H19NO3)+Li]+: 304.1525. Found: 304.1516.
Formic Acid 11,12-Dimethoxy-6-oxo-2,6,8,9-tetrahydro-1H-indolo[7a,1a]-isoquinolin-2-yl Ester (36)
To a solution containing 0.55 g (1.8 mmol) of 35 in 7 mL of 1,4-dioxane at 25 °C was added 2.0 g (18.5 mmol) of selenium dioxide and 0.85 g (18.5 mmol) of formic acid. The reaction mixture was heated at reflux for 7 days with stirring. After cooling to room temperature, 15 mL of a 10% NaOH solution was added and the mixture was extracted with CHCl3. The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Purification of the residue by flash silica gel chromatography provided 0.09 g (14%) of formate 36 as a yellow pale oil: IR (neat) 2933, 1720, 1686 and 1512 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 1.87 (dd, 1H, J = 11.2 and 10.4 Hz), 2.84 (dd, 1H, J = 11.2 and 5.2 Hz), 2.93-3.103 (m, 2H), 3.58 (dt, 1H, J = 12.8 and 6.4 Hz), 3.77 (s, 3H), 3.85 (s, 3H), 4.00 (dt, 1H, J = 12.8 and 7.6 Hz), 5.44-5.52 (m, 1H), 6.08 (s, 1H), 6.17 (d, 1H, J = 10.0 Hz), 6.71 (s, 1H), 6.80 (s, 1H), 6.99 (dd, 1H, J = 10.0 and 2.4 Hz) and 8.02 (d, 1H, J = 1.2 Hz); 13C-NMR (CDCl3, 100 MHz) δ 27.0, 37.3, 41.1, 55.9, 56.1, 65.9, 68.1, 107.8, 112.4, 121.3, 125.6, 126.5, 127.8, 133.5, 147.2, 148.7, 155.9, 159.9 and 170.6; HRMS Calcd. for C19H19NO5: 341.1263. Found: 341.1263.
Another fraction isolated from the column contained 0.09 g (15%) of 2-hydroxy-11,12-dimethoxy-1,2,8,9-tetrahydroindolo[7a,1a]isoquinolin-6-one (37) which was isolated as a tan solid: mp 230-232 °C; IR (KBr) 3419, 2927, 1680 and 1510 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 1.70 (dd, 1H, J = 11.6 and 10.0 Hz), 2.11 (brs, 1H), 2.81 (dd, 1H, J = 11.6 and 4.8 Hz), 2.90-3.12 (m, 2H), 3.60 (ddd, 1H, J = 12.8, 6.8 and 5.2 Hz), 3.75 (s, 3H), 3.85 (s, 3H), 3.91-4.02 (m, 1H), 4.30 (brs, 1H), 6.02 (s, 1H), 6.30 (d, 1H, J = 10.0 Hz), 6.71 (s, 1H), 6.79 (s, 1H) and 6.87 (dd, 1H, J = 10.0 and 2.4 Hz); 13C-NMR (CDCl3, 100 MHz) δ 27.0, 37.4, 45.0, 55.9, 56.1, 66.4, 66.6, 108.0, 112.2, 120.1, 123.5, 126.4, 128.4, 139.4, 147.0, 148.5, 157.3 and 171.0; HRMS Calcd. for C18H19NO4: 313.1314. Found: 313.1305.
Alcohol 37 could also be obtained from the solvolysis of formate 36. To a solution containing 0.002 g (0.064 mmol) of 36 in 3 mL of EtOH was added 0.1 mL of acetyl chloride at 25 °C. The reaction mixture was stirred for 1 h at 25 °C. Removal of the solvent under reduced pressure afforded 0.002 g (100%) of 37 as the exclusive product.
