

l-Methionine γ-lyase 1, a key enzyme in sulfur-containing amino-acid degradation, from the protozoan parasite E. histolytica was crystallized in a form suitable for X-ray structure analysis.
Keywords: l-methionine γ-lyase, EC 4.4.1.11, Entamoeba histolytica
Abstract
l-Methionine γ-lyase (MGL) is a pyridoxal phosphate-dependent enzyme that is involved in the degradation of sulfur-containing amino acids. MGL is an attractive drug target against amoebiasis because the mammalian host of its causative agent Entamoeba histolytica lacks MGL. For the development of anti-amoebic agents based on the structure of MGL, one of two MGL isoenzymes (EhMGL1) was crystallized in the monoclinic space group P21, with unit-cell parameters a = 99.12, b = 85.38, c = 115.37 Å, β = 101.82°. The crystals diffract to beyond 2.0 Å resolution. The presence of a tetramer in the asymmetric unit (4 × 42.4 kDa) gives a Matthews coefficient of 2.8 Å3 Da−1 and a solvent content of 56%. The structure was solved by the molecular-replacement method and structure refinement is now in progress.
1. Introduction
Amoebiasis, which is caused by infection with the enteric protist Entamoeba histolytica, is the second largest parasitic disease after malaria; it affects over 50 million people and results in 70 000 deaths annually (World Health Organization, 1998 ▶). Its major clinical manifestations are amoebic dysentery and extraintestinal abscesses (Stanley, 2003 ▶).
The sulfur-containing amino-acid metabolism of E. histolytica has a number of unique features (Nozaki et al., 2005 ▶; Ali & Nozaki, 2007 ▶). For example, E. histolytica lacks the transsulfuration pathway and thus is unable to convert methionine to cysteine and vice versa. Instead, it possesses l-methionine γ-lyase (MGL; EC 4.4.1.11), which is involved in the degradation of sulfur-containing amino acids (Tokoro et al., 2003 ▶; Sato et al., 2008 ▶). MGL contains pyridoxal 5′-phosphate (PLP) as a cofactor and has been categorized as belonging to the γ-family of PLP-dependent enzymes (Alexander et al., 1994 ▶). MGL catalyzes α,γ- or α,β-elimination of sulfur-containing amino acids and produces ammonia, α-keto acids and volatile thiols such as hydrogen sulfide or methanethiol (Tanaka et al., 1985 ▶). It also catalyzes β- or γ-replacement of cysteine, S-substituted cysteine, methionine and related compounds (Tanaka et al., 1985 ▶). MGL is only present in a limited range of bacteria, protozoa and plants (Nakayama et al., 1984 ▶; Kreis & Hession, 1973 ▶; Yoshimura et al., 2000 ▶; Dias & Weimer, 1998 ▶; Manukhov et al., 2005 ▶; Goyer et al., 2007 ▶; Coombs & Mottram, 2001 ▶; Nozaki et al., 2005 ▶; Rebeille et al., 2006 ▶). Crystal structures have been reported for MGLs from Pseudomonas putida (Motoshima et al., 2000 ▶; Sridhar et al., 2000 ▶), Trichomonas vaginalis (PDB code 1e5f; G. Goodall, J. C. Mottram, G. H. Coombs & A. J. Lapthorn, unpublished work) and Citrobacter freundii (Mamaeva et al., 2005 ▶; Nikulin et al., 2008 ▶).
