

A crystal structure of zebrafish Plk1 reveals the binding of an extended ATP-competitive inhibitor that targets the adaptive pocket and helps to order the activation segment of the kinase. The conformation of the segment resembles that observed in a nonphosphorylated ligand-free wild-type kinase domain.
Keywords: Polo-like kinase 1, small-molecule inhibitors, zebrafish
Abstract
Polo-like kinase 1 (Plk1) is a member of the Polo-like kinase family of serine/threonine kinases involved in the regulation of cell-cycle progression and cytokinesis and is an attractive target for the development of anticancer therapeutics. The catalytic domain of this enzyme shares significant primary amino-acid homology and structural similarity with another mitotic kinase, Aurora A. While screening an Aurora A library of ATP-competitive compounds, a urea-containing inhibitor with low affinity for mouse Aurora A but with submicromolar potency for human and zebrafish Plk1 (hPlk1 and zPlk1, respectively) was identified. A crystal structure of the zebrafish Plk1 kinase domain–inhibitor complex reveals that the small molecule occupies the purine pocket and extends past the catalytic lysine into the adaptive region of the active site. Analysis of the structures of this protein–inhibitor complex and of similar small molecules cocrystallized with other kinases facilitates understanding of the specificity of the inhibitor for Plk1 and documents for the first time that Plk1 can accommodate extended ATP-competitive compounds that project toward the adaptive pocket and help the enzyme order its activation segment.
1. Introduction
Polo-like kinases (Plks) form a small family of multifunctional mitotic serine/threonine protein kinases with essential roles in the regulation of cell-cycle progression and cell division that typically consist of an N-terminal catalytic domain and one or two C-terminal Polo-box sequence motifs that constitute a Polo-box domain (PBD). Four enzymes in this family have been identified to date (Plk1, Plk2/Snk, Plk3/Fnk/Prk and Plk4/Sak); human Plk1 (hPlk1) is the best characterized mammalian representative of the family (van Vugt & Medema, 2005 ▶). It has been shown in vitro that normal cells, but not cancer cells, can survive severe Plk1 depletion (Liu et al., 2006 ▶). Plk1 has been found to be overexpressed in a variety of tumors and its overexpression appears to correlate with a poor prognosis (Eckerdt et al., 2005 ▶).
Given the high interest in Plk1 for the development of anticancer therapeutics, there has been an intense effort to characterize the enzyme structurally and to enable structure-based drug design. Multiple isolated PBD X-ray crystal structures have been described in the literature [PDB codes 1umw (Elia et al., 2003 ▶), 1q4o and 1q4k (Cheng et al., 2003 ▶), 2ogq, 2ojs and 2ojx (Garcia-Alvarez et al., 2007 ▶)]. Only recently, however, have two independent groups reported successful crystallization of the human Plk1 catalytic domain: Pfizer Global Research and Development [inactive Plk1 KD with ligands: the nonhydrolysable ATP analogue adenylylimidodiphosphate (AMPPNP; PDB code 2ou7), the pyrrolo-pyrazole inhibitor PHA-680626 (PDB code 2owb) and the inhibitor BI-2536 (PDB code 2rku); Kothe, Kohls, Low, Coli, Cheng et al., 2007 ▶; Kothe, Kohls, Low, Coli, Rennie et al., 2007 ▶] and Bayer Schering Pharma [wild-type Plk1 KD in complex with a designed ankyrin-repeat protein (DARPin) selective for Plk1 (PDB code 2v5q; Bandeiras et al., 2008 ▶)]. The KD defined by the Pfizer group, which bears an inactivating Thr210→Val substitution in the activation segment, crystallized in a conformation consistent with the active state of other kinases. The activation segment appeared to be ordered, even in the presence of the small-molecule inhibitor, which did not directly interact with it. The KD construct containing the activating Thr210→Asp alteration expressed poorly in insect cells, appeared to be unstable in solution and did not yield diffraction-quality crystals.