(±)-Erysotramidine (2)
To a mixture containing 0.09 g (0.27 mmol) of alcohol 37 in 7 mL of THF and 4 mL of methyl iodide was added 0.18 g (3.2 mmol) of NaOH and 0.17 g (0.8 mmol) of tetra-ethylammonium bromide. The reaction mixture was stirred for 36 h at 25 °C. The solution was poured into ice-water and the resulting mixture was extracted with CHCl3. The combined organic layer was dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. Purification of the residue by preparative TLC provided 0.08 g (91%) of (±)-erysotramidine (2) as a colorless oil19a ; IR (neat) 2930, 1666 and 1518 cm-1; 1H-NMR (CDCl3, 400 MHz) δ 1.71 (dd, 1H, J = 11.6 and 10.4 Hz), 2.80 (dd, 1H, J = 11.6 and 4.4 Hz), 2.90-3.24 (m, 2H), 3.34 (s, 3H), 3.61 (ddd, 1H, J = 12.8, 7.2 and 6.0 Hz), 3.76 (s, 3H), 3.86 (s, 3H), 3.82-3.88 (m, 1H), 4.00 (ddd, 1H, J = 12.8, 8.4 and 7.2 Hz), 6.02 (s, 1H), 6.33 (d, 1H, J = 10.0 Hz), 6.72 (s, 1H), 6.80 (s, 1H) and 6.90 (dd, 1H, J = 10.0 and 2.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 27.0, 37.2, 41.3, 55.8, 56.0, 56.3, 66.3, 74.8, 108.1, 120.2, 124.1, 126.5, 128.6, 136.2, 146.9, 148.5, 157.0 and 170.8; HRMS Calcd. for [(C19H21NO4)+Li]+: 334.1631. Found: 334.1633.
Preparation of (3aR,7aS,8aS)-5-(6-Iodobenzo[d][1,3]dioxole-5-carbonyl)-2,2-dimethyl-7,7a,8,8a-tetrahydro-3aH-[1,3]dioxolo[4,5-f]indol-6(5H)-one (44)
To a solution containing 0.05 g (0.17 mmol) of tert-butyl-2,3,3a,4,5,6-hexahydro-5,6-acetonide-2-oxoindole-1-carboxylate (8)29 in 1 mL of CH3CN was added 8 mg (0.034 mmol) of magnesium perchlorate. The reaction mixture was heated at 40°C for 2 h and cooled to rt. Concentration under reduced pressure and purification by silica gel chromatography provided the deprotected amide as a white solid: mp 151-153°C; IR (neat) 3170, 1683,1324, and 876 cm-1; 1H-NMR (CDCl3 400 MHz) δ 1.37 (s, 3H), 1.52 (q, 1H, J = 11.6 Hz), 1.48 (s, 3H), 2.15 (dt, 1H, J = 11.6 and 5.0 Hz), 2.24 (dd, 1H, J = 16.8 and 9.6 Hz), 2.56 (dd, 1H, J = 16.8 and 9.2 Hz), 2.72-2.81 (m, 1H), 4.23 (dt, 1H, J = 11.6 and 5.0 Hz), 4.61-4.64 (m, 1H), 5.11 (t, 1H, J = 3.2 Hz), and 8.29 (brs, 1H); 13C-NMR (CDCl3, 100 MHz) δ 26.0, 28.6, 32.4, 34.0, 36.0, 71.7, 74.0, 94.9, 109.2, 145.8, and 177.7; Anal. Calcd. for C11H15NO3: C, 63.14; H, 7.23; N, 6.69. Found: C, 63.02; H, 7.21; N, 6.62.
To a solution of 0.03 g (0.12 mmol) of the above amide in 1 mL of CH2Cl2, was added 1.5 mg (0.001 mmol) of dimethylaminopyridine, 0.08 mL (0.58 mmol) of Et3N and 6-iodobenzo[d][1,3]dioxole-5-carbonyl chloride51 in 1 mL of CH2Cl2. The reaction mixture was stirred for 2 h, quenched with water and washed with EtOAc. The combined organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was subjected to flash silica gel chromatography to afford 0.06 g (98%) of 44 as a white solid: mp 82-84°C; IR (neat) 1770, 1698,1240, and 876 cm-1; 1H-NMR (CDCl3 400 MHz) δ 1.42 (s, 3H), 1.52 (q, 1H, J = 11.6 Hz), 1.51 (s, 3H), 2.24 (dt, 1H, J = 11.6 and 4.8 Hz), 2.36 (dd, 1H, J = 17.2 and 10.6 Hz), 2.70 (dd, 1H, J = 17.2 and 8.8 Hz), 2.79-2.88 (m, 1H), 4.29 (dt, 1H, J = 11.6 and 4.8 Hz), 4.75-4.78 (m, 1H), 6.02 (s, 2H), 6.57 (t, 1H, J = 3.2 Hz), 6.75 (s, 1H), and 7.20 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ 25.9, 28.5, 32.3, 33.8, 36.8, 71.4, 73.0, 81.3, 102.4, 106.2, 109.0, 118.9, 134.9, 142.5, 148.5, 150.0, 168.2, and 173.7; Anal. Calcd. for C19H18INO6: C, 47.22; H, 3.75; N, 2.90. Found: C, 47.05; H, 3.79; N, 2.98.
