

Abstract
Adenovirus (Ad) infections stimulate the activation of cellular DNA damage response and repair pathways. Ad early regulatory proteins prevent activation of DNA damage responses by targeting the MRN complex, composed of the Mre11, Rad50 and Nbs1 proteins, for relocalization and degradation. In the absence of these viral proteins, Mre11 colocalizes with viral DNA replication foci. Mre11 foci formation at DNA damage induced by ionizing radiation depends on the Nbs1 component of the MRN complex and is stabilized by the mediator of DNA damage checkpoint protein 1 (Mdc1). We find that Nbs1 is required for Mre11 localization at DNA replication foci in Ad E4 mutant infections. Mre11 is important for Mdc1 foci formation in infected cells, consistent with its role as a sensor of DNA damage. Chromatin immunoprecipitation assays indicate that both Mre11 and Mdc1 are physically bound to viral DNA, which could account for their localization in viral DNA containing foci. Efficient binding of Mre11 to E4 mutant DNA depends on the presence of Nbs1, and is correlated with a significant E4 mutant DNA replication defect. Our results are consistent with a model in which physical interaction of Mre11 with viral DNA is mediated by Nbs1, and interferes with viral DNA replication.
Keywords: Adenovirus, E4 mutant, DNA replication, DNA binding, DNA damage response, MRN complex, Mdc1
Introduction
Adenovirus (Ad) has a 36kbp linear double stranded (ds) DNA genome. Ad infection can potentially trigger DNA damage response cascades that interfere with a productive infectious program (reviewed by Weitzman and Ornelles, 2005). Cellular DNA damage response pathways maintain the genetic integrity and survival of the cell. The surveillance and signal amplification processes induced in response to double strand breaks (DSB), activate DNA repair and cell cycle checkpoint proteins. Ataxia telangiectasia mutated (ATM), AT-and Rad3 related (ATR) and DNA protein kinase (DNA PK) are serine-threonine kinases that are activated in response to unique damage signals. The ATR pathway responds to single stranded DNA (ssDNA) produced as replication proteins stall at the site of damaged DNA, while DNA PK mediates signaling responses that recruit the Ligase IV/Xrcc4 complex to mediate ligation of DSBs in non-homologous end joining (NHEJ) (Shiloh, 2003). The ATM pathway is primarily induced by DNA DSBs and is characterized by the phosphorylation and activation of numerous cellular proteins such as the modified histone protein H2AX, mediator of DNA damage checkpoint protein 1 (Mdc1), Nijmegen break syndrome protein (Nbs1), Breast cancer 1 (BRCA1), the tumor suppressor p53, and checkpoint protein 2 (Chk2) among others, that regulate the cell-cycle checkpoints and DNA repair pathways (Shiloh, 2003). Mdc1 is implicated in the amplification of ATM-dependent DNA damage signals. It is thought to be involved in the accumulation of activated ATM and its various substrates at or near DSBs to facilitate their phosphorylation and regulate the damage response (Lou et al., 2006).
The precise mechanism used to sense a DNA damage event and signal a response is still an active area of research. However, there is substantial evidence that the MRN complex, composed of Mre11, Rad50 and Nbs1, has important roles in the early sensing and activation of ATM. The MRN complex is evolutionarily conserved from yeast to mammals and is involved in homologous recombination, cell-cycle checkpoint regulation and meiotic recombination (Sharples and Leach, 1995; Dolganov et al., 1996; Petrini et al., 1995; Carney et al. 1998). In response to DSBs, MRN complex proteins are some of the earliest proteins associated with the site of DNA damage (Celeste et al., 2003; Lisby et al., 2004). The Mre11/Rad50 heterodimer binds to the ends of the DNA break and Mre11 has nuclease activity that resects the ends to expose regions of microhomology that are important for ligation and repair (Trujillo et al., 1998; Paull and Gellert, 1998). Rad50 tethers the DNA ends together via interactions between its coiled coil domains (Hopfner et al., 2002). Rad50 binds ATP and provides the energy required for the nuclease activity of Mre11 (Paull and Gellert, 2000). The Nbs1 component is essential for recruitment and accumulation of the complex at the site of damage (Falck et al., 2005; Cerosaletti et al., 2006), and the effective activation of ATM in response to damage (Uziel et al., 2003; Carson et al., 2003). Each of these proteins is therefore important for the proper functioning of the MRN complex in the damage signaling and response pathway.
Accumulation of the damage response proteins in foci is a dynamic process and a useful marker to analyze the cellular response to DSBs (Lisby et al., 2004). Foci are formed at DSBs, sites of replication stress, regions of ssDNA, and shortened telomeres (Raderschall et al., 1999; Lisby et al., 2003; D’Adda di Fagagna et al., 2003). It is speculated that accumulation of proteins in foci facilitates the concentration of repair proteins specifically at the lesion. Efficient rejoining of the DSB is then promoted by the relatively high concentration of repair factors in the vicinity of DNA ends within the foci (reviewed by Lisby and Rothstein, 2004).
Ad E4 mutant infections trigger the ATM and ATR damage response cascades (Carson et al, 2003). The products of Ad E4 open reading frame (ORF) 3 (E4-11kDa) and E4 ORF 6 (E4-34kDa) along with the E1b-55kDa protein, target the MRN complex for relocalization and proteasome mediated degradation, and in so doing effectively inhibit the cellular damage response. In the absence of the E4 proteins, the MRN complex accumulates in foci corresponding to viral DNA replication foci, promotes viral genome concatenation, and inhibits viral DNA replication and late gene expression (Stracker et al., 2002; Jayaram and Bridge, 2005; Evans and Hearing, 2005; Mathew and Bridge, 2007). Removal of the Mre11 protein by RNAi (Mathew and Bridge, 2007) or infection of cells that lack Mre11 (ATLD) (Evans and Hearing, 2005), rescues the E4 mutant replication defect, indicating that Mre11 is able to interfere with a productive infection if it is not inactivated by E4 proteins.
Here we have tested the hypothesis that the mechanism of Mre11 interference in E4 mutant DNA replication is a consequence of its physical interaction with viral genomes. We show that Mre11 is physically bound to E4 mutant viral DNA and this depends on the presence of Nbs1. Interfering with Mre11 binding to viral DNA alleviates the E4 mutant DNA replication defect. We also find that Mre11 is important for early Mdc1 foci formation in response to E4 mutant infection, consistent with its role as a sensor of DNA damage events.
RESULTS
Mre11 localizes to E2-72kDa containing viral replication foci in E4 mutant infection, and it binds to E4 mutant viral DNA
E4 mutant H5dl1007 carries a deletion that disrupts all of the E4 ORFs (Bridge and Ketner, 1989), and therefore fails to prevent activation of a DNA damage response and genome concatenation in infected cells (Carson et al., 2003; Jayaram and Bridge, 2005). Mre11 co-localizes with the viral E2-72kDa DNA binding protein (DBP) at E4 mutant DNA replication centers (Stracker et al., 2002). We have examined the distribution of Mre11 in a time course infection with H5dl1007 to further understand the dynamics of the association of Mre11 with DNA replication centers. Initially we see that Mre11 is tightly associated with viral E2-72kDa in the replication foci that start to form early in the infection process (6–12 hours post infection (hpi), Fig. 1B, panels a to f). As the replication centers develop, Mre11 localizes preferentially to the periphery of the E2-72kDa centers at later stages (18–24 hpi, Fig. 1B, panels g to l). The tight association of Mre11 with the E2-72kDa centers early in the infection process suggests that Mre11 might directly or indirectly bind viral DNA in these centers. Mre11 is a member of the MRN complex together with Rad50 and Nbs1, and has DNA binding domains that are likely to be important for its role in DNA repair events. We were therefore interested in investigating the possibility that Mre11 was physically bound to E4 mutant viral DNA and the dynamics of this binding over time. We performed chromatin immunoprecipitation (chIP) experiments on samples prepared from HeLa cells infected with the E4 mutant H5dl1007 for 12 and 24 h to measure binding to viral DNA at intermediate and late times after infection. Pull down of Mre11 using specific antibody (Ab) was confirmed by western blotting. As expected, immunoprecipitation with Mre11 Ab pulled down the ~80kDa protein corresponding to Mre11 in uninfected and E4 mutant infected samples (Fig. 1C, top panel). We performed immunoprecipitation with viral E2-72kDa DNA binding protein and Ab against phosphoinositol-3-kinase (PI3K) as positive and negative controls for the chIP experiments, respectively. These antibodies pulled down the expected proteins in immunoprecipitation experiments as expected (Fig. 1C). We next performed PCR experiments on chromatin samples immunoprecipitated with Abs against Mre11, E2-72kDa, or PI3K from formaldehyde fixed cells. Viral DNA was amplified with primers specific for the Ad E1b region. We see a PCR amplified signal only in E4 mutant infected samples immunoprecipitated with either E2-72kDa Ab or Mre11 Ab at 12 hpi (Fig. 1D). No PCR product was observed in cells that were precipitated with PI3K antibody demonstrating the specificity of Mre11 binding to E4 mutant DNA. We also did not detect viral DNA in samples that were mock immunoprecipitated without the addition of Ab. Binding of Mre11 to E4 mutant DNA was not detected at 24 hpi (Fig. 1D). We have performed similar experiments in Ad5 infected cells and are unable to detect Mre11 binding to Ad5 DNA at either 12 or 24 hpi (data not shown), consistent with the ability of Ad5 to degrade the Mre11 protein in infected cells. E2-72kDa binds to Ad5 DNA at both 12 and 24 hpi (data not shown) as expected. Our UI samples were devoid of any signal as expected. The total input chromatin (TIC) samples were included to show input levels of DNA from UI (TIC1) and E4 mutant infected cells (TIC2). Our results indicate that Mre11 is specifically bound to E4 mutant DNA at 12 hpi.