MGLs from parasitic protozoa are unique in that they exist as two isoenzymes with distinct substrate specificities (e.g. EhMGL1 and EhMGL12 in E. histolytica; 69% amino-acid sequence identity; Tokoro et al., 2003 ▶; Sato et al., 2008 ▶). A previous study demonstrated that EhMGL1, but not EhMGL2, predominantly degrades methionine and cysteine (Sato et al., 2008 ▶). In contrast, trifluoromethionine (TFM), the halogenated analogue of methionine, which exhibits toxicity by being degraded by MGL in Porphyromonas gingivalis, T. vaginalis and E. histolytica (Yoshimura et al., 2002 ▶; Coombs & Mottram, 2001 ▶; Tokoro et al., 2003 ▶), was almost exclusively degraded by EhMGL2. We also showed that the K m of EhMGL1 towards TFM was ninefold lower than that of EhMGL2, while the k cat of EhMGL2 was 22-fold higher than that of EhMGL1 (Sato et al., 2008 ▶), indicating that the catalytic action of MGLs towards TFM might be different. For structure-based design of TFM derivatives, comparison of the tertiary structures of the two EhMGL isotypes should be helpful. We have previously crystallized ligand-free EhMGL2 (Sato et al., 2006 ▶) and determined its X-ray structure as well as those of structures of complexes with substrates and inhibitors (unpublished results). In the present study, we describe the bacterial expression, purification, crystallization and preliminary X-ray diffraction studies of EhMGL1.
2. Materials and methods
2.1. Overexpression and purification of recombinant MGL1
When we attempted to express the EhMGL1 gene (AB094499) in a bacterial expression system (pGEX-6P-1 vector; GE Healthcare Biosciences) as an amino-terminal glutathione S-transferase (GST) fusion protein, a fraction of protein (∼20%) was produced as a truncated form starting from Gly46 (Sato et al., 2008 ▶). This truncation was caused by an inadvertent translation initiation at Met45 which was likely to be a consequence of the similarity of the nucleotide sequence upstream of Met45 to the Shine–Dalgarno sequence. We therefore mutated five nucleotides in this region, which did not cause amino-acid substitution (Sato et al., 2008 ▶). This genetically engineered EhMGL1 construct allowed us to successfully express and purify EhMGL1 protein without truncation. For large-scale preparation, this plasmid was introduced into the Escherichia coli BL21 Star (DE3) strain (Invitrogen) and the transformant was grown in 2 l M9 minimal medium (Sambrook et al., 1989 ▶) supplemented with 1 mM MgSO4, 0.1 mM CaCl2, 3.3 µM FeCl3, 59 µM thiamine–HCl, 82 µM biotin, 20 µM PLP and 50 µg ml−1 ampicillin at 310 K. The medium was prepared with commercially available mineral water to supply trace elements. The induction of expression, affinity purification and on-column digestion of the GST-EhMGL1 fusion protein were carried out as described previously for EhMGL2 (Sato et al., 2006 ▶). The fraction containing GST-removed EhMGL1 was eluted from a GSTrap HP column (1.6 × 2.5 cm; GE Healthcare Biosciences) with 50 mM HEPES–NaOH pH 7.5, 150 mM NaCl and 20 µM PLP and concentrated to 1–2 ml with Amicon Ultra-4 Ultracel-10K. EhMGL1 was further purified by size-exclusion chromatography on a HiPrep 16/60 Sephacryl S-300 column (16 × 60 cm; GE Healthcare Biosciences) equilibrated with 50 mM sodium phosphate pH 7.2 containing 150 mM NaCl and 20 µM PLP. EhMGL1 was eluted as a tetramer as previously described (Tokoro et al., 2003 ▶). The purified EhMGL1 contained ten extra amino-acid residues (GPLGSPEFPG) at the amino-terminus derived from the linker peptide between GST and EhMGL1. The fractions containing the recombinant EhMGL1 were dialyzed against 10 mM HEPES–NaOH pH 7.4 and concentrated using an Amicon Ultra-4 Ultracel-50K to ∼20 mg ml−1. About 10 mg purified EhMGL1 was produced from 2 l culture. The purity was estimated to be >95% pure by densitometric quantitation of the corresponding band on SDS–PAGE (Fig. 1 ▶, lane 4) and the retained activity was comparable to the previous study (Sato et al., 2008 ▶).
Figure 1.