Preparation of (3aS,4aS,4a1R,13bS,13cR)-2,2-Dimethyl-4a,5,13b,13c-tetrahydro-3aH-bis[1,3]dioxolo[4,5-a:4’,5’-j]pyrrolo[3,2,1-de]phenanthridine-6,8(4H,4a1H)-dione (45)
To a solution of containing 0.1 g (0.2 mmol) of imide 44 in 40 mL benzene was added 0.017 g (0.1 mmol) of AIBN and 0.11 mL (0.4 mmol) of n-Bu3SnH. The solution was heated at reflux for 3 h and cooled to rt. Concentration under reduced pressure followed by purification using silica gel chromatography provided 0.04 g (55%) of the major product 45 as a white solid, mp 217-220 °C; IR (neat) 1758, 1481,1275, 927 and 758 cm-1; 1H-NMR (CDCl3 400 MHz) δ 1.43 (s, 3H), 1.53 (s, 3H), 1.60-1.69 (m,1H), 2.28 (dt, 1H, J = 12.4 and 5.2 Hz), 2.44 (dd, 1H, J = 17.2 and 7.2 Hz), 2.46 (m, 1H), 2.94 (dd, 1H, J = 17.2 and 10.0 Hz), 3.13 (dd, 1H, J = 13.2 and 8.8 Hz), 3.97 (dd, 1H, J = 13.2 and 10.0 Hz), 4.24-4.39 (m, 2H), 6.04 (d, 1H, J = 1.2 Hz), 6.05 (d, 1H, J =1.2 Hz), 7.27 (s, 1H) and 7.55 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ 24.4, 25.3, 27.2, 32.8, 38.2, 41.5, 58.5, 73.8, 75.0, 102.3, 106.8, 109.3, 123.4, 137.7, 147.7, 152.7, 161.5, and 172.7; FAB HRMS Calcd for [(C19H19NO6)+Li]+: 364.1372. Found: 364.1382.
epi-Zephyranthine (9)
To a solution containing 0.02 g (0.055 mmol) of compound 45 was added 0.82 mL (0.82 mmol) of BH3 (1.0 M in THF) at 0 °C. The reaction mixture was stirred at 0 °C for 24 h and was then quenched with 3 mL of MeOH. Concentration of the solution under reduced pressure followed by purification using silica gel chromatography provided 0.012 g (72%) of the expected amino-dioxolane as a white solid: mp 170°C (dec); IR (neat) 1483, 1166 and 1053 cm-1; 1H-NMR (CDCl3 600 MHz) δ 1.44 (s, 3H), 1.50 (s, 3H), 1.53 (q, 1H, J = 12.6 Hz), 1.70-1.76 (m, 1H), 2.16 (dt, 1H, J = 12.6 and 6.0 Hz), 2.50-2.58 (m,1H), 2.61 (dd, 1H, J = 12.6 and 9.6 Hz), 2.84 (dd, 1H, J = 12.6 and 9.0 Hz), 3.02 (dt, 1H, J = 11.4 and 7.2 Hz), 3.47 (p, 1H, J = 6.0 Hz), 3.59 (d, 1H, J = 12.6 Hz), 4.14 (d, 1H, J = 12.6 Hz), 4.25 (ddd, 1H, J = 11.4, 7.2 and 4.8 Hz), 4.31 (dd, 1H, J = 9.6 and 7.2 Hz), 5.93 (d, 1H, J = 1.2 Hz), 5.94 (d, 1H, J = 1.2 Hz), 6.74 (s, 1H), and 7.07 (s, 1H); 13C-NMR (CDCl3, 100 MHz) δ 24.4, 27.2,31.2, 31.8, 33.4, 38.7, 62.2, 62.7, 68.9, 73.2, 75.0, 101.3, 106.1, 108.5, 109.3, 127.1, 131.0, 146.5, and 147.9; FAB HRMS Calcd for [(C19H23NO4)+Li]+: 336.1787. Found: 336.1780.