Figure 1. Mre11 localizes to E2-72kDa containing viral replication foci in E4 mutant infection and it is bound to E4 mutant DNA.

HeLa cells were uninfected (UI) or infected with H5dl1007 (1007) at 30 FFU/cell for the times indicated. (A) and (B) Immunofluorescence staining was performed with antibodies specific for Mre11 (1A, panel a; 1B, panels a, d, g, and j) and the viral E2-72kDa protein (1A, panel b; 1B, panels b, e, h, and k). Bar 10µm. Uninfected cells and cells infected for 12 or 24 h with H5dl1007 were treated with formaldehyde and used for chIP experiments as described in materials and methods. (C) Western blotting was performed to confirm immunoprecipitation of Mre11, E2-72kDa and PI3K with their respective antibodies; representative blots are shown. Lanes from samples that were immunoprecipitated with specific Ab or mock immunoprecipitated are labeled (+) and (−), respectively. (D) PCR amplification using primers specific to the E1b region was performed on chromatin samples prepared from uninfected and H5dl1007 infected cells that were immunoprecipitated (+) with Mre11, PI3K, or E2-72kDa Abs as indicated, or mock immunoprecipitated in parallel without the addition of Ab (−). PCR reactions were fractionated on a 1% agarose gel and stained with ethidium bromide to visualize the 400bp expected PCR product. Total input chromatin (TIC) samples prepared from UI (TIC1) and H5dl1007 infected cells (TIC2) were included to indicate input DNA levels. ChIP experiments with the Mre11 and E2-72kDa antibodies were performed 3 times with similar results. ChIP experiments with Mre11 and PI3K antibodies were performed twice with similar results.
Rad50 and Nbs1 are important for Mre11 localization to viral DNA replication centers and inhibition of E4 mutant viral DNA replication
The MRN complex is made up of the Mre11, Rad50 and Nbs1 proteins. The entire complex associates with E4 mutant DNA replication centers and is important for concatenation of viral genomes. However, the role of each member of this complex in E4 mutant infection is not yet clearly understood. We have previously used RNAi mediated knockdown to show that Mre11 interferes with E4 mutant DNA replication (Mathew and Bridge, 2007). We performed siRNA knockdown experiments to target Rad50 and Nbs1 using a pool of 4 duplexes to target each respective mRNA (see Material and Methods) and monitored the effect on Mre11 distribution and E4 mutant DNA replication using immunofluorescence (IF) and Southern blotting experiments, respectively. The results are shown in Fig. 2 and Fig. 3. First we performed western blotting experiments to confirm specific knockdown of the targeted proteins (Fig. 2A and 3A). Transfection with Rad50 or Nbs1 siRNAs specifically knocked down expression of the respective protein in HeLa cells. The level of either Rad50 or Nbs1 was unaffected in cells treated with non-specific siRNA (non-targeting scrambled siRNA), control siRNA against glyceraldehyde-3-phosphate dehydrogenase (GAPD) or lipofection reagent D1.
Figure 2. Rad50 is important for Mre11 stability and the inhibition of E4 mutant DNA replication.
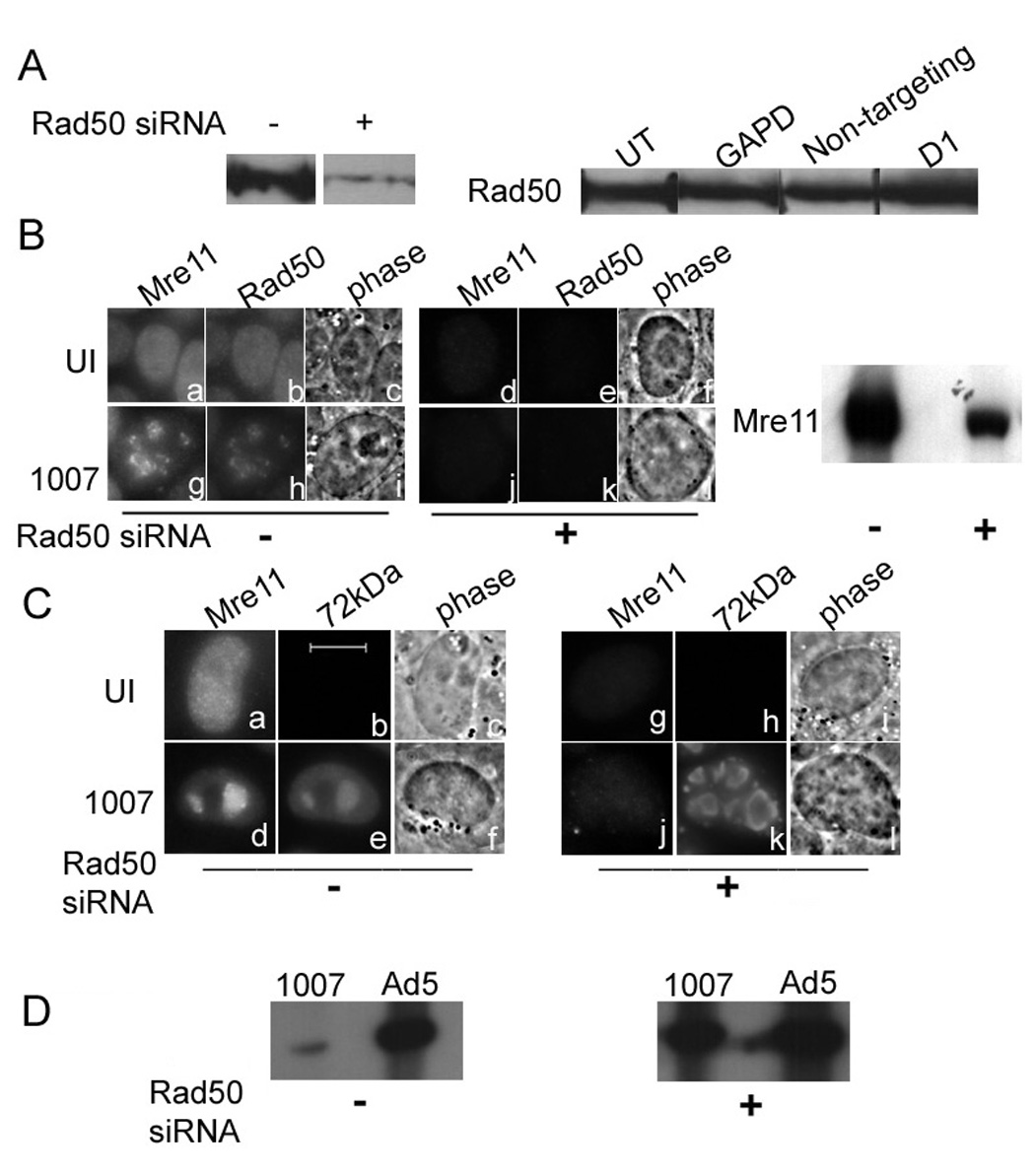
HeLa cells were transfected with control siRNA or Rad50 siRNA prior to infection with Ad5 or H5dl1007 at 3 FFU/cell for 24h. (A) Rad50 knockdown was monitored by western blotting of 75µg of total protein prepared 96 hours after mock (−) or Rad50 specific siRNA (+) transfection, using rabbit polyclonal Ab against Rad50. Additional controls demonstrating the specificity of Rad50 expression knockdown included no treatment (UT), non-targeting siRNA (non targeting), siRNA against GAPD, and treatment with the transfection reagent alone (D1). (B) Mre11 and Rad50 distribution in untransfected uninfected (panels a to c) and cells infected with H5dl1007 (panels g to i) for 24 hpi are shown. The distribution of Mre11 and Rad50 in siRNA transfected HeLa cells that were either uninfected or infected for 24 h with H5dl1007 are shown in panels d to f and j to l respectively. A western blot depicting levels of Mre11 in Rad50 siRNA treated cells is also shown (right panel). (C) Panels a to f show the distribution of host Mre11 and viral E2-72kDa in uninfected and H5dl1007 infected cells in the absence of siRNA transfection. Panels g to l show the distribution of Mre11 and E2-72kDa in cells transfected with Rad50 siRNA that were subsequently uninfected or infected with H5dl1007. Bar 10µm. (D) Levels of viral DNA synthesis in Ad5 and H5dl1007 infected HeLa with (+) and without (−) Rad50 siRNA transfection were quantified by Southern analysis of 10µg of Eco RI digested total DNA prepared at 24 hpi. The Eco R1 C fragment from the DNA digestion was used for comparison between Ad5 and H5dl1007.