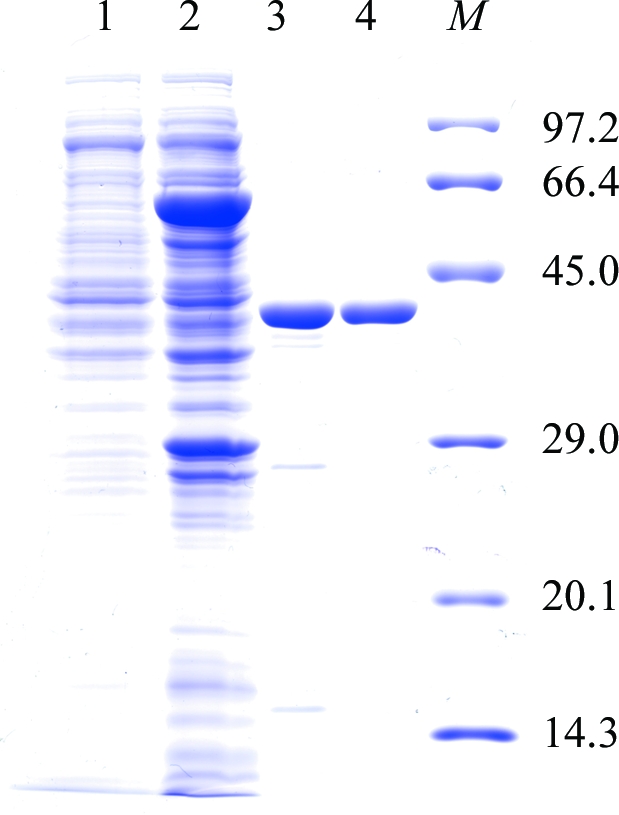
A 12% SDS–PAGE gel stained with Coomassie Brilliant Blue showing the apparent homogeneity of the purified EhMGL1. Lane 1, E. coli soluble extract before IPTG induction; lane 2, E. coli soluble extract after IPTG induction; lane 3, an eluate from a GSTrap HP column after digestion with PreScission Protease; lane 4, the purified EhMGL1 after size-exclusion chromatography using a HiPrep16/60 Sephacryl S-300 column; lane M, molecular-weight markers (kDa).
2.2. Crystallization and X-ray diffraction data collection
Crystallization conditions were screened at 277 and 293 K by the sitting-drop vapour-diffusion method in 96-well CrystalClear Strips (Hampton Research). A 0.5 µl droplet containing about 20 mg ml−1 EhMGL1 dissolved in 10 mM HEPES–NaOH pH 7.4 was mixed with an equal volume of reservoir solution and the droplet was allowed to equilibrate against 100 µl reservoir solution. In the initial screening experiment, Crystal Screen (Jancarik & Kim, 1991 ▶), Crystal Screen II (Hampton Research) and Wizard Screens I and II (Emerald Biostructures) were used as reservoir solutions. Of the 194 conditions used, reagents containing ammonium sulfate as a precipitant gave thin plate-shaped crystals at 277 K. Conditions were further optimized at 277 K by varying the concentration of ammonium sulfate and the buffer pH. The effects of additives were also examined using Additive Screen kits from Hampton Research. The best crystals grew at 277 K from reservoir solution containing 1.8 M ammonium sulfate, 0.1 M cacodylate buffer pH 6.2, 0.1 M lithium citrate and 0.01 M betaine.
For X-ray diffraction experiments at 100 K, a crystal mounted in a nylon loop was transferred into and briefly soaked in a solution containing 2.2 M ammonium sulfate, 0.1 M cacodylate buffer pH 6.2, 0.1 M lithium citrate, 0.01 M betaine and 20%(w/v) glycerol and then frozen by rapidly submerging it in liquid nitrogen. X-ray diffraction data were collected using the rotation method on the BL5A beamline of Photon Factory (Tsukuba, Japan) at a wavelength of 1.000 Å using a CCD ADSC Quantum 315 detector. A total of 180 frames were recorded with an oscillation angle of 1.0°, an exposure time of 5.0 s per frame and a crystal-to-detector distance of 213 mm. Data were processed and scaled with HKL-2000 and SCALEPACK (Otwinowski & Minor, 1997 ▶).