A 0.01 g (0.03 mmol) sample of the above dioxolane in 2 mL of a 3% HCl-MeOH mixture was stirred at 0 °C for 1 h and was then concentrated under reduced pressure. Purification by silica gel chromatography provided 0.008 g (90%)of 9 as a white solid: mp 215-218 °C; IR (neat) 3365, 1483 and 1031 cm-1; 1H-NMR (CDCl3, 600 MHz) δ 1.90 (ddd, 1H, J = 15.0, 6.6 and 3.0 Hz), 2.07 (dt, 1H, J = 12.6 and 6.6 Hz), 2.20 (dt, 1H, J = 15.0 and 3.0 Hz), 2.28 (d, 1H, J = 3.0 Hz), 2.32 (d, 1H, J = 6.6 Hz), 2.60 (qd, 1H, J = 12.6 and 7.8 Hz), 2.68 (dqd, 1H, J = 12.6, 6.6 and 3.0 Hz), 2.85 (dd, 1H, J = 10.8 and 6.6 Hz), 3.08 (dd, 1H, J = 12.6 and 7.8 Hz), 3.15 (t, 1H, J = 10.8 Hz), 3.63 (d, 1H, J = 13.2 Hz), 3.74 (td, 1H, J = 12.6 and 6.6 Hz), 4.04 (ddd, 1H, J = 10.8, 6.6 and 3.0 Hz), 4.15 (p, 1H, J = 3.0 Hz), 4.63 (d, 1H, J = 13.2 Hz), 5.96 (d, 1H, J = 1.8 Hz), 5.97 (d,1H, J = 1.8 Hz), 6.79 (s, 1H), and 7.14 (s, 1H); 13C-NMR (CDCl3, 150 MHz) δ 29.3, 30.7, 34.8, 36.6, 58.3, 62.4, 69.4, 70.4, 71.0, 101.3, 105.6, 109.2, 126.7, and 130.5; HRMS Calcd for C16H19NO4: 289.1314. Found: 289.1312.
Supplementary Material
1H and 13C NMR data of various key compounds lacking CHN analyses together with an ORTEP drawing for compounds 16 and 45 as well as the corresponding CIFs for these compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 2.

Lycorine-type Alkaloids
Acknowledgments
This research was supported by the National Institutes of Health (GM 0539384) and the National Science Foundation (CHE-0450779). We thank our colleague, Dr. Kenneth Hardcastle, for his assistance with the X-ray crystallographic studies.
References and Notes
- 1.(a) Martin SF. In: The Alkaloids. Brossi A, editor. Vol. 30. Academic Press Inc.; New York: 1987. pp. 251–377. [Google Scholar]; (b) Lewis JR. Nat Prod Rep. 1994;11:329. doi: 10.1039/np9941100329. [DOI] [PubMed] [Google Scholar]; Jeffs PW. In: The Alkaloids. Rodrigo RGA, editor. Vol. 19. Academic Press; New York: 1981. pp. 1–80. [Google Scholar]
- 2.(a) Cordell GA. In: The Alkaloids. Manske RHF, Rodrigo RGA, editors. Vol. 17. Academic Press; New York: 1979. pp. 199–384. [Google Scholar]; (b) Saxton JE. Nat Prod Rep. 1993;10:349–395. [Google Scholar]
- 3.(a) Boekelheide V. Alkaloids. 1960;7:201. [Google Scholar]; (b) Hill RK. In: The Alkaloids. Manske RHF, editor. Vol. 9. Academic Press; New York: 1967. p. 483. [Google Scholar]
- 4.(a) Massiot G, Delaude C. In: The Alkaloids. Brossi A, editor. Vol. 34. Academic Press; San Diego, CA: 1988. pp. 211–329. [Google Scholar]; (b) Aimi N, Sakai S, Ban Y. In: The Alkaloids. Brossi A, editor. Vol. 36. Academic Press; San Diego, CA: 1989. pp. 1–68. [Google Scholar]; (c) Bosch J, Bonjoch J, Amat M. In: The Alkaloids. Brossi A, editor. Vol. 48. Academic Press; San Diego, CA: 1996. pp. 75–189. [Google Scholar]
- 5.Dyke SF, Quessy SN. In: The Alkaloids. Rodrigo RGA, editor. Vol. 18. Academic Press; New York: 1981. pp. 1–98. [Google Scholar]
- 6.Chawla AS, Kapoor VK. In: The Alkaloids: Chemical and Biological Perspectives. Pelletier SW, editor. Vol. 9. Pergamon; 1995. pp. 86–153. [Google Scholar]
- 7.Deulofeu V. In: Curare and Curarelike Agents. Bovet D, Bovet-Nitti F, Marini-Bettolo GB, editors. Elsevier; Amsterdam: 1959. p. 163. [Google Scholar]
- 8.For some leading references, see: Banwell MG, Wu AW. J Chem Soc Perkin Trans. 1994;1:2671.Grothjahn DB, Vollhardt KPC. Synthesis. 1993:579.Pearson WH, Schkeryantz JM. J Org Chem. 1992;57:6783.Backvall JE, Andersson PG, Stone GB, Gogoll A. J Org Chem. 1991;56:2988.Jolly RS, Livinghouse T. J Am Chem Soc. 1988;110:7536.Stork G, Mah R. Heterocycles. 1989;28:723.
