

Summary
The anaphase-promoting complex (APC) is a ubiquitin ligase that governs late mitotic events by triggering the destruction of mitotic regulatory proteins. The APC is initially activated in early mitosis by association with the activator Cdc20, which is replaced in late mitosis by Cdh1. Here we show that in budding yeast the activation of APCCdh1 depends in part on regulation of the Cdh1 inhibitor Acm1. We show that Acm1 is a specific, high-affinity inhibitor of APCCdh1 and not APCCdc20, and that inhibition depends on pseudosubstrate and other conserved sequence motifs in Acm1. Consistent with a role as a Cdh1-specific inhibitor, single-cell analyses reveal that Acm1 levels rise in late G1 and drop in early anaphase. The disappearance of Acm1 in anaphase depends on a destruction-box sequence at its N terminus, which targets the protein to APCCdc20 for ubiquitination. Later in mitosis, Acm1 destruction is also promoted by APCCdh1. The localization and destruction of Acm1 are further modulated by phosphorylation at multiple consensus sites for the cyclin-dependent kinase Cdk1. The ability of APCCdc20 to promote the destruction of a Cdh1 inhibitor provides a mechanism ensuring the order of activation of the two APC isoforms. In addition, we speculate that the ability of APCCdh1 to target its own inhibitor enhances the bistability of the late mitotic regulatory system.
Introduction
Chromosome segregation and the final events of mitosis are initiated by the anaphase-promoting complex or cyclosome (APC), a ubiquitin-protein ligase that polyubiquitinates important cell-cycle components and thereby targets them for destruction by the proteasome (Peters, 2006; Thornton and Toczyski, 2006). A key target of the APC is securin, the destruction of which triggers the activation of a protease, separase, that drives sister-chromatid separation (Nasmyth, 2002). The other major targets of the APC are the mitotic cyclins. Cyclin destruction leads to inactivation of cyclin-dependent kinases (Cdks), thereby allowing dephosphorylation of Cdk targets and the completion of late mitotic events (Sullivan and Morgan, 2007).
APC activation in mitosis depends on its sequential association with a pair of related activator proteins, Cdc20 and Cdh1. APC activators are thought to act, at least in part, by recruiting substrates to the APC core for ubiquitination. Cdc20 and Cdh1 both recognize specific sequence motifs, such as the destruction box and KEN box, that are present on APC targets and required for their destruction (King et al., 1996; Pfleger and Kirschner, 2000). The APC is first activated in early mitosis by Cdc20, leading to the destruction of a small group of Cdc20-specific APC substrates, including securin and cyclins, that control anaphase events. After anaphase, Cdc20 is replaced by the second activator, Cdh1, which maintains APC activity for the remainder of mitosis and through the following G1. Cdh1 broadens the substrate specificity of the APC, resulting in the destruction of several late mitotic regulators that are not targeted by APCCdc20.
The two-step activation of the APC, first by Cdc20 and then by Cdh1, depends on fundamental differences in the way that mitotic Cdks govern activator function. Cdks promote Cdc20-dependent APC activation: phosphorylation of APC core subunits by Cdks in early mitosis stimulates Cdc20 binding, leading to APCCdc20 activation in metaphase or earlier (Kraft et al., 2003; Rudner and Murray, 2000; Shteinberg et al., 1999). In contrast, Cdh1 function is inhibited by Cdks, which directly phosphorylate Cdh1 at multiple sites and thereby block its ability to bind the APC (Jaspersen et al., 1999; Zachariae et al., 1998). Thus, when APCCdc20-dependent destruction of cyclins leads to Cdk inactivation in anaphase, the resulting dephosphorylation of Cdh1 promotes its activation. Cdh1 remains active until Cdk activity rises at the beginning of the next cell cycle.