3. Results and discussion
Of the 194 crystallization conditions screened using commercially available screening kits, reagents containing ammonium sulfate gave thin plate-shaped crystals. After optimization of the pH, the concentration of ammonium sulfate and additives, crystals grew to typical dimensions of 0.02 × 0.3 × 0.4 mm (Fig. 2 ▶) in 5 d under the conditions 1.8 M ammonium sulfate, 0.1 M cacodylate buffer pH 6.2, 0.1 M lithium citrate and 0.01 M betaine at 277 K. Diffraction patterns were recorded as 180 frames at 100 K using one crystal. Analysis of the symmetry and systematic absences in the recorded diffraction patterns indicated that the crystals belonged to the monoclinic space group P21, with unit-cell parameters a = 99.12, b = 85.38, c = 115.37 Å, β = 101.82°. Assuming the presence of four EhMGL1 molecules (4 × 42.4 kDa) in the asymmetric unit, the V M value was calculated to be 2.8 Å3 Da−1 with an estimated solvent content of 56%; these values are within the range commonly observed for protein crystals (Matthews, 1968 ▶). A total of 491 182 observed reflections were merged to 140 502 unique reflections in the 50.0–1.93 Å resolution range. The data-collection and processing statistics are summarized in Table 1 ▶.
Figure 2.

A typical crystal of EhMGL1.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 1.000 |
| Space group | P21 |
| Unit-cell parameters | |
| a (Å) | 99.12 |
| b (Å) | 85.38 |
| c (Å) | 115.37 |
| β (°) | 101.82 |
| Solvent content† (%) | 56 |
| Resolution range (Å) | 50.0–1.93 (2.00–1.93) |
| No. of reflections | 491182 |
| Unique reflections | 140502 |
| Completeness (%) | 96.3 (87.0) |
| Rmerge‡ (%) | 7.2 (39.1) |
| I/σ(I) | 8.4 (2.57) |
Assuming the presence of four molecules in the asymmetric unit.
R
merge = 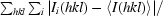
 .
.
An attempt to solve the structure using the molecular-replacement method with the MOLREP program (Navaza, 1994 ▶) as implemented within the CCP4 package (Collaborative Computational Project, Number 4, 1994 ▶) was carried out using the refined coordinates of EhMGL2 (unpublished results; 69% sequence identity to EhMGL1). A promising solution with a homotetramer structure was obtained (correlation coefficient and R factor of 0.557 and 51.3%, respectively) and the model was subsequently subjected to rigid-body refinement, giving an R factor of 46.8%. Amino-acid residues of the initial homotetramer model that differ from those of the search model were replaced with Ala. Using the molecular-replacement solution, refinement of the tetramer structure of EhMGL1 is in progress. In parallel with the refinement, we are now trying to prepare crystals of EhMGL1 complexed with substrate analogues and inhibitors.
Acknowledgments
We are grateful to staff members at the BL5A beamline at Photon Factory for their help with X-ray diffraction data collection. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan to DS (20590429) and TN (18050006, 18073001) and a grant for research to promote the development of anti-AIDS pharmaceuticals from the Japan Health Sciences Foundation to TN.