- 9.(a) Kuehne ME, Matsko TH, Bohnert JC, Motyka L, Oliver-Smith D. J Org Chem. 1981;46:2002. [Google Scholar]; (b) Kuehne ME, Earley WG. Tetrahedron. 1983;39:3707. [Google Scholar]; (c) Kuehne ME, Brook CS, Xu F, Parsons R. Pure Appl Chem. 1994;66:2095. [Google Scholar]; (d) Nkiliza J, Vercauteren J. Tetrahedron Lett. 1991;32:1787. [Google Scholar]; (e) Rawal VH, Michoud C, Monestel RF. J Am Chem Soc. 1993;115:3030. [Google Scholar]
- 10.(a) Overman LE, Sugai S. Helv Chim Acta. 1985;68:745. [Google Scholar]; (b) Overman LE, Robertson G, Robichaud AJ. J Org Chem. 1989;54:1236. [Google Scholar]; (c) Overman LE, Sworin M, Burk RM. J Org Chem. 1983;48:2685. [Google Scholar]
- 11.(a) Magnus P, Gallagher T, Brown P, Pappalardo P. Acc Chem Res. 1984;17:35. [Google Scholar]; (b) Gallagher T, Magnus P, Huffman JC. J Am Chem Soc. 1983;105:4750. [Google Scholar]; (c) Desmaële D, d’Angelo J. J Org Chem. 1994;59:2292. [Google Scholar]
- 12.(a) Kraus GA, Thomas PJ, Bougie D, Chen L. J Org Chem. 1990;55:1624. [Google Scholar]; (b) Sole D, Bonjoch J. Tetrahedron Lett. 1991;32:5183. [Google Scholar]; (c) Bonjoch J, Sole D, Bosch J. J Am Chem Soc. 1993;115:2064. [Google Scholar]; (d) Schultz AG, Guzzo PR, Nowak DM. J Org Chem. 1995;60:8044. [Google Scholar]; (e) Schultz AG, Holoboski MA, Smyth MS. J Am Chem Soc. 1993;115:7904. [Google Scholar]; (f) Schultz AG, Holoboski MA, Smyth MS. J Am Chem Soc. 1996;118:6210. [Google Scholar]
- 13.(a) Padwa A, Price AT. J Org Chem. 1995;60:6258. [Google Scholar]; (b) Pearson WH, Postich MJ. J Org Chem. 1994;59:5662. [Google Scholar]
- 14.(a) Rigby JH, Qabar M, Ahmed G, Hughes RC. Tetrahedron. 1993;49:10219. [Google Scholar]; (b) Rigby JH, Laurent S, Cavezza A, Heeg MJ. J Org Chem. 1998;63:5587. [Google Scholar]; (c) Rigby JH, Hughes RC, Heeg MJ. J Am Chem Soc. 1995;117:7834. [Google Scholar]; (d) Rigby JH, Mateo ME. Tetrahedron. 1996;52:10569. [Google Scholar]; (e) Rigby JH, Qabar MH. J Org Chem. 1993;58:4473. [Google Scholar]
- 15.(a) Mori M, Kuroda S, Zhang C, Sato Y. J Org Chem. 1997;62:3263. doi: 10.1021/jo9701187. [DOI] [PubMed] [Google Scholar]; (b) Uesaka N, Saitoh F, Mori M, Shibasaki M, Okamura K, Date T. J Org Chem. 1994;59:5633. [Google Scholar]; (c) Takano S, Inomata K, Ogasawara K. Chem Lett. 1992:443. [Google Scholar]
- 16.Tsuda Y, Sano T. In: The Alkaloids. Cordell GA, editor. Vol. 48. Academic Press; San Diego: 1996. pp. 249–337. [Google Scholar]
- 17.Kawasaki T, Onoda N, Watanabe H, Kitahara T. Tetrahedron Lett. 2001;42:8003. [Google Scholar]
- 18.(a) Belleau B. J Am Chem Soc. 1953;75:5765. [Google Scholar]; (b) Mondon A, Hansen KF. Tetrahedron Lett. 1960:5. [Google Scholar]; (c) Ishibashi H, Sato T, Takahashi M, Hayashi M, Ikeda M. Heterocycles. 1988;27:2787. [Google Scholar]; (d) Tsuda Y, Hosoi S, Ishida K, Sangai M. Chem Pharm Bull. 1994;42:204. [Google Scholar]; (e) Cassayre J, Quiclet-Sire B, Saunier J–B, Zard SZ. Tetrahedron Lett. 1998;39:8995. [Google Scholar]; (f) Rigby JH, Deur C, Heeg MJ. Tetrahedron Lett. 1999;40:6887. [Google Scholar]; (g) Parsons AF, Williams DA. Tetrahedron. 2000;56:7217. [Google Scholar]; (h) Toyao A, Chikaoka S, Takeda Y, Tamura O, Muraoka O, Tanabe G, Ishibashi H. Tetrahedron Lett. 2001;42:1729. [Google Scholar]; (i) Miranda LD, Zard SZ. Org Lett. 2002;4:1135. doi: 10.1021/ol025534e. [DOI] [PubMed] [Google Scholar]; (j) Allin SM, James SL, Elsegood MRJ, Martin WP. J Org Chem. 2002;67:9464. doi: 10.1021/jo0205661. [DOI] [PubMed] [Google Scholar]