The activation of Cdh1 by dephosphorylation depends not only on inactivation of Cdks but also on phosphatases that remove the inhibitory phosphates. The phosphatase responsible for Cdh1 activation is best understood in the budding yeast Saccharomyces cerevisiae. In this species, the phosphatase Cdc14 is activated during late mitosis and is required for dephosphorylation of Cdh1 and other Cdk targets (D'Amours and Amon, 2004; Stegmeier and Amon, 2004; Sullivan and Morgan, 2007).
Given the central importance of the APC in the initiation of chromosome segregation, it is essential that APC activation by Cdc20 and Cdh1 be robustly regulated. Part of this regulation depends on inhibitory proteins that bind APC activators at certain cell-cycle stages and thereby ensure the correct timing of APC activation (Peters, 2006). When kinetochores fail to attach to the mitotic spindle, for example, the spindle assembly checkpoint proteins Mad2 and Mad3 bind Cdc20 and thereby block activation of APCCdc20 until spindle assembly is complete (Musacchio and Salmon, 2007). The mechanism of inhibition by Mad2 remains unclear, but Mad3 acts as a pseudosubstrate that occludes the substrate-binding site of Cdc20 (Burton and Solomon, 2007). Another well understood APC inhibitor is the vertebrate protein Emi1 and its homolog Rca1 of Drosophila (Grosskortenhaus and Sprenger, 2002; Reimann et al., 2001). Emi1 binds APC activators and competitively inhibits substrate binding (Miller et al., 2006). In the cell, Emi1 levels first rise in late G1 and help suppress APCCdh1 activity as the cell enters a new cycle (Hsu et al., 2002); Emi1 also suppresses the activity of APCCdc20 complexes when they first form in early mitosis. In prometaphase, Emi1 phosphorylation triggers its ubiquitination by the ubiquitin ligase SCF, and the resulting Emi1 destruction contributes to APCCdc20 activation (Hansen et al., 2004).
Acm1 is an APC inhibitor that was recently identified in studies of Cdh1-associated proteins from S. cerevisiae (Dial et al., 2007; Martinez et al., 2006). These studies suggested that Acm1 is an inhibitor of Cdh1 function in vivo and APCCdh1 activity in vitro, and that Acm1 may act by blocking the binding of Cdh1 to its substrates. Acm1 levels oscillate in the cell cycle, rising in late G1 and declining in mitosis, consistent with a function as an APC inhibitor. However, the precise function of Acm1 in the regulatory network governing late mitosis remains unclear.
Through a combination of biochemical and cytological analyses, we find that Acm1 is a specific inhibitor of Cdh1 and not Cdc20, and that it contains pseudosubstrate sequences that are required for its inhibitory effects. Most importantly, we find that Acm1 contains a destruction box sequence that targets it for ubiquitination by APCCdc20, resulting in its disappearance in anaphase cells. The ability of Cdc20 to promote destruction of a Cdh1 inhibitor helps ensure the sequential activation of Cdc20 and Cdh1 in late mitosis.
Results
Acm1 is a specific inhibitor of APCCdh1
Acm1 is known to inhibit APCCdh1 in vitro and in vivo, but its effects on APCCdc20 are not known. We therefore measured the effects of Acm1 on the activities of APCCdh1 and APCCdc20 in vitro, using budding yeast securin (Pds1) as substrate. We found that Acm1, produced by translation in reticulocyte lysates, was a potent inhibitor of APCCdh1 activity in vitro but had no effect on ubiquitination by APCCdc20, suggesting that it is a specific inhibitor of Cdh1 (Figure 1A).
Figure 1. Acm1 is a Cdh1-specific APC inhibitor.
