

A combination of spectroscopic and crystallographic data have been used to identify the native metal-ion preference of GpdQ. These data suggest that GpdQ is a homobinuclear non-haem iron enzyme.
Keywords: glycerophosphodiesterases, metallohydrolases, binuclear, malonate, anomalous scattering, nonhaem
Abstract
The structure of a malonate-bound form of the glycerophosphodiesterase from Enterobacter aerogenes, GpdQ, has been refined at a resolution of 2.2 Å to a final R factor of 17.1%. The structure was originally solved to 2.9 Å resolution using SAD phases from Zn2+ metal ions introduced into the active site of the apoenzyme [Jackson et al. (2007 ▶), J. Mol. Biol. 367, 1047–1062]. However, the 2.9 Å resolution was insufficient to discern significant details of the architecture of the binuclear metal centre that constitutes the active site. Furthermore, kinetic analysis revealed that the enzyme lost a significant amount of activity in the presence of Zn2+, suggesting that it is unlikely to be a catalytically relevant metal ion. In this communication, a higher resolution structure of GpdQ is presented in which malonate is visibly coordinated in the active site and analysis of the native metal-ion preference is presented using atomic absorption spectroscopy and anomalous scattering. Catalytic implications of the structure and its Fe2+ metal-ion preference are discussed.
1. Introduction
The glycerophosphodiesterases (EC 3.1.4.46) constitute a ubiquitous family of proteins that are involved in hydrolysis of the 3′–5′ phosphodiester bond between sn-glycerol-3-phosphate and a leaving alcohol (such as ethanolamine or choline) in glycerophosphodiesters (Larson et al., 1983 ▶). Recently, we crystallized and solved the structure of a novel glycerophosphodiesterase from Enterobacter aerogenes (Jackson, Carr, Kim, Liu & Ollis, 2006 ▶; Jackson et al., 2007 ▶). In addition to its native function, GpdQ has received attention because of its potential utility in bioremediation, either as part of a catabolic pathway for the degradation of phosphotriesters (McLoughlin et al., 2004 ▶) or in isolation for the breakdown of toxic phosphodiesters (Ghanem et al., 2007 ▶).
Previous structures of GpdQ have revealed that the enzyme is a metallophosphoesterase of the α/β-sandwich structural fold (Mitić et al., 2006 ▶) homologous to the purple acid phosphatases (Klabunde et al., 1996 ▶), the serine/threonine protein phosphatases (Rusnak & Mertz, 2000 ▶), Mre11 nuclease (Höpfner et al., 2001 ▶) and 5′-nucleotidase (Knöfel & Sträter, 1999 ▶). Unfortunately, the low resolution (2.9 Å) of the previous structure meant that much of the fine detail of the active site could not be distinguished beyond the characterization of the relative affinities of the two metal-ion-binding sites. It was found that the metal ion in the α site was bound at full occupancy, whereas the β site was only partially occupied, suggesting that GpdQ contains tight (α) and loose (β) metal-ion-binding sites. Other important features of binuclear active sites, such as the water coordination and the identification of a bridging ligand, could not be observed. Thus, in the absence of a high-resolution structure, little information relevant to the catalytic mechanism could be obtained.
Metalloenzymes typically have particular metal-ion preferences, which are usually reflected in their physiological function. For instance, the purple acid phosphatases have a preference for the strong Lewis acid Fe3+ at the α site and are thus able to efficiently generate a nucleophile at low pH values (Schenk et al., 2005 ▶, 2008 ▶). Other binuclear metalloenzymes have been shown to have heterobinuclear metal centres in which each site has a preference for a different metal ion. For instance, the bacterial phosphotriesterases have a preference for Fe2+ at the α site and a preference for Zn2+ at the β site, consistent with the roles of these metals in nucleophile generation and substrate binding, respectively (Jackson, Carr, Kim, Liu, Herrald et al., 2006 ▶). Thus, characterization of the native metal-ion preference of GpdQ is an important step towards understanding its physiological function and catalytic mechanism. Spectroscopic methods are useful for investigating the presence and quantity of metals in enzymes. However, in binuclear metalloenzymes the relative populations of metal ions at each position can best be understood in detail by measuring their anomalous diffraction with tuneable synchrotron X-ray sources (Sommerhalter et al., 2005 ▶).