- 19.(a) Toda J, Niimura Y, Takeda K, Sano T, Tsuda Y. Chem Pharm Bull. 1998;46:906. [Google Scholar]; (b) Jousse C, Demaële D. Eur J Org Chem. 1999:909. [Google Scholar]
- 20.Wasserman HH, Amici RM. J Org Chem. 1989;54:5843. [Google Scholar]
- 21.(a) Sano T, Toda J, Kashiwaba N, Ohshima T, Tsuda Y. Chem Pharm Bull. 1987;35:479. [Google Scholar]; (b) Tsuda Y, Hosoi S, Katagiri N, Kaneko C, Sano T. Heterocycles. 1992;33:497. [Google Scholar]
- 22.(a) Danishefsky SJ, Panek JS. J Am Chem Soc. 1987;109:917. [Google Scholar]; (b) Ahmed-Schofield R, Mariano PS. J Org Chem. 1987;52:1478. [Google Scholar]; (c) Irie H, Shibata K, Matsuno K, Zhang Y. Heterocycles. 1989;29:1033. [Google Scholar]; (d) Kawasaki T, Onoda N, Watanabe H, Kitahara T. Tetrahedron Lett. 2001;42:8003. [Google Scholar]; (e) Yasui Y, Suzuki K, Matsumoto T. Synlett. 2004:619. [Google Scholar]
- 23.(a) Gervay JE, McCapra F, Money T, Sharma GM. J Chem Soc Chem Commun. 1966:142. [Google Scholar]; (b) Tanaka H, Shibata M, Ito K. Chem Pharm Bull. 1984;32:1578. doi: 10.1248/cpb.33.1419. [DOI] [PubMed] [Google Scholar]; (c) Chou CT, Swenton JS. J Am Chem Soc. 1987;109:6898. [Google Scholar]
- 24.(a) Mondon A, Ehrhardt M. Tetrahedron Lett. 1966:2557. [Google Scholar]; (b) Haruna M, Ito K. J Chem Soc Chem Commun. 1976:345. [Google Scholar]; (c) Ishibashi H, Sato K, Ikeda M, Maeda H, Akai S, Tamura Y. J Chem Soc Perkin Trans. 1985;1:605. [Google Scholar]; (d) Westling M, Smith R, Livinghouse T. J Org Chem. 1986;51:1159. [Google Scholar]; (e) Chikaoka S, Toyao A, Ogasawara M, Tamura O, Ishibashi H. J Org Chem. 2003;68:312. doi: 10.1021/jo020573p. [DOI] [PubMed] [Google Scholar]
- 25.(a) Stanislawski PC, Willis AC, Banwell MG. Org Lett. 2006;8:2143. doi: 10.1021/ol060642c. [DOI] [PubMed] [Google Scholar]; (b) Fukumoto H, Esumi T, Ishihara J, Hatakeyama S. Tetrahedron Lett. 2003;44:8047. [Google Scholar]; (c) Reimann E, Ettmayr C. Monatsh Chem. 2004;135:1143. [Google Scholar]; (d) Allin SM, Streetly GB, Slater M, James SL, Martin WP. Tetrahedron Lett. 2004;45:5493. [Google Scholar]; (e) Kim G, Kim JH, Lee KY. J Org Chem. 2006;71:2185. doi: 10.1021/jo052500m. [DOI] [PubMed] [Google Scholar]; (f) Blake AJ, Gill C, Greenhalgh DA, Simpkins NS, Zhang F. Synthesis. 2005:3287. [Google Scholar]
- 26.(a) Padwa A, Ginn JD. J Org Chem. 2005;70:5197. doi: 10.1021/jo050515e. [DOI] [PubMed] [Google Scholar]; (b) Padwa A, Bur SK, Zhang H. J Org Chem. 2005;70:6833. doi: 10.1021/jo0508797. [DOI] [PubMed] [Google Scholar]; (c) Zhang H, Padwa A. Org Lett. 2006;8:247. doi: 10.1021/ol052524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barton DHR, Gunatilaka AAL, Letcher RM, Lobo AMFT, Widdowson DA. J Chem Soc Perkin Trans. 1973;1:874. [Google Scholar]