References
- Alexander, F. W., Sandmeier, E., Mehta, P. K. & Christen, P. (1994). Eur. J. Biochem.219, 953–960. [DOI] [PubMed]
- Ali, V. & Nozaki, T. (2007). Clin. Microbiol. Rev.20, 164–187. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Coombs, G. H. & Mottram, J. C. (2001). Antimicrob. Agents Chemother.45, 1743–1745. [DOI] [PMC free article] [PubMed]
- Dias, B. & Weimer, B. (1998). Appl. Environ. Microbiol.64, 3327–3331. [DOI] [PMC free article] [PubMed]
- Goyer, A., Collakova, E., Shachar-Hill, Y. & Hanson, A. D. (2007). Plant Cell Physiol.48, 232–242. [DOI] [PubMed]
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411.
- Kreis, W. & Hession, C. (1973). Cancer Res.33, 1862–1865. [PubMed]
- Mamaeva, D. V., Morozova, E. A., Nikulin, A. D., Revtovich, S. V., Nikonov, S. V., Garber, M. B. & Demidkina, T. V. (2005). Acta Cryst. F61, 546–549. [DOI] [PMC free article] [PubMed]
- Manukhov, I. V., Mamaeva, D. V., Rastorguev, S. M., Faleev, N. G., Morozova, E. A., Demidkina, T. V. & Zavilgelsky, G. B. (2005). J. Bacteriol.187, 3889–3893. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Motoshima, H., Inagaki, K., Kumasaka, T., Furuichi, M., Inoue, H., Tamura, T., Esaki, N., Soda, K., Tanaka, N., Yamamoto, M. & Tanaka, H. (2000). J. Biochem.128, 349–354. [DOI] [PubMed]
- Nakayama, T., Esaki, N., Sugie, K., Beresov, T. T., Tanaka, H. & Soda, K. (1984). Anal. Biochem.138, 421–424. [DOI] [PubMed]
- Navaza, J. (1994). Acta Cryst. A50, 157–163.
- Nikulin, A., Revtovich, S., Morozova, E., Nevskaya, N., Nikonov, S., Garber, M. & Demidkina, T. (2008). Acta Cryst. D64, 211–218. [DOI] [PubMed]
- Nozaki, T., Ali, V. & Tokoro, M. (2005). Adv. Parasitol.60, 1–99. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Rebeille, F., Jabrin, S., Bligny, R., Loizeau, K., Gambonnet, B., Van Wilder, V., Douce, R. & Ravanel, S. (2006). Proc. Natl Acad. Sci. USA, 103, 15687–15692. [DOI] [PMC free article] [PubMed]
- Sambrook, J, Fritsch, E. F. & Maniatas, T. (1989). Molecular Cloning: A Laboratory Manual, p. A.3. New York: Cold Spring Harbor Laboratory Press.
- Sato, D., Yamagata, W., Harada, S. & Nozaki, T. (2008). FEBS J.275, 548–560. [DOI] [PubMed]
- Sato, D., Yamagata, W., Kamei, K., Nozaki, T. & Harada, S. (2006). Acta Cryst. F62, 1034–1036. [DOI] [PMC free article] [PubMed]
- Sridhar, V., Xu, M., Han, Q., Sun, X., Tan, Y., Hoffman, R. M. & Prasad, G. S. (2000). Acta Cryst. D56, 1665–1667. [DOI] [PubMed]
- Stanley, S. Jr (2003). Lancet, 361, 1025–1034. [DOI] [PubMed]
- Tanaka, H., Esaki, N. & Soda, K. (1985). Enzyme Microb. Technol.7, 530–537.
- Tokoro, M, Asai, T., Kobayashi, S., Takeuchi, T. & Nozaki, T. (2003). J. Biol. Chem.43, 42717–42727. [DOI] [PubMed]
- World Health Organization (1998). The World Health Report 1998 Geneva: World Health Organization.
- Yoshimura, M., Nakano, Y. & Koga, T. (2002). Biochem. Biophys. Res. Commun.292, 964–968. [DOI] [PubMed]
- Yoshimura, M., Nakano, Y., Yamashita, Y., Oho, T., Saito, T. & Koga, T. (2000). Infect. Immun.68, 6912–6916. [DOI] [PMC free article] [PubMed]