2. Materials and methods
2.1. Enzyme purification and crystallography
The protein was expressed, purified and crystallized as described previously (Jackson, Carr, Kim, Liu & Ollis, 2006 ▶; Jackson et al., 2007 ▶), except that the protein was not incubated in the presence of metal chelators prior to crystallization and atomic absorption spectroscopy (AAS). The protein concentration was determined by measuring the absorbance at 280 nm in 6 M urea using the extinction coefficient 39 880 M −1 cm−1, which was calculated using the ProtParam tool as implemented in the ExPASy web interface (http://www.expasy.org/tools/protparam.html; Gasteiger et al., 2005 ▶). Thus, the protein contained the metals that it incorporated during expression. Crystals were isomorphous to those previously used in crystallographic experiments, although they were ∼2–3 weeks old, in contrast to the previous crystals, which were 3–4 months old.
Diffraction data were recorded under cryogenic conditions (100 K) at the Advanced Photon Source (Argonne National Laboratory, Illinois, USA) on beamline 19-ID using an ADSC Q315 area detector (Table 1 ▶). A fluorescence scan was performed to investigate the presence of metals within the crystal. In order to determine whether the α or β sites were preferentially occupied by Zn2+, data sets were collected from isomorphous crystals from the same drop at 1.2947 and 1.2804 Å, corresponding to wavelengths immediately before and after the Zn K absorption edge (1.2837 Å). Radiation damage to the first crystal precluded its use for data collection at both wavelengths. Data were scaled and integrated using DENZO and SCALEPACK (Otwinowski & Minor, 1997 ▶), copying the free R flags from a set of reflections from a previously solved data set (Jackson et al., 2007 ▶). Refinement was carried out using REFMAC (Murshudov et al., 1997 ▶; Table 1 ▶) between rounds of manual model building and water location using Coot (Emsley & Cowtan, 2004 ▶). Malonate was added to the structure based on electron density present in mF o − DF c maps (Fig. 1 ▶). Anomalous electron-density maps were calculated for both data sets and used to ascertain the relative occupancies of Fe and Zn metal ions within the active site. The B factor of the malonate ion was higher than its coordinating ligands, which initially suggested that the molecule was present at low occupancy. However, when the occupancy was lowered positive density arose at the site. Thus, the high B factor may indicate genuine mobility, as observed previously for other ligands bound at the active sites of metalloenzymes (Jackson et al., 2005 ▶). Structures were validated using MOLPROBITY (Davis et al., 2007 ▶). All rendered images were produced with PyMOL (http://pymol.sourceforge.net/; DeLano, 2003 ▶) using subunit B of the dimer.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Remote | Zn K edge | |
|---|---|---|
| Space group | P213 | P213 |
| Unit-cell parameters (Å) | a = 164.16 | a = 164.34 |
| Matthews coefficient (Å3 Da−1) | 5.98 | 6.00 |
| Solvent content (%) | 79.46 | 79.53 |
| Data collection | ||
| Temperature (K) | 100 | 100 |
| Wavelength (Å) | 1.2947 | 1.2804 |
| Resolution (Å) | 30.00–2.20 (2.34–2.20) | 25.00–2.40 (2.55–2.40) |
| Total reflections | 284663 (31379) | 339743 (39534) |
| Unique reflections | 73371 (11622) | 57620 (9413) |
| Multiplicity | 3.9 (2.7) | 5.9 (4.2) |
| 〈I/σ(I)〉 | 20.9 (1.6) | 23.1 (2.4) |
| Completeness (%) | 98.2 (94.1) | 99.6 (98.5) |
| Rmerge† (%) | 6.9 (47.4) | 10.6 (53.1) |
| Anomalous completeness (%) | 89.8 | 96.6 |
| Refinement | ||
| No. of atoms | 4445 | 4438 |
| Average B values (Å2) | ||
| Protein | 50.4 | 45.8 |
| Metals | 46.4 | 49.8 |
| Ligands | 65.0 | 65.8 |
| Waters | 48.1 | 43.0 |
| Rwork/Rfree‡ | 17.0/18.9 (28.5/29.9) | 15.9/17.9 (26.1/26.5) |
| E.s.u. (maximum likelihood) (Å2) | 6.81 | 7.86 |
| Ramachandran outliers | 0.0 | 0.0 |
| Ramachandran favoured | 97.0 | 97.0 |
| R.m.s.d. from standard geometry | ||
| Bond lengths (Å) | 0.014 | 0.014 |
| Bond angles (°) | 1.602 | 1.607 |
| PDB code | 2zo9 | 2zoa |
R
merge = 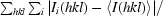
 . 〈I(hkl)〉 is the average intensity of the i observations.