- 28.(a) Tsuda Y, Sakai Y, Nakai A, Kaneko M, Ishiguro Y, Isobe K, Taga J, Sano T. Chem Pharm Bull. 1990;38:1462. [Google Scholar]; (b) Ishibashi H, Sato T, Takahashi M, Hayashi M, Ishikawa K, Ikeda M. Chem Pharm Bull. 1990;38:907. [Google Scholar]; (c) Irie H, Shibata K, Matsuno K, Zhang Y. Heterocycles. 1989;29:1033. [Google Scholar]
- 29.Padwa A, Wang Q. J Org Chem. 2006;71:3210. doi: 10.1021/jo060238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.(a) Padwa A, Brodney MA, Dimitroff M. J Org Chem. 1998;63:5304. doi: 10.1021/jo010020z. [DOI] [PubMed] [Google Scholar]; (b) Bur SK, Lynch SM, Padwa A. Org Lett. 2002;4:473. doi: 10.1021/ol016804g. [DOI] [PubMed] [Google Scholar]; (c) Ginn JD, Padwa A. Org Lett. 2002;4:1515. doi: 10.1021/ol025746b. [DOI] [PubMed] [Google Scholar]
- 31.Dramatic effects on the rate of the Diels-Alder reaction were previously noted to occur when an amido group was used to anchor the diene and dienophile, see: Oppolzer W, Fröstl W. Helv Chim Acta. 1975;58:590. doi: 10.1002/hlca.19750580232.White JD, Demnitz FWJ, Oda H, Hassler C, Snyder JP. Org Lett. 2000;2:3313. doi: 10.1021/ol000200f.Padwa A, Ginn JD, Bur SK, Eidell CK, Lynch SM. J Org Chem. 2002;67:3412. doi: 10.1021/jo0111816.Tantillo DJ, Houk KN, Jung ME. J Org Chem. 2001;66:1938. doi: 10.1021/jo001172h.
- 32.(a) Hayashi T. Synlett. 2001:879. [Google Scholar]; (b) Sakai M, Hayashi H, Miyaura N. Organometallics. 1997;16:4229. [Google Scholar]; (c) Senda T, Ogasawara M, Hayashi T. J Org Chem. 2001;66:6852. doi: 10.1021/jo0103930. [DOI] [PubMed] [Google Scholar]
- 33.Takaya Y, Ogasawara M, Hayashi T, Sakai M, Miyaura N. J Am Chem Soc. 1998;120:5579. [Google Scholar]
- 34.Hayashi T, Senda T, Takaya Y, Ogasawara M. J Am Chem Soc. 1999;121:11591. [Google Scholar]
- 35.Hayashi T, Senda T, Ogasawara M. J Am Chem Soc. 2000;122:10716. [Google Scholar]
- 36.Murakami M, Igawa H. Chem Commun. 2002:390. doi: 10.1039/b108808d. [DOI] [PubMed] [Google Scholar]
- 37.(a) Lautens M, Fagnou K, Rovis T. J Am Chem Soc. 2000;122:5650. [Google Scholar]; (b) Lautens M, Fagnou K, Taylor M. Org Lett. 2000;2:1677. doi: 10.1021/ol005729r. [DOI] [PubMed] [Google Scholar]; (c) Lautens M, Fagnou K. Tetrahedron. 2001;57:5067. [Google Scholar]; (d) Lautens M, Chiu P, Ma S, Rovis T. J Am Chem Soc. 1995;117:532. [Google Scholar]; (e) Lautens M, Chiu P, Colucci JT. Angew Chem Int Ed Engl. 1993;32:281. [Google Scholar]; (f) Lautens M, Di Felice C, Huboux A. Tetrahedron Lett. 1989;30:6817. [Google Scholar]
- 38.Bertounesque E, Florent J-C, Monneret C. Synthesis. 1991:270. [Google Scholar]
- 39.Vyvyan JR, Meyer JA, Meyer KD. J Org Chem. 2003;68:9144. doi: 10.1021/jo035112y. [DOI] [PubMed] [Google Scholar]
- 40.(a) Ito K, Suzuki F, Haruna M. J Chem Soc Chem Commun. 1978:733. [Google Scholar]; (b) Tsuda Y, Hosoi S, Katagiri N, Kaneko C, Sano T. Chem Pharm Bull. 1993;41:2087. [Google Scholar]; (c) Hosoi S, Nagao M, Tsuda Y, Isobe K, Sano T, Ohta T. J Chem Soc Perkin Trans. 2000;1:1505. [Google Scholar]