Considering the poor reproducibility of the crystallization conditions on standard vapor-diffusion plates documented by Pfizer and Bayer Schering with hPlk1, we undertook a screen of Plk1s from other species and identified a minimal KD fragment of zPlk1 that expressed robustly in Escherichia coli, was easy to purify and reliably yielded crystals of the enzyme in a ligand-free form or in complex with small-molecule inhibitors in less than a week following the commencement of crystallization trials and thus became a tool suitable for high-throughput structure-based drug design. Using this construct, we screened a library of ATP-competitive Aurora A inhibitors and discovered a compound with low affinity for mouse Aurora A but with submicromolar potency for zebrafish and human Plk1. In this report, we present the crystal structure of a zPlk1 KD–inhibitor complex and show that the inhibitor interacts with the purine-binding pocket and extends into the adaptive region of the active site; by doing so, it is likely to stabilize the conformation of the enzyme’s activation segment. The tools and structural insights described in this study might help to develop new small-molecule Plk1 inhibitors with improved selectivity that target the adaptive region of the enzyme.
2. Materials and methods
2.1. Cloning, expression and purification of zPlk1
The zPlk1 (GenBank entry 68533569) KD sequence used for crystallography (Fig. 1 ▶), encompassing residues 17–312, was amplified by Tgo polymerase (Roche Diagnostics) with GAGCTGGATCCGATCCCAAATCTGCTCCTCTGAAAGAGATTCCCG as the 5′ primer containing a BamHI cloning site (bold), GTACGCTCG AGTCAGGAGAACCTGGGAGGAACAGTGAGACAGG as the 3′ primer containing an XhoI cloning site (bold) and full-length zPlk1 cDNA (Open BioSystems) as template DNA. The activating mutation in codon 196 (ACC→GAC), which changes the phosphorylatable Thr196 to Asp, was introduced by site-directed mutagenesis using the QuikChange Site-Directed Mutagenesis Kit from Stratagene as recommended by the manufacturer with the primers GATGGGGAGCGAAAGAAGGACCTTTGTGGCACGCCAAACTAC (5′ primer) and GTAGTTTGGCGTGCCACAAAGGTCCTTCTTTCGCTCCCCATC (3′ primer). The DNA inserts were gel-purified, digested with XhoI and BamHI and cloned into the corresponding sites of the pGEX6P-1 plasmid (GE Healthcare) for expression as N-terminal glutathione S-transferase (GST) fusion proteins in E. coli. The expected molecular weight of the protein with the N-terminal Gly-Pro-Leu-Gly-Ser linker residues and a Thr196→Asp substitution in the activation segment was 34 386.0 Da.
Figure 1.

Alignment of full-length zebrafish and human Plk1 amino-acid sequences with the amino-acid sequence of the zPlk1 crystallography construct (residues 12–312 with the activating Thr196→Asp substitution) used in this study. The Gly-Pro-Leu-Gly-Ser linker residues that separate the GST tag from the zPlk1 KD polypeptide are underlined. Amino acids shown in blue represent the N-terminal-most and C-terminal-most residues of the zPlk1 crystallography construct. The phosphorylatable Thr196 in zPlk1 and the corresponding Thr210 in hPlk1 are shown in red. Asterisks signify identity; colons indicate conserved residues.
Expression and purification were performed essentially as described previously for mouse Aurora A and different variants of zPlk1 (Elling et al., 2007 ▶, 2008 ▶). Once purified, the samples were typically concentrated to 5–6 mg ml−1, centrifuged to remove the precipitate, aliquoted for crystallization experiments and activity assays and snap-frozen in liquid nitrogen for storage at 193 K. Protein purity was assessed by Coomassie-stained SDS–PAGE gels, its concentration was estimated by measuring the absorbance of the samples at 280 nm and protein modifications were analyzed with electrospray mass spectrometry as described previously (Hansen et al., 2005 ▶).