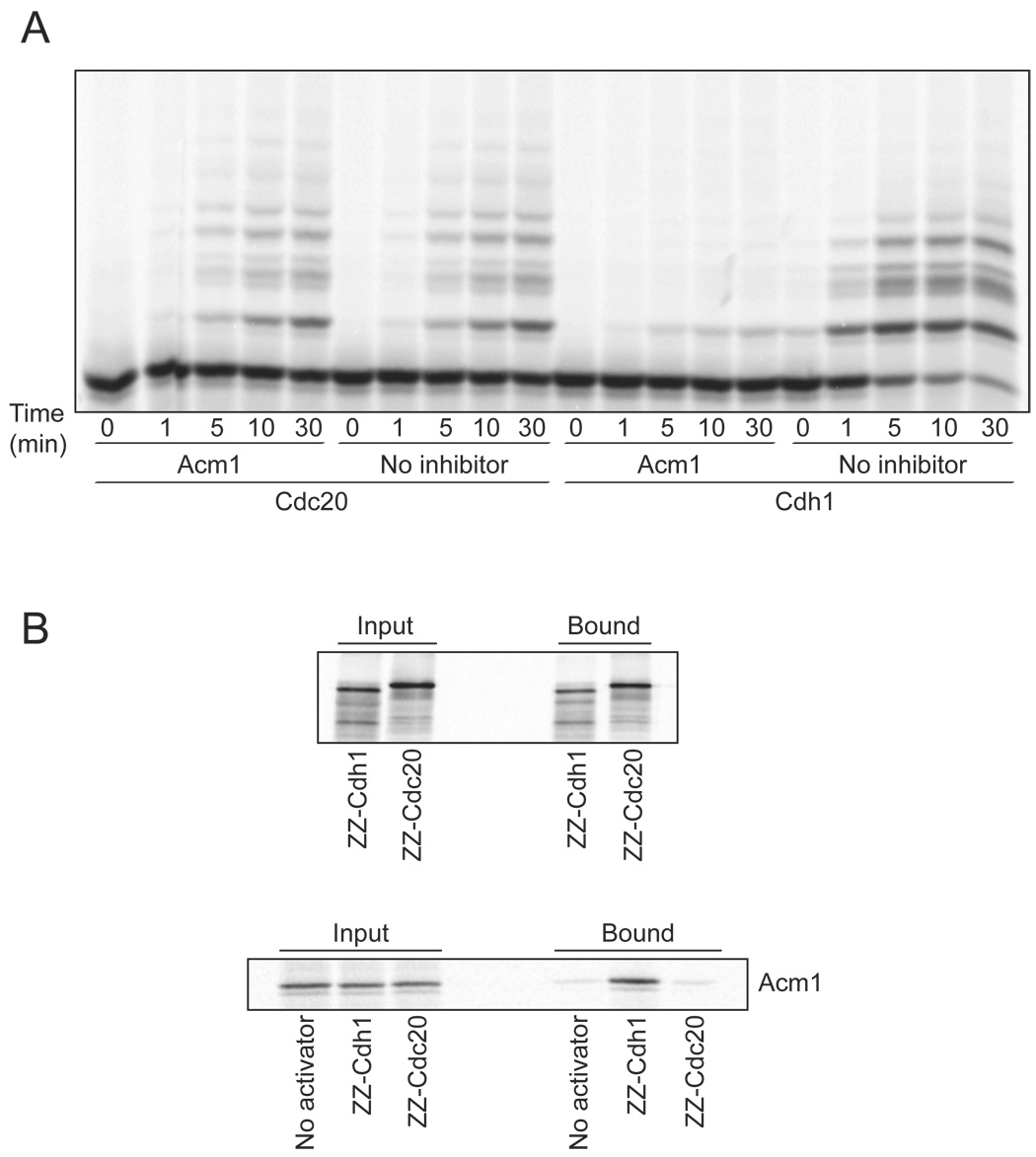
(A) APC reactions were performed with 35S-methionine-labeled securin (Pds1), using either Cdc20 or Cdh1 as activator, for the indicated times. In vitro translated Acm1 was added where indicated, and an equivalent amount of lysate was added to the control (no inhibitor) reactions.