. 〈I(hkl)〉 is the average intensity of the i observations.
R
work = 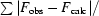
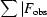 |; 5% of data that were excluded from refinement were used to calculate R
free.
|; 5% of data that were excluded from refinement were used to calculate R
free.
Figure 1.

The coordination of malonate in the active site of GpdQ, shown in stereo. OMIT electron density (mF o − DF c) corresponding to malonate within the active site is shown contoured at 3σ. Carboxylic acid groups at either end of the malonate molecule are coordinated in a μ-1,1 bridging mode between the metals and by the NH groups of Asn80 and His81 in the second shell of the active site.
2.2. Substrate docking
The substrate-binding mode of the native substrate of GpdQ, glycerophosphoethanolamine (GPE), was investigated using the program CDOCKER (Wu et al., 2003 ▶) as implemented in the Accelrys Discovery Studio. H atoms were added to GpdQ and GPE and the CHARMm force field was used to assign partial charges to the ligands. The malonate molecule and waters close to the active site were removed. To search for chelating binding modes, no bridging hydroxide or terminal hydroxides were included in the model. To search for terminal binding modes, a terminal hydroxide and a bridging hydroxide were added to the model based on the positions of the hydroxyl groups of malonate. For both searches GPE was initially manually docked into GpdQ using Coot (Emsley & Cowtan, 2004 ▶). The CHARMm forcefield was used throughout the docking procedure (Brooks et al., 1983 ▶). Ten random ligand conformations of GPE were generated and docked into the active site, and the best ten poses were refined.
2.3. Atomic absorption spectroscopy (AAS)
The quantity of protein-bound metal ions was determined in triplicate by AAS using a Varian Spectra AA 220FS. Standard solutions for Fe, Co, Zn, Ni, Mn and Cu ranging in concentration from 20 to 100 p.p.b. were prepared from analytical stock solutions (Merck, Germany) using MilliQ water. Protein samples were diluted with desalted buffer (50 mM HEPES pH 7.0). This buffer and Milli-Q were used as controls in AAS measurements; no measurable quantities of metal ions were detected. The estimated error for each metal ion was less than 5%.
2.4. Enzyme kinetics
Determination of the steady-state kinetic parameters of purified GpdQ was achieved by monitoring the production of p-nitrophenolate at 410 nm (∊410 = 16 600 M −1 cm−1) at various concentrations of bis(p-nitrophenyl phosphate) (Sigma) ranging from 100 µM to 20 mM. The reaction was buffered with 50 mM HEPES pH 8.4 and contained 100 µM of either FeSO4 or ZnSO4, both of which were freshly prepared. The values of k cat and K m were determined by fitting the data to the Michaelis–Menten equation using the program Kaleidagraph.
3. Results and discussion
3.1. Active-site architecture and coordination of malonate: implications for substrate binding
Previous structures indicated the presence of two metal ions in the active site with differing occupancies, indicating that the α site has a significantly higher affinity than the β site. No other notable features were evident. The structures presented here also show higher occupancy at the α site (0.75) than the β site (0.55). In addition, the higher resolution allows the coordination of malonate to the binuclear metal centre to be observed. Malonate was identified at the active site based on mF o − DF c electron density and its presence in the crystallization buffer (Fig. 1 ▶). The malonate-ion coordinates primarily to the α metal ion, with O atoms from one of the carboxylic acid groups coordinated terminally to the metal ion and in a tridentate mode between the α (2.6 Å) and β (2.4 Å) metal ions and the main-chain carbonyl group of His195 (2.7 Å). These positions were expected to be occupied by a solvent molecule based on homology to other members of this family (see, for example, Guddat et al., 1999 ▶; Höpfner et al., 2001 ▶). The other carboxylic acid group of the malonate ion is shown to coordinate to Asn80 (2.6 Å) and weakly to His81 (3.2 Å).