2.2. Compound synthesis
The inhibitor used for cocrystallization experiments (Compound 902) was synthesized as described in Fig. 2 ▶(a). Firstly, the purine mimetic was assembled by heating a mixture of 4,6-dichloropyrimidine (2.0 g, 13.43 mmol), 1H-[1,2,4]triazole-3-ylamine (1.24 g, 14.77 mmol) and caesium carbonate (6.56 g, 20.13 mmol) in 20 ml dimethylacetamide at 353 K for 2 h. The solvent was removed and the residue was dissolved in 200 ml dichloromethane. The resulting mixture was washed with 100 ml each of water and brine. The organic portion was dried over sodium sulfate and concentrated under vacuum to give 1.15 g (44% yield) 6-chloro-4-amino[1,2,4]triazolyl-pyrimidine (1) as a yellowish solid that was used without further purification. To a solution of 1 (0.405 g, 2.07 mmol) in 10 ml t-butyl alcohol was added [5-(2-aminoethyl)-thiazol-2-yl]-carbamic acid t-butyl ester (0.5 g, 2.07 mmol; the synthetic preparation of this reagent will be published elsewhere) and di-isopropylethylamine (0.740 ml, 4.14 mmol). This mixture was then heated at 353 K for 12 h. The solvent was removed to provide the condensation product 2 as crude material. Flash column chromatography (dichloromethane/ethyl acetate/methanol in a 2:1:0.2 ratio), performed with Merck Kieselgel 60 silica gel, afforded 0.479 g (58% yield) of the t-butyl carbamate-protected compound 2 as a light yellow solid.
Figure 2.

(a) Synthesis and chemical structure of Compound 902: 1-(5-methyl-2-trifluoromethyl-furan-3-yl)-3-(5-{2-[6-(1H-[1,2,4]triazol-3-ylamino)-pyrimidin-4-ylamino]-ethyl}-thiazol-2-yl)-urea. a, 1H-[1,2,4]triazole-3-ylamine, Cs2CO3, DMA; b, [5-(2-aminoethyl)thiazol-2-yl]carbamic acid t-butyl ester, DIEA, t-BuOH; c, HCl, dioxane, DCM; d, 3-isocyanato-5-methyl-2-trifluoromethylfuran, DIEA, DMF. (b) Chemical structure of Compound 065: 1-{5-[2-(thieno[3,2-d]pyrimidin-4-ylamino)-ethyl]-thiazol-2-yl}-3-(3-trifluoromethylphenyl)-urea.
To ummask the thiazole amino group, a solution of 2 (0.03 g, 0.088 mmol) in 10 ml dichloromethane was treated with 1 ml hydrochloric acid as a 4.0 N solution in dioxane. The reaction mixture was stirred at room temperature for 1 h, after which the solvents were removed and the residue was dried under vacuum for 1 h. This deprotected material was directly dissolved in 2 ml dimethylformamide and treated with di-isopropylethylamine (0.076 ml, 0.440 mmol) and 3-isocyanato-5-methyl-2-trifluoromethylfuran (0.022 g, 0.115 mmol) to form the urea. The reaction mixture was stirred at room temperature for 10 h, after which the solvent was removed. Preparative HPLC afforded 0.019 g (44% yield) Compound 902 as a white solid. This powder was dissolved in DMSO and used in a 2:1 molar ratio with the protein for crystallization experiments. The identities of all the intermediates and the final product were confirmed by 1H NMR and LC/MS.
2.3. Plk1 biochemical activity assays
Plk1 IC50 values were determined using the IMAP fluorescence polarization-based assay (Molecular Devices) according to the manufacturer’s instructions. Assay conditions consisted of 15 nM Plk1, 25 µM ATP and 100 nM substrate peptide based on human Cdc25C (5FAM-RALMEASFADQAR-NH2) diluted in assay buffer composed of 10 mM Tris pH 7.2, 10 mM MgCl2, 2 mM DTT, 100 µM Na3VO4 and 0.01%(v/v) Triton X-100. Compounds were diluted in DMSO to generate serial dilutions containing stock compounds at 20 times the final concentration. Assay reactions contained 5%(v/v) DMSO. Reactions were terminated after incubation at 303 K for 1 h by addition of 60 µl 60%/40% Progressive Binding Buffer A/B and 1:1000 IMAP nanoparticles (Molecular Devices). Following an additional 1 h incubation step, kinase activity was measured by the change in fluorescence polarization (mP) units detected using an LJL Analyst (LJL BioSystems). IC50 values were calculated using a four-parameter fit and GraphPad Prism software (GraphPad Software). Aurora A biochemical assays were conducted as described previously (Elling et al., 2007 ▶).