(B) ZZ-tagged versions of Cdc20 and Cdh1 were translated in vitro with 35S-methionine and mixed with IgG beads. After washing, binding to the beads was assessed (top panel), confirming that the two activators were synthesized and bound to the beads in equal amounts. To assess the binding of Acm1 to the activators, ZZ-Cdc20 and ZZ-Cdh1 were translated in vitro without radiolabel and incubated with 35S-methionine-labeled Acm1 and IgG beads. After washing, bound activator protein was determined by SDS-PAGE and PhosphorImager analysis (bottom panel).
We explored the specificity of Acm1 further by analyzing its binding to APC activators. Cdh1 and Cdc20 were translated in vitro with ZZ-tags at their N termini, and then immobilized on IgG beads. In control reactions, activators were labeled with 35S-methionine to confirm that they were synthesized and bound to the beads in equal amounts (Figure 1B, top). We then synthesized unlabeled activators and immobilized them on IgG beads in the presence of 35S-methionine-labeled Acm1. After washing the beads, analysis of the immunoprecipitates revealed that Acm1 interacted specifically with Cdh1 and not Cdc20, consistent with our APC assays showing Cdh1-specific inhibition (Figure 1B, bottom). These results also demonstrated direct binding of Acm1 to Cdh1 in the absence of the APC. The affinity of the Acm1-Cdh1 interaction is clearly much higher than that of the interaction between Cdh1 and its substrates. For example, securin binding to Cdh1 was not detectable in parallel experiments (data not shown).
A pseudosubstrate region of Acm1 is required for its inhibitory activity
We next examined the contribution of different regions of Acm1 to its inhibitory activity and binding to Cdh1. Although only 209 residues in length, Acm1 contains many potential regulatory sequences, including three RxxL motifs that resemble the minimal destruction (D)-box sequence of APC targets, a KEN box sequence also found in APC targets, and five full Cdk phosphorylation consensus sites (S/T*-P-x-K/R) (Figure S1). Alignment of the Acm1 sequence with those of closely related proteins in other Saccharomyces species suggests that the KEN motif and the first and third RxxL motifs have been conserved in evolution (Figure S1).
Previous work suggested that Acm1 inhibits the binding of Cdh1 to its targets (Dial et al., 2007; Martinez et al., 2006). Given the abundance of destruction motifs in its sequence, we hypothesized that Acm1 is a pseudosubstrate inhibitor that binds tightly to substrate-binding sites on Cdh1. To address this possibility, we produced various deletion mutants of Acm1 by translation in vitro and tested their ability to inhibit APCCdh1 activity (Figure S2; summary of results in Figure 2). Deletion of the N-terminal 45 residues had no effect, indicating that the first RxxL motif (residues 8–11) is not required for the inhibitory activity of Acm1. A fragment including only residues 46 to 134 retained full inhibitory activity, focusing our attention in this region. Additional deletion analyses revealed that one critical inhibitory site in this region lies between residues 98 and 122, which contains the two other RxxL motifs and KEN box (Figure 2). Deletion of the poorly conserved second RxxL motif (residues 111–114) had no effect, but the inhibitory activity of Acm1 was abolished by replacement of the KEN box with three alanines or by deletion of the well-conserved third RxxL motif (residues 119–122) (Figure 3A). Point mutations in the KEN box and third RxxL motif also caused major losses of Acm1 inhibitory activity (Figure 2).
Figure 2. Summary of Acm1 mutant analyses.

This figure provides a qualitative summary of results obtained with Acm1 deletion and point mutants as diagrammed in the left column; the three RxxL sequences and the KEN box are indicated. Mutants were analyzed in four groups by one or more of the five assays shown in the columns at right. The approximate amount of activity in each assay is indicated by + and − symbols; blanks indicate that the mutant was not tested in that assay.
(A) Inhibition of APCCdh1 activity in vitro was assessed by adding the indicated mutant (synthesized by unlabeled translation in vitro of PCR-amplified DNA) to an APCCdh1 assay, as described in Figure 1A. See Figure S2 for representative data.