Although malonate is not itself a good mimic of the phosphodiester substrates of GpdQ, the carboxylic acid groups of malonate may approximate the delocalized monoanionic charge around the two phosphoryl O atoms of phosphodiesters at physiological pH. Thus, the coordination of the carboxylic acid groups of malonate may be informative regarding possible modes of substrate binding. The first possibility revealed by this structure is that the substrates may displace the μ-hydroxo bridge, as observed previously when phosphodiester substrates bound to a binuclear metallo-phosphotriesterase (Jackson et al., 2008 ▶) and an exonuclease (Hamdan et al., 2002 ▶). The native substrate of GpdQ, GPE, has been manually docked in this mode previously (Jackson et al., 2007 ▶). The second possibility (Fig. 2 ▶) is that the carboxyl groups of malonate have displaced the terminal and bridging solvent molecules and that substrates may adopt a coordination mode terminal to the β metal ion, involving hydrogen bonding to the NH groups of Asn80 and His81. This substrate-binding mode has been proposed for some purple acid phosphatases, supported by kinetic and structural data (Funhoff et al., 2005 ▶; Lindqvist et al., 1999 ▶).
Figure 2.

The best docking pose for GPE within the active site of GpdQ, shown in stereo. The coordination of malonate (MAL) in the crystal structure is shown by lines. Solvent molecules are modelled based on the positions of the carboxyl groups of malonate. The phosphoryl O atoms of the substrate are within hydrogen-bonding distance of the NH groups of Asn80 and His81 and the terminal water/hydroxide is aligned for SN2 nucleophilic attack at the electrophilic phosphorus of the substrate.
To test these hypotheses, GPE was docked into the active site of GpdQ using CDOCKER, a molecular-dynamics (MD) based docking program that involves the utilization of a CHARMm-based MD simulation scheme in which random ligand conformations generated from high-temperature MD are translated into the binding site. Binding poses are then searched for using random rigid-body rotations and simulated annealing with a grid potential before minimization with a full force-field potential is used to refine the ligand poses. No suitable poses were found that involved the substrate displacing the bridging hydroxide. In contrast, the best pose, shown in Fig. 2 ▶, involves the terminal coordination of GPE at the β metal ion with the phosphoryl O atoms of the substrate within hydrogen-bonding distance of the NH groups of Asn80 and His81. Additionally, the alignment of the terminal solvent molecule with the electrophilic phosphorus and leaving group is consistent with the SN2 nucleophilic displacement mechanism proposed for other binuclear metallophosphoesterases (Höpfner et al., 2001 ▶; Jackson et al., 2008 ▶).
3.2. Metal-ion preference at the α and β sites
The metal-ion content of purified GpdQ was analysed using AAS. The results, listed in Table 2 ▶, reveal that Fe constitutes the majority of the metal ions within the binuclear active site (1.3 metals per binuclear active site), although a significant amount of zinc was also present (0.3 metals per binuclear active site). The content of other transition-metal ions was below the detection limit of the instrument. The overall metal content was less than two metal ions per active site, which is consistent with the partial occupancy of metals within the structure. However, it is not clear from the AAS data whether there is a particular preference of either metal-binding site for Fe or Zn.
Table 2. Metal:monomer stochiometry from atomic absorption spectroscopy.