Figure 3. Nbs1 is important for Mre11 localization and the inhibition of E4 mutant DNA replication.
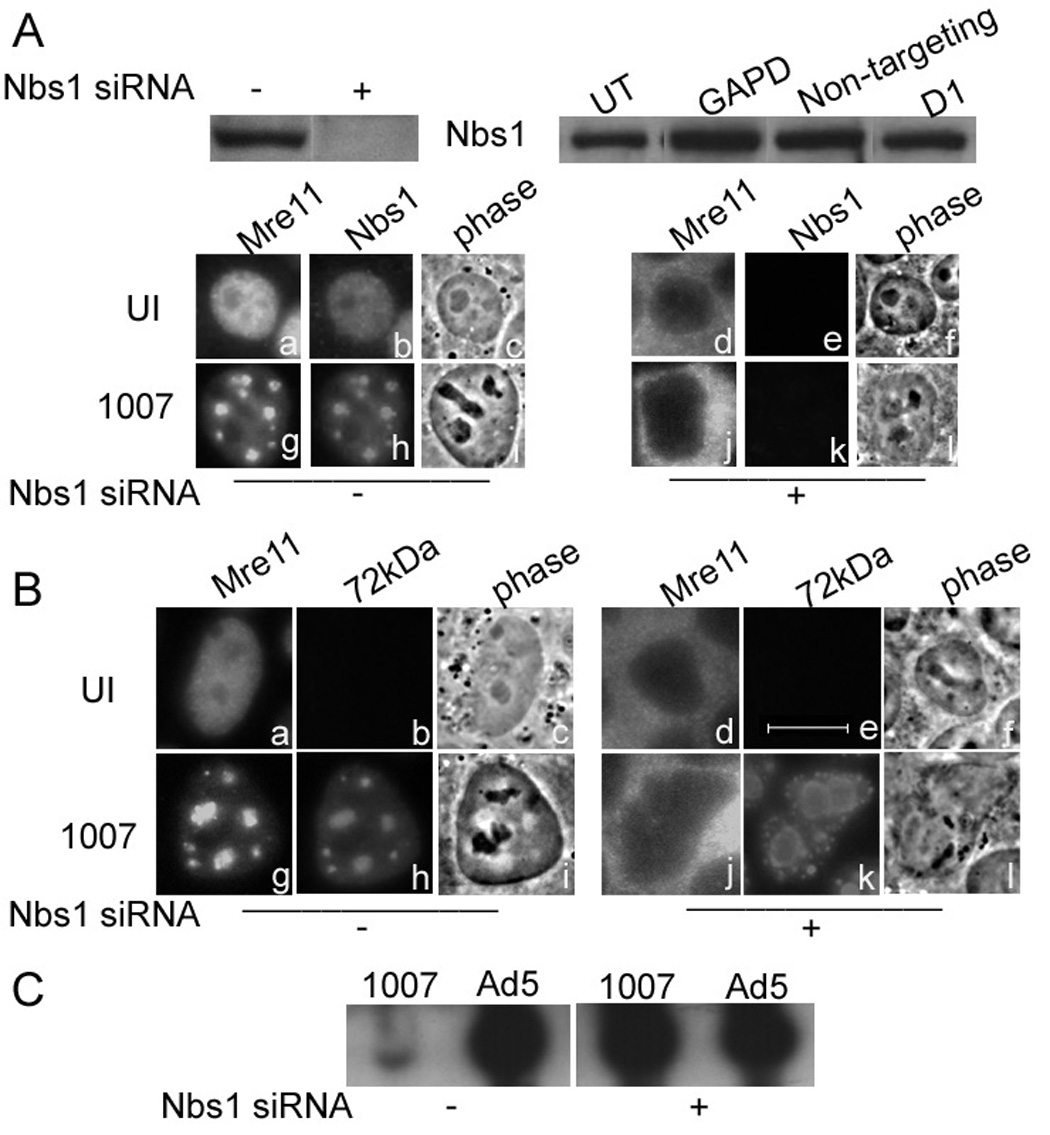
HeLa cells were transfected with control siRNA or Nbs1 siRNA prior to infection with Ad5 or H5dl1007 at 3 FFU/cell for 24h. (A) Nbs1 knockdown was monitored by western blotting using 75µg of total protein prepared 96 hours after mock (−) or Nbs1 specific siRNA (+) transfection, using goat polyclonal Ab against Mre11. Additional controls demonstrating the specificity of Nbs1 expression knockdown included no treatment (UT), non-targeting siRNA (non targeting), siRNA against GAPD, and treatment with the transfection reagent alone (D1). Mre11 and Nbs1 distribution in untransfected, uninfected cells (panels a to c) and cells infected with H5dl1007 (Panels g to i) for 24 h are shown. The distribution of Mre11 and Nbs1 in siRNA transfected HeLa cells that were either uninfected or infected for 24 h with H5dl1007 are shown in panels d to f and j to l respectively. (B) Mre11 and viral E2-72kDa protein distribution in untransfected cells that were either uninfected (panels a–c) or infected (panels g to I) are shown. The distribution of Mre11 and E2-72kDa distribution in Nbs1 siRNA transfected cells that were either uninfected or infected with H5dl1007 is shown in panels d to f and j to l, respectively. Bar 10µm. (C) Levels of viral DNA synthesis in Ad5 and H5dl1007 infected HeLa with (+) and without (−) Nbs1 siRNA transfection were quantified by Southern analysis of 10µg of Eco RI digested total DNA prepared at 24 hpi. The C fragment from the DNA digestion was used to compare Ad5 and H5dl1007 DNA levels.