2.4. Crystallization, data collection and structure determination
Diffraction-quality crystals of the zPlk1 KD–inhibitor complex were obtained by hanging-drop vapor diffusion at 277 K against a reservoir consisting of 0.1 M HEPES pH 7.5, 0.2 M (NH4)2SO4, 22.5%(w/v) PEG 3350, 15%(v/v) glycerol. The crystal for data collection was cryoprotected with 15%(v/v) ethylene glycol and flash-frozen in liquid nitrogen (see Table 1 ▶ for space group and unit-cell parameters).
Table 1. Crystallographic data and refinement statistics.
| Amino-acid substitution | Thr196→Asp |
| Residue boundary | 17–312 |
| Ligand | Compound 902 |
| PDB code | 3db6 |
| Space group | I23 |
| Unit-cell parameter (Å) | a = 135.1 |
| X-ray source | SSRL BL 9-1 |
| Wavelength (Å) | 0.979 |
| Resolution (Å) | 30–2.85 |
| No. of observations† | 46300 (6796) |
| No. of reflections† | 9717 (1413) |
| Completeness† (%) | 99.8 (100.0) |
| Mean I/σ(I)† | 8.2 (2.0) |
| Rmerge on I†‡ | 0.081 (0.396) |
| Cutoff criteria | I < −3σ(I) |
| Model and refinement statistics | |
| Resolution range (Å) | 30–2.85 |
| No. of reflections§ | 8622 (1090) |
| Completeness (%) | 99.7 |
| Cutoff criterion | |F| > 0.0 |
| No. of residues | 287 |
| No. of water molecules | 16 |
| R.m.s.d. bond lengths (Å) | 0.006 |
| R.m.s.d. bond angles (°) | 1.028 |
| Luzzati error (Å) | 0.413 |
| Correlation factor¶ | 0.839 |
| Rcryst†† | 22.1 |
| Rfree | 25.7 |
| Ramachandran plot statistics‡‡ | |
| Most favored | 219 (87.6%) |
| Additional allowed | 29 (11.6%) |
| Generously allowed | 2 (0.8%) |
| Disallowed | 0 (0%) |
| Overall G factor§§ | 0.2 |
Values in parentheses are for the high-resolution shell (3–2.85 Å). Missing residues: GPLGS, 17–23 and 311–312.
R
merge = 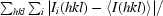
 .
.
Values in parentheses indicate the numbers of reflections used to calculate the R free factor.
Correlation factor between the structure factors and the model as calculated using SFCHECK (Vaguine et al., 1999 ▶).
R
cryst = 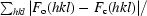
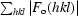 , where F
o and F
c are observed and calculated structure factors, respectively.
, where F
o and F
c are observed and calculated structure factors, respectively.
Computed with PROCHECK (Laskowski et al., 1993 ▶).
The overall G factor is a measure of the overall normality of the structure and is obtained from an average of all the different G factors for each residue in the structure. The factor is computed for torsion angles as well as main-chain bond lengths and angles using the Engh and Huber small-molecule means and standard deviations (Engh & Huber, 1991 ▶). It is essentially a log-odds score based on the observed distributions of these stereochemical parameters (Laskowski et al., 1993 ▶).