| Metal | No. per monomer |
|---|---|
| Fe | 1.3 |
| Zn | 0.3 |
| Cu | <0.05 |
| Co | <0.05 |
| Mn | <0.05 |
| Ni | <0.05 |
A fluorescence scan was performed on a crystal of GpdQ. Although we did not obtain quantitative data from the fluorescence scan, it was consistent with the AAS results in that the only significant peaks corresponded to Fe and Zn and the Fe peak was the highest (data not shown). To investigate the location of the Zn ions, X-ray data sets were collected at wavelengths before and after the Zn K edge from isomorphous crystals from the same crystallization drop. An anomalous difference electron-density map calculated from the 1.295 Å wavelength data set (where the theoretical anomalous scattering from Fe is ∼60% of that obtained at the Fe K edge) demonstrates that the α and β metal-ion sites are occupied by approximately equal amounts of Fe (Fig. 3 ▶ a). On moving past the Zn K edge, a significant peak is seen in the anomalous difference density map (contoured at 5σ), solely at the α site (Fig. 3 ▶ b). Thus, it appears from this analysis that the majority of the enzyme exists in a homobinuclear Fe–Fe form, with a minority of the protein in either heterobinuclear Zn–Fe or Fe or Zn mononuclear forms. When overexpressed, metalloenzymes can often promiscuously incorporate metal ions other than those preferred naturally (Jackson, Carr, Kim, Liu, Herrald et al., 2006 ▶), which may explain the small amount of Zn that has been incorporated in this study. Based on these data, it is clear that the metal-ion preference at both the α and β sites is for Fe2+. The effects of these metals on activity was tested kinetically (Table 3 ▶), demonstrating that the turnover of bis(p-nitrophenyl phosphate) by GpdQ in the presence of Fe2+ is ∼16-fold higher than in the presence of Zn2+ (k cat = 0.176 s−1 for GpdQ–Zn2+ versus k cat = 2.78 s−1 for GpdQ–Fe2+), supporting the notion that Fe2+ is the native metal ion. Additionally, several close relatives of GpdQ are known to exist natively as Fe–Fe non-haem iron phosphatases, including mammalian purple acid phosphatases (Wang et al., 1992 ▶; Mitić et al., 2006 ▶) and the Ser/Thr protein phosphatase from bacteriophage λ (Reiter & Rusnak, 2004 ▶).
Figure 3.

Anomalous difference electron-density maps contoured at 5σ. (a) At a wavelength preceding the Zn K edge (1.295 Å) the residual scattering from the Fe present in the samples indicates that both the α and β sites are occupied at similar levels. (b) Upon moving past the Zn K edge (1.280 Å) the anomalous scattering of Zn reveals that Zn is mostly coordinated at the α site.
Table 3. Kinetic parameters for the hydrolysis of bis(p-nitrophenyl phosphate) by GpdQ in the presence of 100 µM Fe2+ or Zn2+ .
| Protein | kcat (s−1) | Km (mM) | kcat/Km (s−1 M−1) |
|---|---|---|---|
| GpdQ–Fe2+ | 2.78 ± 0.26 | 7.0 ± 1.6 | 400.0 |
| GpdQ–Zn2+ | 0.176 ± 0.008 | 3.5 ± 0.4 | 49.8 |
4. Conclusion
A combination of spectroscopic and X-ray crystallographic data have been used to identify the native metal-ion preference of GpdQ. These data suggest that GpdQ is a homobinuclear non-haem iron enzyme. The structure presented here reveals the active-site architecture of GpdQ in detail for the first time and the binding mode of the malonate ion points to two possible substrate-binding modes. The second binding mode, involving coordination by the NH groups of Asn80 and His81 in the second shell of the active site, is consistent with a model for substrate binding proposed for the homologous enzyme purple acid phosphatase and nucleophilic attack by a terminally coordinated solvent molecule. Further work is currently under way using spectroscopy, kinetic analysis and X-ray crystallography to fully elucidate the substrate-binding mode and catalytic mechanism of GpdQ.
Supplementary Material
PDB reference: glycerophosphodiesterase, 2zo9, r2zo9sf
PDB reference: 2zoa, r2zoasf
Acknowledgments
We thank the staff of the SBC-CAT beamline for their technical support during data collection. Use of the SBC-CAT beamline was supported by a grant from the Australian Synchrotron Radiation Project (ASRP) to AO and CJ. This research was supported by an Australian Research Council (DP0664039) grant to DO and GS.
References
- Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. & Karplus, M. (1983). J. Comput. Chem.4, 187–217.
- Davis, I. W., Leaver-Fay, A., Chen, V. B., Block, J. N., Kapral, G. J., Wang, X., Murray, L. W., Arendall, W. B., Snoeyink, J., Richardson, J. S. & Richardson, D. C. (2007). Nucleic Acids. Res.35, W375–W383. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2003). PyMOL Reference Manual. DeLano Scientific LLC, San Carlos, USA.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Funhoff, E. G., Wang, Y., Andersson, G. & Averill, B. A. (2005). FEBS J.272, 2968–2977. [DOI] [PubMed]
- Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D. & Bairoch, A. (2005). The Proteomics Protocols Handbook, edited by J. M. Walker, pp. 571–607. Totowa: Humana Press.