Rad50 is distributed in foci that contain Mre11 in H5dl1007 infected, untreated cells (Fig. 2B, panels g and h). When we knock down Rad50 with its specific siRNA, levels of Mre11 are also significantly reduced when measured by IF (Fig. 2B compare panels a and g with d and j) and western blotting (Fig. 2B right panel). Phosphorimager quantitation indicated at least a 3-fold reduction in levels of the Mre11 protein in Rad50 siRNA treated cells, indicating that Rad50 is important for stabilizing the Mre11 protein. The E4 mutant replication centers that form in Rad50 siRNA treated cells are larger and more numerous than those in the untreated HeLa cells (Fig. 2C compare panels e and k). The level of E4 mutant DNA replication was measured by Southern blotting experiments (Fig. 2D). In HeLa cells that have not been transfected with Rad50 siRNA, E4 mutant DNA replication is severely defective compared to wild-type Ad5 (50–70 fold). However in HeLa cells knocked down for Rad50, the DNA replication defect is significantly rescued (to within 2-fold of Ad5), as evidenced by the relative increase in levels of E4 mutant DNA. Our results indicate that knockdown of Rad50 moderates the E4 mutant DNA replication defect. However, this could be due to a secondary effect on the stability of Mre11, rather than a direct effect of Rad50 knockdown.
Nbs1 is distributed in foci containing Mre11 in E4 mutant infected, untransfected cells (Fig. 3A, panels g and h). RNAi knockdown of Nbs1 dramatically affects the distribution of Mre11 within the cell. In the presence of Nbs1, Mre11 is primarily in the nucleus. However, knockdown of Nbs1 mis-localizes Mre11 to the cytoplasm (Fig. 3A, panels j and k). This is an expected result because the Nbs1 protein provides a nuclear localization signal (NLS) that is required for the translocation of Mre11 to the nucleus (Desai-Mehta et al., 2001). We investigated the effect of this mis-localization on E4 mutant DNA replication by IF. E4 mutant replication centers were generally larger in cells treated with Nbs1 siRNA when compared to untreated cells (Fig. 3B, compare panel h with k). We further investigated the effect of Nbs1 knockdown and Mre11 mis-localization on E4 mutant DNA replication, by Southern blotting experiments. Our results are shown in Fig. 3C. In the cells that were not transfected with Nbs1 siRNA, levels of E4 mutant DNA are significantly lower than that of Ad5 (50- to 70-fold). However, knock down of Nbs1 by siRNA dramatically increases the level of E4 mutant DNA which is now similar to wild type Ad5 (within 2-fold). These results show that knocking down Nbs1 rescues the E4 mutant DNA replication defect; this could be due to mis-localization of Mre11 in the cytoplasm, and/or to a more direct role of Nbs1 in interfering with E4 mutant DNA replication.
Nuclear Mre11 is unable to localize to E4 mutant replication centers and does not bind viral DNA in the absence of Nbs1
Mre11 requires the Nbs1 protein for translocation into the nucleus. Nbs1 is also important for the localization of Mre11 in foci in response to DNA damage induced by ionizing radiation (IR) (Cerosaletti et al., 2006). We were interested in investigating the role of Nbs1 in the accumulation of nuclear Mre11 at E4 mutant DNA replication foci. For this purpose we obtained cells lacking Nbs1 (NBS-ILB1 LXIN), and cells that lack Nbs1 but stably express Mre11 engineered with an exogenous nuclear localization signal to direct it to the nucleus independently of Nbs1 (NBS-ILB1 NLS.12, hereafter referred to as NLS.12) from Drs. Cerosaletti and Concannon. Cerosaletti et al., (2006) have shown that the Mre11-NLS protein produced in NLS.12 cells interacts with Rad50 and is able to activate ATM dependent damage responses when introduced into ATLD cells lacking Mre11. These data indicate that the engineered Mre11-NLS is functional. We used this experimental system to determine if Nbs1 is important for the formation of Mre11 foci in E4 mutant infections. NBS-ILB1 LXIN, NLS.12, and HeLa cells were infected with Ad5 or E4 mutant H5dl1007. The ability of Mre11 to localize in viral DNA replication centers was assessed by IF and binding to viral DNA was measured in chIP experiments. Our results are represented in Fig. 4. We see that in HeLa cells (Fig. 4A, panels a to i), infection with H5dl1007 causes the formation of Mre11 foci that co-localize with the viral E2-72kDa foci. In dramatic contrast, infection of NLS.12 cells (Fig. 4A, panels j to r) shows a clear inhibition of Mre11 foci formation, although Mre11 is detected in the nucleus in these cells as expected. Immunofluorescence experiments confirmed that Rad50 was also localized in the nucleus in uninfected and E4 mutant infected NLS.12 cells (data not shown). In late phase infected NLS.12 cells, Mre11 surrounds but is still excluded from viral E2-72kDa-containing centers (compare panels p and q). The confocal overlay (panel r) shows no overlap of the red and green staining patterns. E4 mutant infected NBS-ILB1 LXIN cells, which have Mre11 mis-localized in the cytoplasm, also do not show Mre11 foci formation (data not shown). We subsequently performed chip experiments on NLS.12 cells as described for Fig. 1 to analyze the DNA binding capacity of nuclear Mre11 in the absence of Nbs1. Immunoprecipitation controls are shown in Fig. 4B and indicate that antibodies against Mre11 and E2-72kDa pull down the expected proteins. We show in Fig. 4C that the absence of Nbs1 alters the DNA binding capacity of Mre11 as evidenced by the absence of a specific PCR amplified signal in chromatin immunoprecipitated from E4 mutant infected samples with Mre11 Ab. Our uninfected controls are devoid of any signal as expected (Fig. 4C), we also saw no PCR product in samples that were precipitated with PI3K Ab (data not shown). The positive control experiments in which chromatin from E4 mutant infected samples was immunoprecipitated with the viral E2-72kDa Ab showed a specific PCR product (Fig. 4C). Our results indicate that in E4 mutant infections, efficient binding of Mre11 to viral DNA and localization at replication centers requires the Nbs1 protein.
Figure 4. Nbs1 is important for Mre11 localization in E4 mutant replication centers and Mre11 binding to viral DNA.

(A) HeLa and NLS.12 cells were infected with H5dl1007 for 10 h (early) and 24 h (late) at 3 FFU/cell. Confocal microscopy was used to analyze the distribution of host Mre11 (panels a,d,g,j,m,p) and viral E2-72kDa (panels b,e,h,k,n,q) proteins in uninfected and infected cells. Merged images of these staining patterns are shown in panels c, f, i, and l, o, r. ChIP experiments were performed as described in Fig. 1 using samples prepared from NLS.12 cells that were uninfected or infected with H5dl1007 for 12h at 30FFU/cell. (B) Western blotting was performed to confirm immunoprecipitation of Mre11 and E2-72kDa from chromatin prepared from H5dl1007 infected and uninfected NLS.12 cells, in preparation for chIP analysis. Lanes from samples that were immunoprecipitated with specific Ab or mock immunoprecipitated are labeled (+) and (−), respectively. (C) PCR amplification using primers specific to the E1b region was performed on UI and H5dl1007 infected samples prepared at 12 hpi and immunoprecipitated with Mre11 or E2-72kDa. PCR products were analyzed as described in Fig. 1. Lanes from samples that were immunoprecipitated with specific Ab or mock immunoprecipitated in parallel are labeled (+) and (−), respectively. Total input chromatin samples from uninfected (TIC1) and H5dl1007-infected (TIC2) cells were included to indicate input DNA levels. The chIP experiments were performed twice with similar results. (D) Levels of viral DNA synthesis in HeLa and NLS.12 cells infected with Ad5 and H5dl1007 at 3FFU/cellwere quantified by Southern analysis of 10µg of Eco RI digested total DNA prepared at 24 hpi. The C fragment from the DNA digestion was used for comparison between Ad5 and H5dl1007.
The inability of Mre11 to localize to viral replication foci and bind viral DNA rescues E4 mutant replication
Our earlier work showed that Mre11 interferes with E4 mutant DNA replication (Mathew and Bridge, 2007). Our results with NLS.12 cells indicate that in the absence of Nbs1, Mre11 does not efficiently associate with viral replication centers or bind viral DNA even when it is directed to the nucleus with an independent NLS. We next determined if the inability of Mre11 to physically associate with viral genomes in NLS.12 cells affects E4 mutant DNA replication. H5dl1007 infected NLS.12 cells have larger DNA replication centers than those present in HeLa cells (Fig. 4A compare h with q). Southern blotting (Fig. 4D) shows that H5dl1007 DNA levels were significantly increased in NLS.12 cells to within 2 fold of Ad5. The 50- to 70-fold defect in E4 mutant DNA accumulation seen in HeLa cells, is substantially rescued in NLS.12 cells (Fig. 4D), indicating that the failure of Mre11 to associate with the E4 mutant DNA replication centers and bind viral DNA in NLS.12 cells (Fig. 4A and C) correlates with dramatically improved DNA replication.