Diffraction data were collected under standard cryogenic conditions on beamline 9-1 at the Stanford Synchrotron Research Laboratory, processed using MOSFLM (Leslie, 1992 ▶) and scaled with SCALA (Evans, 1993 ▶). The structures were determined from single-wavelength native diffraction experiments by molecular replacement with MOLREP (Lebedev et al., 2008 ▶) using a search model from the previously determined structure of an activated zPlk1 KD variant (PDB code 3d5v; Elling et al., 2008 ▶). The refinement of the initial solutions with REFMAC (Murshudov et al., 1999 ▶) yielded experimental electron-density maps suitable for model building with O (Jones et al., 1991 ▶). Residues 17–23 and 311–312 were not visible in the electron-density maps and were omitted from refinement. The final atomic model of the protein–inhibitor complex was refined to an R factor of 22.1% and an R free value of 25.7% at 2.85 Å resolution. PROCHECK (Laskowski et al., 1993 ▶) revealed no disallowed (ϕ, ψ) combinations and excellent stereochemistry (see Table 1 ▶ for a summary of X-ray data-collection and refinement statistics). All proteins and small-molecule inhibitors in the figures were rendered with PyMOL (DeLano, 2002 ▶).
3. Results and discussion
While screening full-length hPlk1 against an in-house library of compounds likely to inhibit Aurora A, we identified an extended urea-containing inhibitor, Compound 902, that displayed low affinity for mouse Aurora A but a submicromolar IC50 for hPlk1. We then obtained crystals of the complex between zPlk1 KD and Compound 902 and discovered that in spite of the fairly low 2.85 Å resolution of the structure, there was unambiguous electron density for the compound in the active site of the enzyme as shown in the OMIT maps (Fig. 3 ▶ and Supplementary Fig. 11). One of the pyrimidine N atoms hydrogen bonds directly to the amine N atom of Cys119 (distance = 2.9 Å) located in the hinge region (Fig. 4 ▶ a), while the amide N atom in the three-atom linker between the purine mimetic and the thiazole group interacts weakly with the backbone carbonyl of Glu117 (distance = 3.3 Å). The affinity of the compound for the protein is further enhanced by a direct hydrogen bond between the urea O atom and the catalytic Lys68 (distance = 3.1 Å) and between the trifluoromethyl furan-proximal urea N atom and one of the carboxylate O atoms of Glu87 (distance = 2.7 Å) in the αC helix. Although the triazole moiety of the purine mimetic, as well as the thiazole N atom and its proximal urea N atom, do not appear to participate in any direct hydrogen-bonding interactions with protein atoms, given the resolution of the structure we cannot eliminate the possibility that the binding of the inhibitor to the enzyme is further stabilized by additional water-mediated hydrogen bonds.
Figure 3.

Electron density for Compound 902 and key interactions between the compound and the active-site residues of zPlk1. (a) 2F o − F c OMIT electron-density map (2.85 Å resolution; PDB code 3db6) within a 2.0 Å distance of Compound 902. The map was contoured at the 1σ level andis displayed as a blue mesh with C atoms shown in yellow, N atoms in blue, O atoms in red, S atoms in orange and F atoms in grey. (b) Key direct hydrogen-bonding interactions between the active-site residues of zPlk1 and Compound 902. The inhibitor hydrogen bonds to residues in the hinge (Cys119), αC helix (Glu87) and strand β5 (catalytic Lys68). The DFG-loop, displayed in marine blue, adopts an active ‘in’ conformation.
Figure 4.

IC50 measurements against full-length hPlk1. Each data point represents three independent experiments; vertical bars indicate the standard error of the mean.