(B) Inhibition of APCCdh1 activity in vivo was assessed by testing whether overexpression of the mutant suppressed the lethality of overexpressed CDH1-m11, as shown in Figure 3B.
(C) Cdh1 binding was determined by incubating 35S-methionine-labeled Acm1 mutant (synthesized by translation in vitro of PCR-amplified DNA) with bead-immobilized Cdh1 protein as described in Figure 1B. See Figure S3 for representative data.
(D) Ubiquitination by APCCdh1 was measured by incubating 35S-methionine-labeled Acm1 mutant (synthesized by translation in vitro of PCR-amplified DNA) in an APCCdh1 reaction, followed by analysis of reaction products by SDS-PAGE. See Figure S4 for representative data.
(E) Ubiquitination by APCCdc20 was measured by incubating 35S-methionine-labeled Acm1 mutant (synthesized by translation in vitro of PCR-amplified DNA) in an APCCdc20 reaction as described in Figure 5A.
Figure 3. Inhibition of Cdh1 by Acm1 requires a central pseudosubstrate region.
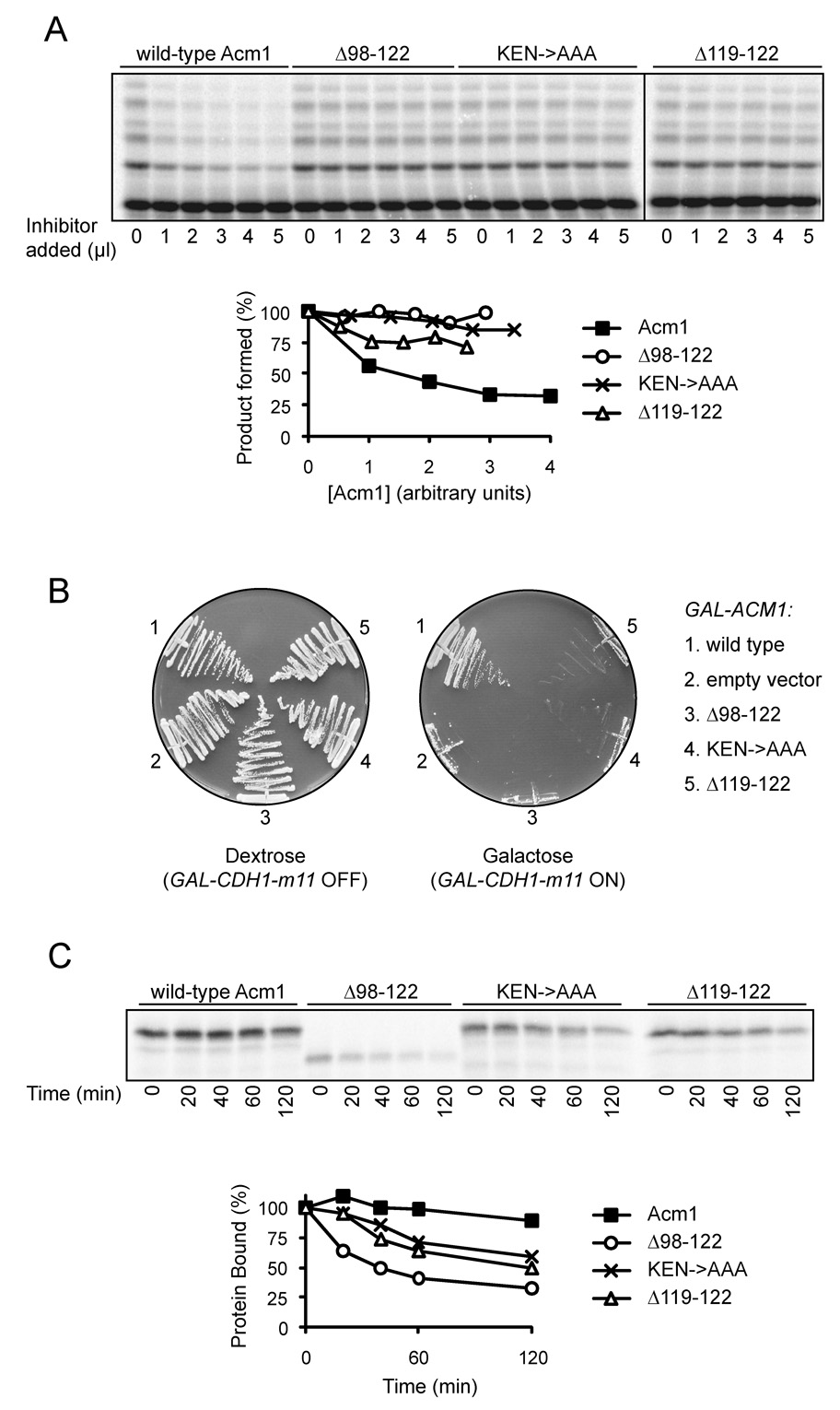
(A) APCCdh1 reactions were performed with 35S-methionine-labeled securin in the presence of increasing amounts of translation mixture containing Acm1 or Acm1 mutants as indicated. To control for small differences in the translation efficiency of different mutants, mutants were also synthesized in parallel reactions with 35S-methionine, and in the quantification at bottom the amounts of lysate were adjusted accordingly.
(B) Strains expressing CDH1-m11 from the galactose-inducible GAL1-10 promoter were transformed with plasmids expressing galactose-inducible ACM1-TAP or the indicated ACM1 mutants. Expression of CDH1-m11 and the ACM1 mutants is repressed on dextrose medium but induced on galactose medium. Expression of the Acm1 mutants alone had no impact on cell growth, and equivalent expression of the Acm1 variants was confirmed by Western blotting (data not shown).