Mre11 is required for the formation of Mdc1 foci in response to E4 mutant infections
Mdc1 accumulates in foci in response to IR induced damage as well as following Ad infection. In IR induced damage, Mdc1 is required to sustain MRN foci formation at sites of DNA damage (Goldberg et al., 2003). Although Ad infections induce the accumulation of Mdc1 in early foci that correspond to viral E2-72kDa-containing centers, we have found that the Mdc1 protein is not necessary for the accumulation of the Mre11 protein to these same foci (Mathew and Bridge, 2007). The MRN complex has been implicated as a sensor of DNA damage events that signal the activation of early response proteins such as ATM and γH2AX, both of which potentially act upstream of Mdc1 in the damage cascade (Stewart et al., 2003; Lou et al., 2006). We were therefore interested in understanding if Mdc1 accumulation in foci in Ad infections was dependent on the presence of Mre11. HeLa cells were either treated or not treated with siRNA specific for Mre11, and then infected with H5dl1007 at 3 FFU/cell. The results are shown in Fig. 5. Control experiments were performed to ensure that the siRNA used specifically knocked down Mre11 (Mathew and Bridge 2007; data not shown). Immunofluorescence staining for Mre11 and Mdc1 was performed and representative results from a 6hr time point are shown in Fig. 5A. We see that in the presence of Mre11, E4 mutant infection results in Mdc1 accumulation in distinct foci. However when Mre11 is knocked down in these cells, there is a reduction in the number of cells with Mdc1 foci in E4 mutant infections. To quantify this observation, cells were scored blind to assess early Mdc1 foci formation in the presence and absence of Mre11 (Fig. 5B). In untreated infected cells, we see an increase in the number of cells that show Mdc1 foci from 2 to 6 hpi. However, when Mre11 is knocked down by its specific siRNA, there is a significant decrease in the number of infected cells that show distinct Mdc1 foci relative to the untreated cells, indicating that Mre11 is important for formation of early Mdc1 foci in E4 mutant infection.
Figure 5. Mre11 is required for the formation of Mdc1 foci in response to E4 mutant infections.

HeLa cells were transfected with control siRNA or Mre11 siRNA prior to infection with H5dl1007 at 3 FFU/cell for 24h. (A) Immunofluorescence staining was performed to analyze the distribution of Mre11 and Mdc1. Panels a to f show Mre11 and Mdc1 distribution in untransfected HeLa cells that were either uninfected (a–c) or infected with H5dl1007 (d–f). Panels g to l show Mre11 and Mdc1 distribution in Mre11 siRNA transfected cells that were either uninfected (g–i) or infected with H5dl1007 (j–l). Bar 10µm. Immunostained cells from the time course were scored blind for Mdc1 foci formation in the presence and absence of Mre11. The graph presented in (B) shows the percentage of cells with Mdc1 foci in uninfected and infected cells prepared at the times indicated, that were untransfected (black bars) or transfected (white bars) with Mre11 siRNA.
Mdc1 binds to E4 mutant viral DNA
We see Mdc1 in foci corresponding to early replication foci in response to E4 mutant infections and this process depends on the presence of Mre11. This suggests that Mdc1 may be aggregating in regions where the incoming viral genomes are located. Mdc1 is not known to directly bind DNA; however, it is present in protein complexes containing γH2AX that tightly associate with damaged DNA (Stewart et al., 2003; Lou et al., 2006). We performed chIP experiments as described in Fig. 1 to determine if Mdc1 was physically interacting with viral genomes. Chromatin immunoprecipitation with Abs against Mdc1 and E2-72kDa was carried out on uninfected and E4 mutant infected samples prepared at 6 hpi and the results are shown in Fig. 6. As expected, in both UI and E4 mutant infected samples immunoprecipitation with Mdc1 Ab pulled down a protein identified as Mdc1 in western analysis (Fig. 6A; top panel). Positive control experiments with viral E2-72kDa specific antibodies also showed pull down of the viral E2-72kDa protein in E4 mutant infected cells (Fig. 6A; bottom panel). Chromatin pulled down with the Mdc1 Ab was amplified using viral specific primers corresponding to E1b, and yielded a DNA fragment of the expected size (Fig. 6B). We do not see a similar band in chromatin immunoprecipitated from uninfected cells, or from samples that were mock immunoprecipitated in the absence of Ab. Control experiments using viral E2-72kDa Ab showed a similar amplification of viral DNA using the same primers (Fig. 6B). We have also found that immunoprecipitation with PI3K Ab does not pull down E4 mutant DNA in parallel experiments (data not shown). Our results indicate that cellular Mdc1 is bound to E4 mutant viral DNA at early times after infection and suggest that Mdc1 may be bound to viral DNA when it is redistributed to foci.
Figure 6. Mdc1 binds to E4 mutant viral DNA.
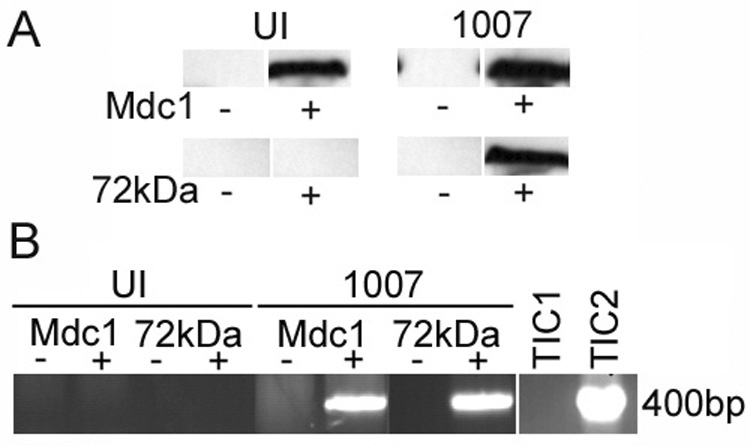
HeLa cells were infected with H5dl1007 at 30 FFU/cell for 6h and processed for chIP as described in Fig. 1. (A) Western blotting was performed to confirm immunoprecipitation of Mdc1 and E2-72kDa and representative blots are shown. Lanes from samples that were immunoprecipitated with specific Ab or mock immunoprecipitated are labeled (+) and (−), respectively. (B) PCR amplification using primers specific to the E1b region was performed on uninfected (UI) and H5dl1007 (1007) infected samples immunoprecipitated with Mdc1 or E2-72kDa Abs (+) or mock immunoprecipitated in parallel (−). PCR products were analyzed as described in Fig. 1. Total input chromatin (TIC) samples were included to indicate input DNA levels in uninfected (TIC1) and H5dl1007 (TIC2) infected cells. The Mdc1 chIP experiment was performed 3 times with similar results.
DISCUSSION
The MRN complex is redistributed to foci that co-localize with E2-72kDa in E4 mutant DNA replication centers (Stracker et al., 2002). We have performed a time course with H5dl1007 and find that Mre11 foci are tightly associated with viral DNA replication centers until around 12–15 hpi, after which the localization of Mre11 becomes peripheral to the E2-72kDa containing centers. This change in the localization of Mre11 is accompanied by changes in the physical binding of Mre11 to E4 mutant DNA (Fig. 1). Mre11 binds E4 mutant DNA at 12hpi. Interestingly, later in the infection when Mre11 is no longer tightly co-localized with the E2-72kDa centers, it also fails to bind E4 mutant DNA in chIP assays. Although the reason for this is as yet unclear, it could reflect a change in the efficiency of Mre11 binding to viral DNA late in the infection. As viral replication proteins accumulate they could out-compete Mre11 for efficient binding to viral DNA. Alternatively, the loss of binding at late times in infection could reflect an end point in the cellular response to the viral DNA damage signal, where the proteins are deactivated and return a diffuse nuclear distribution once they have finished "repairing" viral DNA to form concatemers.