Given the similarity between Compound 902 and a previously reported related mouse and human Aurora kinase inhibitor, Compound 065 (PDB code 3d14), we were surprised to discover that Compound 902 displayed good potency against full-length hPlk1 with an IC50 of 0.73 µM (Fig. 4 ▶) but had no detectable activity up to 20 µM concentration on mouse Aurora A. The reverse held true for Compound 065, which is a potent low-nanomolar inhibitor of mouse Aurora A with an IC50 of 10.9 nM but is inactive towards hPlk1 (IC50 > 20 µM). It is possible that the triazolylamino pyrimidine moiety, the purine mimetic in Compound 902, is not compatible with the ATP pocket in mouse Aurora A. In Plk1, this purine pocket-binding element may pull the compound toward the hinge and slightly away from the αC helix such that only one of the urea N atoms participates in a direct hydrogen-bonding interaction with the carboxylate O atoms of Glu87 (Fig. 5 ▶ a). In mouse Aurora A, the urea N atoms make bidentate hydrogen bonds to both carboxylate O atoms of Glu181 (Fig. 5 ▶ b) and there is an additional water-mediated hydrogen-bonding interaction that involves the thiazole N atom. Compound 902 adopts a V-shaped conformation in zPlk1, whereas Compound 065 is more rod-like in the active site of mouse Aurora A (Fig. 5 ▶ c). The V-shaped conformation of Compound 902 allows the projection of the trifluoromethyl furan group into a well defined adaptive region of the active site, while the trifluorophenyl group of Compound 065 backs up directly against the αC helix. Based on these observations, it would be interesting to determine whether extending the linker between the purine mimetic and the thiazole ring in Compound 902 would permit the urea N atoms to form a bidentate hydrogen bond to both carboxylate O atoms of Glu87, thus increasing the potency of the inhibitor for zPlk1.
Figure 5.

Comparison of the interactions of Compound 902 with zPlk1 and of the related Compound 065 with mouse Aurora A. C atoms of zPlk1 are shown in green and those of mouse Aurora A in violet. N atoms are displayed in blue, O atoms in red, S atoms in orange and F atoms in grey. (a) Compound 902 in the active site of zPlk1. The binding of the inhibitor to the protein appears to contribute to the ordering of the activation segment. The trifluoromethyl furan group fits tightly into the adaptive region of the active site. There is only one direct hydrogen bond between Glu87 in the αC helix and one of the urea N atoms. (b) Compound 065 in the active site of mouse Aurora A (1.9 Å resolution; PDB code 3d14). The numbering of the mouse residues is consistent with human Aurora A to facilitate cross-comparison with the multiple human Aurora-A crystal structures already available in the Protein Data Bank. The urea moiety of Compound 065 moves closer to the αC helix to allow a direct bidentate hydrogen-bonding interaction between the carboxylate O atoms of Glu181 and the urea N atoms. The pyrimidine ring of the purine mimetic forms a single hydrogen bond to the amide N atom of Ala213 (equivalent to Cys119 in zPlk1). (c) Overlay of zPlk1 (green) in complex with Compound 902 and mouse Aurora A (violet) in complex with Compound 065. The divergent paths that the inhibitors follow in the active sites of the two related enzymes, particularly in the purine-binding pocket and the adaptive region, are illustrated. Key secondary structural elements are labeled. The DFG-loop is displayed as sticks.
The crystal structure presented in this communication reveals that the small molecule not only occupies the purine-binding pocket of the enzyme, but also extends deep into the adaptive region of the active site. This is somewhat surprising since we know that in the activated zPlk1 KD the activation segment and parts of the αC helix, which delineate the adaptive pocket in kinases (Liao, 2007 ▶), are highly disordered (PDB code 3d5v). Consistent with this observation, no structures of compounds that target the adaptive region of Plk1 have been reported to date and all of the inhibitors for which structural information does exist appear to bind exclusively in the purine-binding pocket of the protein and the immediately adjacent spaces (Fig. 6 ▶). It is our hope that the data presented in this study will aid in the discovery of potent and selective inhibitors of Plk1 and Plk1-related enzymes.
Figure 6.

The active site of zPlk1 KD Thr196→Asp with overlaid Plk1 inhibitors. The activation segment of the enzyme is well ordered and the structure reveals a well defined adaptive region. The previously crystallized Plk1 inhibitors occupy the purine-binding pocket and the region immediately adjacent to it. The C atoms of PHA-680626 (2.1 Å resolution; PDB code 2owb) are shown in green, those of BI-2536 (1.95 Å resolution; PDB code 2rku) in violet, those of wortmannin (2.8 Å resolution; PDB code 3d5x) in marine blue and those of Compound 902 in yellow (2.85 Å resolution; PDB code 3db6). The surface of the active site is displayed in shades of grey.