(C) ZZ-tagged Cdh1 was synthesized by unlabeled translation in vitro, mixed with 35S-methionine-labeled wild-type or mutant Acm1, and incubated with IgG beads. After 3 rapid washes, samples were taken at the indicated times over a 2-hour time course at room temperature, and after each sample collection another wash was performed. To control for dissociation of ZZ-Cdh1 from the IgG beads, a parallel experiment was performed by incubating 35S-methionine-labeled ZZ-Cdh1 with IgG beads and carrying out the same series of washes. For the quantification in the bottom panel, the amount of bound Acm1 was adjusted to account for the slight dissociation of Cdh1-ZZ from the IgG beads.
To confirm that loss of Acm1 inhibitory activity in vitro corresponds to a loss of function in vivo, we tested the ability of Acm1 mutants to inhibit APCCdh1 in yeast cells. Overexpression of a Cdh1 variant bearing mutations in its Cdk phosphorylation sites (Cdh1-m11) is lethal (Zachariae et al., 1998). This lethality can be suppressed by co-overexpression of ACM1 (Martinez et al., 2006). We found that the ability of Acm1 to suppress lethality was abolished by deletion of residues 98–122, mutation of the KEN box, or deletion of the third RxxL motif (Figure 3B), confirming the critical function of this region.
We also tested the effects of mutations in Acm1 on its ability to bind Cdh1, using the binding assay described earlier (see Figure 1B). Interestingly, binding was not greatly affected by mutation of the KEN or third RxxL motifs, and deletion of the entire inhibitory region (98–122) had only moderate effects on Acm1-Cdh1 binding (Figure S3; summarized in Figure 2). Analysis of dissociation rates with these mutants confirmed that the KEN and RxxL mutants retained much of the affinity of the wild-type Acm1 protein, which did not dissociate significantly over a 2-hour time course. Deletion of residues 98–122 increased the dissociation rate only moderately (Figure 3C). In addition, an Acm1 fragment containing the central inhibitory region and adjacent Cterminal sequences (residues 89–168) failed to bind Cdh1 tightly or have a significant effect on APCCdh1 activity (Figure S3, Figure 2).