- Ghanem, E., Li, Y., Xu, C. & Raushel, F. M. (2007). Biochemistry, 46, 9032–9040. [DOI] [PubMed]
- Guddat, L. W., McAlpine, A. S., Hume, D., Hamilton, S., de Jersey, J. & Martin, J. L. (1999). Structure, 15, 757–767. [DOI] [PubMed]
- Hamdan, S., Carr, P. D., Brown, S. E., Ollis, D. L. & Dixon, N. E. (2002). Structure, 10, 535–546. [DOI] [PubMed]
- Höpfner, K. P., Kärcher, A., Craig, L., Woo, T. T., Carney, J. P. & Tainer, J. A. (2001). Cell, 105, 473–485. [DOI] [PubMed]
- Jackson, C. J., Carr, P. D., Kim, H.-K., Liu, J.-W., Herrald, P., Mitic, N., Schenk, G., Smith, C. A. & Ollis, D. L. (2006). Biochem. J.397, 501–508. [DOI] [PMC free article] [PubMed]
- Jackson, C. J., Carr, P. D., Kim, H.-K., Liu, J.-W. & Ollis, D. L. (2006). Acta Cryst. F62, 659–661. [DOI] [PMC free article] [PubMed]
- Jackson, C. J., Carr, P. D., Liu, J.-W., Watt, S. J., Beck, J. L. & Ollis, D. L. (2007). J. Mol. Biol.367, 1047–1062. [DOI] [PubMed]
- Jackson, C. J., Foo, J.-L., Kim, H.-K., Carr, P. D., Liu, J.-L., Salem, G. & Ollis, D. L. (2008). J. Mol. Biol.375, 1189–1196. [DOI] [PubMed]
- Jackson, C. J., Kim, H.-K., Carr, P. D., Liu, J.-W. & Ollis, D. L. (2005). Biochem. Biophys. Acta, 1752, 56–64. [DOI] [PubMed]
- Klabunde, T., Sträter, N., Fröhlich, R., Witzel, H. & Krebs, B. (1996). J. Mol. Biol.259, 737–748. [DOI] [PubMed]
- Knöfel, T. & Sträter, N. (1999). Nature Struct. Biol.6, 448–453. [DOI] [PubMed]
- Larson, T. J., Ehrmann, M. & Boos, W. (1983). J. Biol. Chem.258, 5428–5432. [PubMed]
- Lindqvist, Y., Johansson, E., Kaija, H., Vihko, P. & Schneider, G. (1999). J. Mol. Biol.291, 135–147. [DOI] [PubMed]
- McLoughlin, S. Y., Jackson, C. J., Liu, J.-W. & Ollis, D. L. (2004). Appl. Environ. Microbiol.70, 404–412. [DOI] [PMC free article] [PubMed]
- Mitić, N., Smith, S. J., Neves, A., Guddat, L. W., Gahan, L. R. & Schenk, G. (2006). Chem. Rev.106, 3338–3363. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Reiter, T. A. & Rusnak, F. (2004). Biochemistry, 43, 782–790. [DOI] [PubMed]
- Rusnak, F. & Mertz, P. (2000). Physiol. Rev.80, 1483–1521. [DOI] [PubMed]
- Schenk, G., Elliot, T. W., Leung, E., Carrington, L. E., Mitic, N., Gahan, L. R. & Guddat, L. W. (2008). BMC Struct. Biol.8, 6. [DOI] [PMC free article] [PubMed]
- Schenk, G., Gahan, L. R., Carrington, L. E., Mitić, N., Valizadeh, M., Hamilton, S. E., de Jersey, J. & Guddat, L. W. (2005). Proc. Natl Acad. Sci. USA, 102, 273–278. [DOI] [PMC free article] [PubMed]
- Sommerhalter, M., Lieberman, R. L. & Rosenweig, A. C. (2005). Inorg. Chem.44, 770–778. [DOI] [PubMed]
- Wang, Z., Ming, L. J., Que, L., Vincent, J. B., Crowder, M. W. & Averill, B. A. (1992). Biochemistry, 31, 5263–5268. [DOI] [PubMed]
- Wu, G., Robertson, D. H., Brooks, C. L. III & Vieth, M. (2003). J. Comput. Chem.24, 1549–1562. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: glycerophosphodiesterase, 2zo9, r2zo9sf
PDB reference: 2zoa, r2zoasf