All three members of the MRN complex contribute to the functioning of the complex (Luo et al., 1999; Yamaguchi-Iwai et al., 1999; Zhu et al., 2001; reviewed in Assenmacher and Hopfner, 2004). Mre11 is a DNA binding protein with exo- and endonuclease activity and is an essential component of the DNA DSB repair machinery (Trujillo et al., 1998; Paull and Gellert, 1998). Rad50 is required for the stable binding of Mre11 to DNA (Connelly et al., 2003; deJager et al., 2002; Paull and Gellert, 1999; Trujillo and Sung, 2001) and provides the energy needed for the enzymatic activities of Mre11 (Paull and Gellert, 2000). The Nbs1 component is essential for proper localization of the MRN complex to the nucleus and its association with damaged DNA (Desai-Mehta et al., 2001; Cerosaletti et al., 2006). RNAi mediated knockdown of Mre11 (Mathew and Bridge, 2007) and either Rad50 or Nbs1 (Fig. 2 & Fig. 3) dramatically rescues E4 mutant DNA levels, suggesting that all the members of the MRN complex are important for inhibiting E4 mutant DNA replication. However, Rad50 RNAi affects the stability of Mre11 while Nbs1 RNAi mis-localizes Mre11 in the cytoplasm. Therefore these results do not determine if the primary role of Nbs1 and Rad50 in inhibiting E4 mutant DNA synthesis is through their effect on Mre11, or due to independent functions.
Cerosaletti et al., (2006), established a simian virus 40 (SV40) transformed cell line using NBS fibroblasts from a patient homozygous for the 657del5 mutation (Kraakman van der Zwet et al., 2003), engineered to express an Mre11 gene with an artificial C-terminal nuclear localization signal (NLS) sequence. These cells have a defective Nbs1 protein but Mre11 is still able to localize to the nucleus where it exists in a complex with Rad50. IR treatment of these cells does not induce foci formation or activation of the ATM response pathway, suggesting a critical role for Nbs1 in the cellular response to DNA damage independent of its ability to translocate Mre11 to the nucleus (Cerosaletti et al., 2006). This experimental system provides us with a unique opportunity to analyze the role of Nbs1 in E4 mutant DNA replication in cells where Mre11 is independently directed to the nucleus. The distinct Mre11 foci seen in E4 mutant infected HeLa cells are not observed in the NLS.12 cells that lack Nbs1 but have Mre11 localized in the nucleus (Fig. 4). Interestingly, we cannot detect Mre11 binding to viral DNA in NLS.12 cells in chIP assays, and the E4 mutant DNA replication defect is alleviated (Fig. 4). These observations suggest that the physical binding of Mre11 to E4 mutant viral DNA is dependent on Nbs1 and is important for the ability of the MRN complex to interfere in E4 mutant DNA replication. Nbs1 is not thought to bind DNA directly (reviewed in Zhou et al., 2006), and in the absence of Mre11, neither Rad50 nor Nbs1 is able to interfere with E4 mutant DNA replication (Mathew and Bridge, 2007). Nbs1 binds to γH2AX and recruits Mre11 and Rad50 to the proximity of IR induced DNA damage (Tauchi et al., 2001). It is possible that it plays a similar role in E4 mutant infections by loading Mre11 onto viral DNA to perform repair functions.
Mdc1 co-precipitates with the MRN complex and is required for the stability of MRN foci in IR induced damage (Goldberg et al., 2003; Mochan et al., 2003). However, Mdc1 knockdown does not affect Mre11 localization to E4 mutant replication foci (Mathew and Bridge, 2007). Conversely, we find that Mre11 is required for recruiting Mdc1 to E4 mutant replication foci (Fig. 5). This suggests that Mre11 functions upstream of Mdc1 in the cellular response to E4 mutant infection, consistent with the idea that Mre11 is a sensor of DNA damage (Carson et al., 2003; reviewed by Petrini and Stracker, 2003). We find that Mdc1 is physically bound to E4 mutant viral DNA (Fig. 6), but since RNAi mediated knockdown of Mdc1 does not rescue E4 mutant DNA replication (Mathew and Bridge, 2007), the significance of this interaction is not clear. It is possible that Mre11 still binds to E4 mutant DNA in the absence of Mdc1, and that this is sufficient to inhibit viral DNA replication. The observation that Mre11 localization to E4 mutant DNA replication foci does not require Mdc1 (Mathew and Bridge, 2007) supports this suggestion. Alternatively, RNAi knockdown of Mdc1 may not sufficiently reduce levels of the protein to affect Mre11 localization and E4 mutant DNA replication. Although we have not identified a direct role for Mdc1 in regulating E4 mutant DNA replication, it could nevertheless be important for recruiting additional factors to E4 mutant replication foci. DNA PK and Rad51 are essential components of the NHEJ and HR pathways of DSBR, respectively, and are known to interact with Mdc1. (Lou et al., 2004; Zhang et al., 2005). The interaction of Mdc1 with viral DNA could also be important for amplifying DNA damage responses initiated by Ad infection. The ability of Mdc1 to simultaneously bind γH2AX and ATM may facilitate ATM-dependent phosphorylation of H2AX and other DNA damage response proteins at sites of DNA damage (Lou et al., 2006, reviewed in Kim et al, 2006)
How does the MRN complex interfere in E4 mutant DNA replication? Our data suggests that Nbs1 is important for recruiting Mre11 to viral DNA. Mre11 could then potentially interfere with viral DNA replication by physically hindering DNA replication proteins from accessing viral DNA termini. Alternatively or additionally, physical binding could be necessary for the nuclease activity of Mre11, to destroy DNA ends that contain the viral origin of replication. In this scenario, the binding of Mre11 to viral DNA would be a pre-requisite for directing its enzymatic activity to viral DNA. In conclusion, our results indicate that Mre11 binds E4 mutant viral DNA in an Nbs1 dependent manner and in so doing, interferes with viral DNA replication. Mre11 is also important for re-localization of Mdc1 in response to E4 mutant infection, which is consistent with its potential role as a sensor of DNA damage within cells.
MATERIALS AND METHODS
Cells and viruses
HeLa and E4 mutant-complementing W162 (Weinberg and Ketner, 1983) monolayer cell cultures were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2mM L-glutamine, 10U/mL penicillin and 10 µg/mL streptomycin. NBS-ILB1 cells stably transfected with pLXIN retroviral vector alone or pLXIN Mre11-NLS (NLS.12) were provided by K. Cerosaletti and P. Concannon (Cerosaletti et al., 2006), and were maintained in DMEM with 15% FBS, 500µg/mL G418 (Invitrogen), 10U/mL penicillin and 10µg/mL streptomycin. Ad5 and E4 mutant H5dl1007 (Bridge and Ketner, 1989) were propagated on HeLa and W162 cells, respectively. Their titers were determined in W162 cells and expressed as fluorescent focus forming units (FFU)/mL (Philipson, 1961). Cells were infected at a multiplicity of infection (MOI) of 3 or 30 FFU/cell. E4 mutant H5dl1007 carries a deletion that extends from the Sma I site in E4 ORF1 to map unit 93.3 in the middle of ORF6. This mutant lacks ORFs 2, 3, 3/4, and 4, deletes the N-terminus of ORFs 6 and 6/7, and deletes the C-terminus of ORF 1 (Bridge and Ketner, 1989).
Immunofluorescence analysis
Infected and uninfected HeLa cells were seeded on cover slips in 35mm dishes and fixed as described previously (Mathew and Bridge, 2007). Cells on cover slips were fixed and stained for immunofluorescence as described (Aspegren and Bridge, 1998) using the following primary antibodies: rabbit polyclonal anti-Rad50 (Genetex), rabbit polyclonal anti-Nbs1 (Cell signaling), goat polyclonal anti-Mre11 (Santa Cruz Biotechnology), and rabbit polyclonal anti-Mdc1 (Bethyl Labs), at dilutions recommended by the manufacturers. Rabbit polyclonal anti-72kDa (T. Linné) was used at a 1:2000 dilution. Secondary antibodies included Donkey anti-goat and anti-rabbit Alexafluor tagged antibodies.