- 41.(a) Padwa A, Kappe CO, Reger TS. J Org Chem. 1996;61:4888. [Google Scholar]; (b) Padwa A, Hennig R, Kappe CO, Reger TS. J Org Chem. 1998;63:1144. [Google Scholar]; (c) Padwa A, Wang Q. Org Lett. 2006;8:601. doi: 10.1021/ol0527330. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lee HI, Cassidy MP, Rashatasakhon P, Padwa A. Org Lett. 2003;5:5067. doi: 10.1021/ol036106r. [DOI] [PubMed] [Google Scholar]
- 42.The interesting switch in chemoselectivity of 13 as a function of the acid used is not at all obvious. Its possible that the specific rearrangement pathway followed may be related to subtle differences in the site(s) of protonation.
- 43.(a) Neumann WP. Synthesis. 1987:665. [Google Scholar]; (b) Curran DP. Synthesis. 1988:417–489. [Google Scholar]; (c) Giese B. Radicals in Organic Synthesis: Formation of Carbon-Carbon Bonds. Pergamon Press; Oxford: 1986. [Google Scholar]; (d) Studer A, Bossart M. Tetrahedron. 2001;57:9649. [Google Scholar]; (e) Aldabbagh F, Bowman WR. Contemp Org Synth. 1997;4:261. [Google Scholar]; (f) Curran DP. Radical Addition Reactions. In: Trost BM, Fleming I, editors. Comprehensive Organic Synthesis. Vol. 4. Pergamon; Oxford: 1991. p. 715. [Google Scholar]; (g) Renaud P, Sibi ME, editors. Radicals in Organic Synthesis. 1 & 2 Wiley-VCH; Weinheim: 2001. [Google Scholar]
- 44.Beckwith ALJ, Ingold KU. In: Rearrangement in Ground and Excited States. de Mayo P, editor. Academic Press; New York: 1980. [Google Scholar]
- 45.(a) Barton DHR, Togo H, Zard SZ. Tetrahedron. 1985;41:5507. [Google Scholar]; (b) Attardi ME, Taddei M. Tetrahedron Lett. 2001;42:3519. [Google Scholar]
- 46.Lee YS, Lee JY, Kim DW, Park H. Tetrahedron. 1999;55:4631. [Google Scholar]
- 47.(a) Hoshino O. In: The Alkaloids. Cordell GA, editor. Vol. 51. Academic Press; New York: 1998. pp. 323–424. [Google Scholar]; (b) Lewis JR. Nat Prod Rep. 1998:107–110. [Google Scholar]
- 48.(a) Ozeki S. Chem Pharm Bull. 1964;12:2533. [PubMed] [Google Scholar]; (b) Tsuda Y, Sano T, Taga J, Isobe K, Toda J, Takagi S, Yamaki M, Murata M, Irie H, Tanaka H. J Chem Soc Perkin Trans. 1979;1:1358. [Google Scholar]; (c) Yamaki M, Murata M, Takagi S, Tsuda Y, Sano T, Taga J, Isobe K, Tanaka H, Irie H, Uyeo S. Heterocycles. 1976;5:163. [Google Scholar]; (d) Oppolzer W, Spivey AC, Bochet CG. J Am Chem Soc. 1994;116:3139. doi: 10.1021/ja910184j. [DOI] [PubMed] [Google Scholar]
- 49.Rigby JH, Qabar M. J Am Chem Soc. 1991;113:8795. [Google Scholar]
- 50.Schultz AG, Holoboski MA, Smyth MS. J Am Chem Soc. 1996;118:6210. [Google Scholar]
- 51.McIntosh MC, Weinreb SM. J Org Chem. 1993;56:4823. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H and 13C NMR data of various key compounds lacking CHN analyses together with an ORTEP drawing for compounds 16 and 45 as well as the corresponding CIFs for these compounds. This material is available free of charge via the Internet at http://pubs.acs.org.