These results indicated that regions outside residues 98–122 of Acm1 make important contributions to Cdh1 binding. Studies of additional deletion mutants (Figure 2) suggested that one potentially important region lies amino-terminal of the KEN box sequence. An N-terminal fragment containing residues 1–97 exhibited a low level of inhibitory and binding activity, suggesting that a Cdh1-binding site exists in this region. Deletion of the 59 N-terminal residues of Acm1 did not reduce its inhibitory activity, while deletion of 69 residues greatly reduced activity (Figure 2). We therefore conclude that the high-affinity binding of Acm1 to Cdh1 depends on at least two interactions: one involving the central KEN/RxxL region between residues 98 and 122, and another interaction that involves the region around residues 60–70. Interestingly, the latter region, like the former, has been well conserved in evolution (Figure S1).
Acm1 localization is regulated during the cell cycle by Cdk1
Acm1 protein levels are known to oscillate during the cell cycle, rising first in late G1 (when ACM1 is first transcribed) and then decreasing during mitosis, slightly earlier than the cyclin Clb2 (Dial et al., 2007; Martinez et al., 2006). We explored the regulation of Acm1 further by using single-cell analyses to asses its levels and subcellular localization at different cell-cycle stages. We constructed an acm1Δ strain with an integrated plasmid carrying ACM1 (tagged at its C terminus with Myc epitopes) under the control of its own promoter. Interestingly, when Acm1 first appeared in late G1 cells it was localized primarily in the nucleus, but then became diffusely localized throughout the cell during progression through S phase and early mitosis (Figure 4A, B). Acm1 was abundant in cells containing short pre-anaphase spindles but was absent from anaphase cells, suggesting that most of the protein is destroyed in anaphase.
Figure 4. Acm1 localization during the cell cycle is controlled by Cdk1.
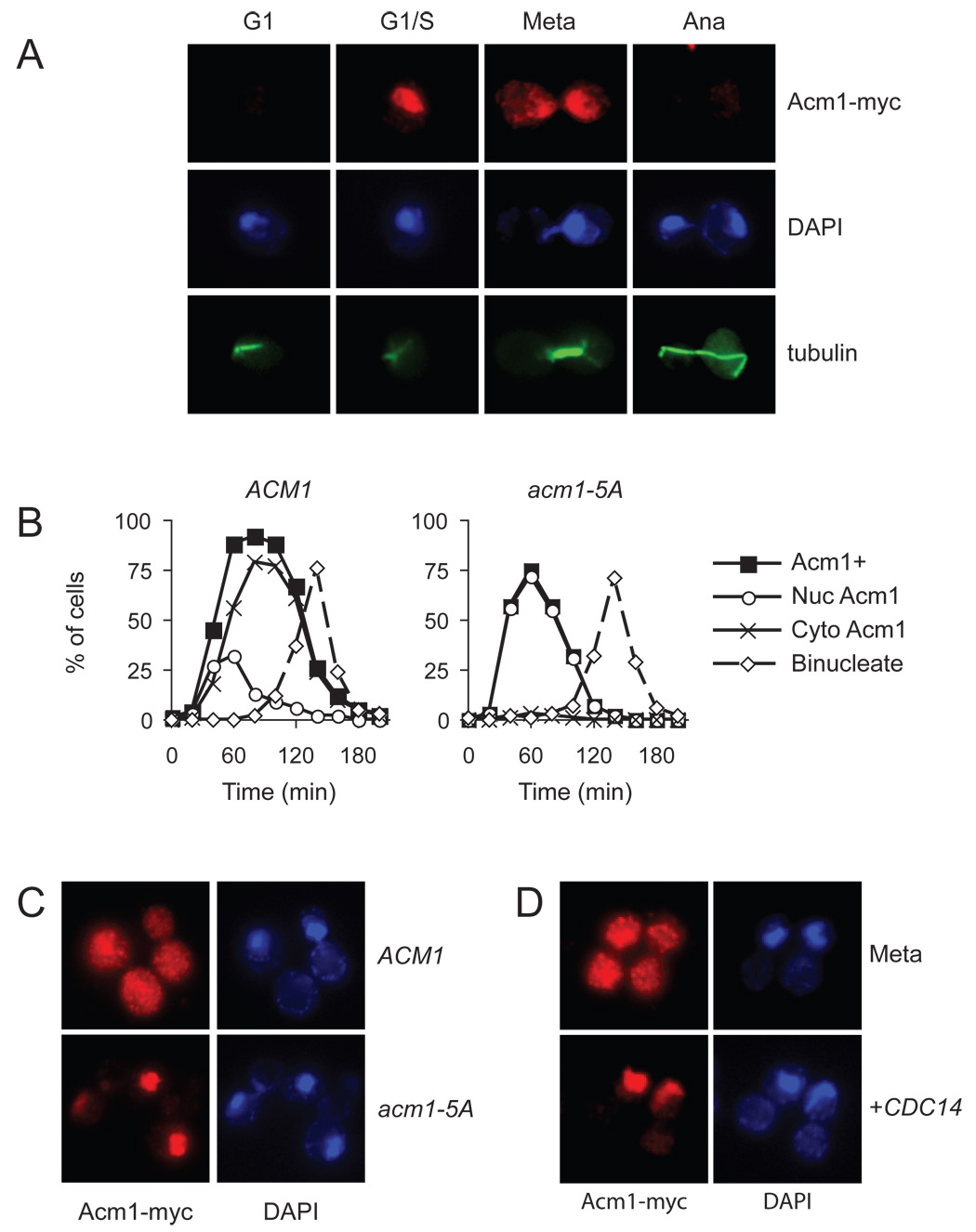