Microscopy
Images of cells were visualized and scored by conventional fluorescence microscopy with a Nikon eclipse E-400 microscope using a 100X objective. Images were obtained and recorded using a SPOT-2 charge-coupled device and capture software provided by the manufacturer (Diagnostic Instruments Inc.). Confocal microscopy (Fig. 1 and Fig. 4) was performed with an Olympus FV500 Fluoview using a 100X objective. Cells labeled with a single fluorochrome (either Alexa 488/FITC or Alexa 594/TR) were checked in both optical channels for cross talk. No leakage of signal was observed between the two channels. The images were assembled using Adobe photoshop 6.0/7.0 software.
Western blotting analysis
Infected or uninfected HeLa cells were processed for western blotting as described previously (Mathew and Bridge, 2007). Equal amounts of total protein were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis using 8% polyacrylamide gels. Proteins were transferred to enhanced chemiluminescence (ECL) nitrocellulose (Amersham Pharmacia) overnight and the membranes were probed with primary antibodies diluted in 5% nonfat dry milk. Goat polyclonal Ab against Mre11 (Santa Cruz Biotechnology) was used at a dilution of 1:250. Rabbit polyclonal Ab against Mdc1 (Bethyl Laboratories) was diluted to 1:2500 for use. Rabbit polyclonal Ab against Rad50 (Genetex) or Nbs1 (Cell Signaling) was used at a dilution of 1:500. The mouse monoclonal Ab against E2-72kDa (A. Levine) was diluted 1:100 for use. Protein blots were incubated with 1:1500 dilution of horseradish peroxidase conjugated anti-goat (Santa Cruz Biotechnology), anti-mouse or anti-rabbit IgG secondary antibodies in 5% nonfat dry milk. Detection of proteins was performed by incubating blots with ECL reagent and subsequently exposing them to ECL hyperfilm (Amersham). For phosphorimaging analysis of proteins, a 1:2000 dilution of alkaline phosphatase conjugated goat anti-rabbit IgG secondary Ab (Sigma Aldrich) was used with enhanced chemifluorescence (ECF) substrate (Amersham) and detected on a STORM 860 phosphorimager (Molecular Dynamics). Images were analyzed using ImageQuant® 5.2 (Molecular Dynamics) software to quantify the amount of protein.
Viral DNA analysis
Isolation of total DNA was performed from infected and uninfected cells as described previously (Bridge and Ketner, 1989). 15 µg of total DNA from each sample was digested with Eco RI and electrophoresed through a 1% agarose gel. DNA was transferred to Hybond-N nylon membrane (GE Healthcare/Amersham) according to manufacturer’s specifications, and used for Southern blotting. A 32P-labeled probe was synthesized from Ad5 genomic DNA using the multiprime DNA labeling system (GE Healthcare/Amersham). Hybridization with 5 × 106 cpm/ml probe was performed at 65°C for 20h as described (Sambrook et al., 1989). Levels of viral DNA were quantified by phosphorimaging analysis of the Ad Eco RI C fragment. Scanned images were analyzed using ImageQuant 5.2® (Molecular Dynamics) software to quantify the amount of DNA.
RNAi analysis
HeLa cells seeded onto 35mm tissue culture dishes at 50% confluency were transfected with siRNA according to manufacturer’s specifications (Dharmacon Technologies) at 200pmoles/plate. siRNA against GAPD (positive control) (siCONTROL Human GAPD Duplex D-001140-01-05) and non-targeting scrambled siRNA (negative control) (siCONTROL non targeting siRNA pool D-001206-13-05) and a lipofection reagent (D1) control were also included as controls for the experiments. siRNA pools specific to human Rad50 (siGENOME smart pool M-005232-01) or Nbs1 (siGENOME smart pool M-009641-01) were used to knockdown the expression of these proteins. Levels of the Rad50 or Nbs1 protein after siRNA transfection were analyzed using Western blotting and immunofluorescence. HeLa cells were cultured with siRNA for a minimum of 96 hours before infecting with Ad5 or H5dl1007 viruses for an additional 24h. Viral DNA analysis was then performed as described above.
Chromatin immunoprecipitation (chIP)
ChIP experiments were performed using a variation of the methods described in Ostapchuk et al., 2005 and Rosenke et al., 2006. Briefly, 4-5 × 108 cells were infected for various times. Cell monolayers in 10cm dishes were incubated with 4mls of 1% formaldehyde in DMEM lacking fetal bovine serum for 10 min. Following this 125mM glycine was added and incubated for 5 min to stop the formaldehyde treatment. The cells were washed with ice-cold PBS twice, scraped into PBS supplemented with a cocktail of protease inhibitors (0.1 µM PMSF, 20 µg/mL Leupeptin and 20 µg/mL Aprotinin) and centrifuged for 4 min at 1000Xg at 4°C. The cells were lysed in SDS lysis buffer (50 mM Tris-HCl [pH 8.0], 10 mM EDTA, 1% SDS) and then sonicated 8X for 15 seconds. Samples were clarified by centrifugation, and pre-cleared by incubating with Gammabind sepharose (Amersham Pharmacia) followed by centrifugation to remove proteins bound non specifically to the sepharose. The supernatants were then used for immunoprecipitation with the following antibodies: goat polyclonal anti-Mre11 (4µg) (Santa Cruz biotechnology), mouse monoclonal anti-PI3K (4ug) (Santa Cruz biotechnology), mouse monoclonal anti-E2-72kDa (1:50 dilution) (A. Levine) or rabbit polyclonal anti-Mdc1 (2µg) (Bethyl Labs) using manufacturer’s specifications. Samples were incubated overnight with the specific antibodies at 4°C with gentle rotation. Gammabind was added and the samples were incubated for 1 hr at 4°C. Washes were performed as described in Ostapchuk et al., 2005. The samples were eluted by incubating with freshly prepared elution buffer (50mM sodium deoxycholate, 1% SDS) for 15 min at room temperature with rotation. The gammabind was removed by centrifugation and supernatants were collected. The elution step was performed two more times. Formaldehyde crosslinks were reversed by adding 150mM NaCl and incubating the samples overnight at 65°C. Proteinase K digestion was performed at 45°C for 1 hour, followed by two phenol-chloroform extraction steps and precipitation with ethanol. The precipitated DNA was resuspended in nuclease free water and used in PCR reactions with primers (E1b forward – 5’ taatgagcttgatctgctggcgc 3’; E1b reverse – 5’ accatgttatgcttaatcacagc 3’) specific to the Ad E1b region.