Supplementary Material
PDB reference: Polo-like kinase 1 catalytic domain, complex with Compound 902, 3db6, r3db6sf
Supplementary material file. DOI: 10.1107/S1744309108019623/wd5099sup1.pdf
Acknowledgments
We would like to thank Dr Robert S. McDowell for critical reading of the manuscript.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: WD5099).
References
- Bandeiras, T. M. et al. (2008). Acta Cryst. D64, 339–353. [DOI] [PubMed]
- Cheng, K. Y., Lowe, E. D., Sinclair, J., Nigg, E. A. & Johnson, L. N. (2003). EMBO J.22, 5757–5768. [DOI] [PMC free article] [PubMed]
- DeLano, W. L. (2002). The PyMOL User’s Manual. DeLano Scientific, San Carlos, California, USA.
- Eckerdt, F., Yuan, J. & Strebhardt, K. (2005). Oncogene, 24, 267–276. [DOI] [PubMed]
- Elia, A. E., Rellos, P., Haire, L. F., Chao, J. W., Ivins, F. J., Hoepker, K., Mohammad, D., Cantley, L. C., Smerdon, S. J. & Yaffe, M. B. (2003). Cell, 115, 83–95. [DOI] [PubMed]
- Elling, R. A., Fucini, R. V. & Romanowski, M. J. (2008). Acta Cryst. D64, doi:10.1107/S0907444908019513. [DOI] [PubMed] [Google Scholar]
- Elling, R. A., Tangonan, B. T., Penny, D. M., Smith, J. T., Vincent, D. E., Hansen, S. K., O’Brien, T. & Romanowski, M. J. (2007). Protein Expr. Purif.54, 139–146. [DOI] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Evans, P. R. (1993). Proceedings of the CCP4 Study Weekend. Data Collection and Processing, edited by L. Sawyer, N. Isaacs & S. Bailey, pp. 114–122. Warrington: Daresbury Laboratory.
- Garcia-Alvarez, B., de Carcer, G., Ibanez, S., Bragado-Nilsson, E. & Montoya, G. (2007). Proc. Natl Acad. Sci. USA, 104, 3107–3112. [DOI] [PMC free article] [PubMed]
- Hansen, S. K., Cancilla, M. T., Shiau, T. P., Kung, J., Chen, T. & Erlanson, D. A. (2005). Biochemistry, 44, 7704–7712. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Kothe, M., Kohls, D., Low, S., Coli, R., Cheng, A. C., Jacques, S. L., Johnson, T. L., Lewis, C., Loh, C., Nonomiya, J., Sheils, A. L., Verdries, K. A., Wynn, T. A., Kuhn, C. & Ding, Y. H. (2007). Biochemistry, 46, 5960–5971. [DOI] [PubMed]
- Kothe, M., Kohls, D., Low, S., Coli, R., Rennie, G. R., Feru, F., Kuhn, C. & Ding, Y. H. (2007). Chem. Biol. Drug Des.70, 540–546. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lebedev, A. A., Vagin, A. A. & Murshudov, G. N. (2008). Acta Cryst. D64, 33–39. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Liao, J. J. (2007). J. Med. Chem.50, 409–424. [DOI] [PubMed]
- Liu, X., Lei, M. & Erikson, R. L. (2006). Mol. Cell. Biol.26, 2093–2108. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999). Acta Cryst. D55, 247–255. [DOI] [PubMed]
- Vaguine, A. A., Richelle, J. & Wodak, S. J. (1999). Acta Cryst. D55, 191–205. [DOI] [PubMed]
- Vugt, M. A. van & Medema, R. H. (2005). Oncogene, 24, 2844–2859. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Polo-like kinase 1 catalytic domain, complex with Compound 902, 3db6, r3db6sf
Supplementary material file. DOI: 10.1107/S1744309108019623/wd5099sup1.pdf