Acknowledgements
We are very grateful to Drs. Karen Cerosaletti and Patrick Concannon for the NBS-ILB1 LXIN (vector only) and Mre11-NLS (cl.12) cells, and Arnold Levine and Thomas Linné for providing E2-72kDa antibodies used in this study. We also wish to thank all the members of our laboratory for their suggestions and support. This research was supported by the National Cancer Institute (grant CA82111), and awards from Miami University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aspegren A, Rabino C, Bridge E. Organization of splicing factors in adenovirus-infected cells reflects changes in gene expression during the early to late phase transition. Exp. Cell Res. 1998;245:203–213. doi: 10.1006/excr.1998.4264. [DOI] [PubMed] [Google Scholar]
- Assenmacher N, Hopfner KP. MRE11/RAD50/NBS1: complex activities. Chromosoma. 2004;63:631–638. doi: 10.1007/s00412-004-0306-4. [DOI] [PubMed] [Google Scholar]
- Bridge E, Ketner G. Redundant control of adenovirus late gene expression by early region 4. J. Virol. 1989;63:631–638. doi: 10.1128/jvi.63.2.631-638.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR, 3rd, Hays L, Morgan WF, Petrini JH. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double strand break repair to the cellular DNA damage response. Cell. 1998;93:477–486. doi: 10.1016/s0092-8674(00)81175-7. [DOI] [PubMed] [Google Scholar]
- Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003;22:6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for initial recognition of DNA breaks. Nat. Cell Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- Cerosaletti K, Wright J, Concannon P. Active role for nibrin the kinetics of ATM activation. Mol. Cell Biol. 2006;26:1691–1699. doi: 10.1128/MCB.26.5.1691-1699.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly JC, deLeau ES, Leach DR. Nucleolytic processing of a protein-bound DNA end by the E. coli SbcCD (MR) complex. DNA Rep. 2003;2:795–807. doi: 10.1016/s1568-7864(03)00063-6. [DOI] [PubMed] [Google Scholar]
- D’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- de Jager M, Wyman C, van Gent DC, Kanaar R. DNA end binding specificity of human Rad50/Mre11 is influenced by ATP. Nucleic Acids Res. 2002;30:4425–4431. doi: 10.1093/nar/gkf574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai-Mehta A, Cerosaletti KM, Concannon P. Distinct functional domains of nibrin mediate Mre11 binding, focus formation and nuclear localization. Mol. Cell Biol. 2001;21:2184–2191. doi: 10.1128/MCB.21.6.2184-2191.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolganov GM, Maser RS, Novikov A, Tostov L, Chong S, Bressan DA, Petrini JH. Human Rad50 is physically associated with human Mre11:identification of a conserved multiprotein complex implicated in recombinational DNA repair. Mol. Cell Biol. 1996;16:4832–4841. doi: 10.1128/mcb.16.9.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JD, Hearing P. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 2005;7:6207–6215. doi: 10.1128/JVI.79.10.6207-6215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434:605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- Goldberg M, Stucki M, Falck J, D’Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. MDC1 is required for the intra S phase DNA damage checkpoint. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, Karcher A, Henderson B, Bodmer JL, McMurray CT, Carney JP, Petrini JH, Trainer JA. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002;418:562–566. doi: 10.1038/nature00922. [DOI] [PubMed] [Google Scholar]
- Jayaram S, Bridge E. Genome concatenation contributes to the late gene expression defect of an adenovirus E4 mutant. Virology. 2005;342:286–296. doi: 10.1016/j.virol.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Kim JE, Minter-Dykhouse K, Chen J. Signaling networks controlled by the MRN complex and MDC1 during early DNA damage responses. Mol Carcinog. 2006;4:403–408. doi: 10.1002/mc.20221. [DOI] [PubMed] [Google Scholar]
- Kraakman-van der Zwet M, Overkamp WJ, Friedl AA, Klein B, Verhaegh GW, Jaspers NG, Midro AT, Eckardt-Schupp F, Lohman PH, Zdzienicka MZ. Immortalization and characterization of Nijmegen Break syndrome fibroblasts. Mutat. Res. 2003;434:17–27. doi: 10.1016/s0921-8777(99)00009-9. [DOI] [PubMed] [Google Scholar]
- Lisby M, Mortensen UH, Rothstein R. Colocalization of multiple DNA double strand breaks at a single Rad52 repair centre. Nat. Cell Biol. 2003;5:572–577. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- Lisby M, Rothstein R. DNA repair: keeping it together. Curr. Biol. 2004;14:R994–R996. doi: 10.1016/j.cub.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Lou Z, Chen BP, Asaithamby A, Minter-Dykhouse K, Chen DJ, Chen J. MDC1 regulates DNA PK autophosphorylation in response to DNA damage. J. Biol. Chem. 2004;279:46359–46362. doi: 10.1074/jbc.C400375200. [DOI] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis J, van Deursen J, Nussenzweig A, Paull T, Alt FW, Chen J. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell. 2006;21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Luo G, Yao MS, Bender CF, Mills M, Bladl AR, Bradley A, Petrini JH. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc. Natl. Acad. Sci. U S A. 1999;96:7376–7381. doi: 10.1073/pnas.96.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew SS, Bridge E. The cellular Mre11 protein interferes with Adenovirus E4 mutant DNA replication. Virology. 2007;365:346–355. doi: 10.1016/j.virol.2007.03.049. [DOI] [PubMed] [Google Scholar]
- Mochan TA, Venere M, DiTullio RA, Halazonetis TD. 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res. 2003;63:8586–8591. [PubMed] [Google Scholar]
- Ostapchuk P, Yang J, Auffarth E, Hearing P. Functional interaction of the adenovirus IVa2 protein with adenovirus type 5 packaging sequences. J. Virol. 2005;79:2831–2838. doi: 10.1128/JVI.79.5.2831-2838.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Gellert M. The 3’ to 5’ exonuclease activity of Mre11 facilitates repair of DNA double strand breaks. Mol. Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- Paull TT, Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Gellert M. A mechanistic basis for Mre11-directed DNA joining at microhomologies. Proc. Natl. Acad. Sci. U S A. 2000;97:6409–6414. doi: 10.1073/pnas.110144297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 2003;13:458–462. doi: 10.1016/s0962-8924(03)00170-3. [DOI] [PubMed] [Google Scholar]
- Petrini JH, Walsh ME, DiMare C, Chen XN, Korenberg JR, Weaver DT. Isolation and characterization of the human MRE11 homologue. Genomics. 1995;29:80–86. doi: 10.1006/geno.1995.1217. [DOI] [PubMed] [Google Scholar]
- Philipson L. Adenovirus assay by the fluorescent cell counting procedure. Virology. 1961;15:263–268. doi: 10.1016/0042-6822(61)90357-9. [DOI] [PubMed] [Google Scholar]
- Raderschall E, Golub EI, Haaf T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc. Natl. Acad. Sci. U S A. 1995;96:1921–1926. doi: 10.1073/pnas.96.5.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenke K, Samuel MA, McDowell ET, Toerne MA, Fortunato EA. An intact sequence-specific DNA-binding domain is required for human cytomegalovirus-mediated sequestration of p53 and may promote in vivo binding to the viral genome during infection. Virology. 2006;348:19–34. doi: 10.1016/j.virol.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fitsch EF, Maniatis T. Molecular Cloning: A laboratory manual. Cold Spring Harbor: Cold Spring Harbor Press; 1989. [Google Scholar]
- Sharples GJ, Leach DR. Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast. Mol. Microbiol. 1995;17:1215–1217. doi: 10.1111/j.1365-2958.1995.mmi_17061215_1.x. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- Tauchi H, Kobayashi J, Morishima K, Matsuura S, Nakamura A, Shiraishi T, Ito E, Masnada D, Delia D, Komatsu K. The forkhead-associated domain of NBS1 is essential for nuclear foci formation after irradiation but not essential for hRAD50[middle dot]hMRE11[middle dot]NBS1 complex DNA repair activity. J. Biol. Chem. 2001;276:12–15. doi: 10.1074/jbc.C000578200. [DOI] [PubMed] [Google Scholar]
- Trujillo KM, Yuan SS, Lee EY, Sung P. Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11, and p95. J. Biol. Chem. 1998;273:21447–21450. doi: 10.1074/jbc.273.34.21447. [DOI] [PubMed] [Google Scholar]
- Trujillo KM, Sung P. DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50*Mre11 complex. J. Biol. Chem. 2001;276:35458–35464. doi: 10.1074/jbc.M105482200. [DOI] [PubMed] [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003;22:5612–5621. doi: 10.1093/emboj/cdg541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DH, Ketner G. A cell line that supports the growth of a defective early region 4 deletion mutant of human adenovirus type 2. Proc. Natl. Acad. Sci. USA. 1983;80:5383–5386. doi: 10.1073/pnas.80.17.5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman MD, Ornelles DA. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene. 2005;24:7686–7696. doi: 10.1038/sj.onc.1209063. [DOI] [PubMed] [Google Scholar]
- Yamaguchi-Iwai Y, Sonoda E, Sasaki MS, Morrison C, Haraguchi T, Hiraoka Y, Yamashita YM, Yagi T, Takata M, Price C, Kakazu M, Takeda S. Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J. 1999;18:6619–6629. doi: 10.1093/emboj/18.23.6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ma Z, Treszezamsky A, Powell SN. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat. Struct. Mol. Biol. 2005;12:902–909. doi: 10.1038/nsmb991. [DOI] [PubMed] [Google Scholar]
- Zhou J, Lim CU, Li JJ, Cai L, Zhang Y. The role of NBS1 in the modulation of PIKK family proteins ATM and ATR in the cellular response to DNA damage. Cancer Lett. 2006;243:9–15. doi: 10.1016/j.canlet.2006.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Petersen S, Tessarollo L, Nussenzweig A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol. 2001;11:105–109. doi: 10.1016/s0960-9822(01)00019-7. [DOI] [PubMed] [Google Scholar]