(A) Asynchronous ACM1-myc9 cells were processed for immunofluorescence analysis with anti-Myc and anti-tubulin antibodies, and stained with DAPI for detection of DNA. Cell-cycle position was determined on the basis of cell and spindle morphology and the separation of DNA masses.
(B) ACM1-myc9 and acm1-5A-myc9 cells were arrested in G1 with alpha factor and released into a synchronous cell cycle. Samples were collected every 20 min for analysis of budding, binucleate formation and the presence of Acm1-myc immunofluorescence. Acm1-positive cells were further characterized to determine if Acm1 was exclusively nuclear (Nuc Acm1) or dispersed throughout the cell (Cyto Acm1). For clarity, the time course of budding is not shown here but was very similar to that shown in Figure 5C.
(C) ACM1-myc9 and acm1-5A-myc9 cells were arrested in mitosis by nocodazole treatment, and immunofluorescence performed to assess Acm1 localization.
(D) ACM1-myc9 cells, carrying an integrated plasmid with CDC14 under the control of the GAL1-10 promoter, were blocked in mitosis by nocodazole treatment under noninducing conditions, followed by immunofluorescence analysis of Acm1 localization (top). CDC14 expression was induced by addition of galactose to the culture medium and immunofluorescence performed to assess Acm1 localization (bottom).
The dispersal of Acm1 from the nucleus as cells progressed from G1 to S phase suggested that its localization might be controlled by cell-cycle regulators. An interesting possibility was that Acm1 localization is governed by Cdk1 activity, which rises in late G1 and remains elevated until cyclins are destroyed in late mitosis. As mentioned earlier, Acm1 contains five highly conserved Cdk1 consensus phosphorylation sites (Figure S1), and in our previous work we identified Acm1 as an excellent Cdk1 substrate in vitro (Ubersax et al., 2003). We therefore analyzed the localization of a nonphosphorylatable Acm1 mutant (Acm1-5A) in which all five Cdk1 sites were changed to alanine (Figure 4B, C). Like the wild-type protein, Acm1-5A first appeared in late G1. Strikingly, however, it remained in the nucleus throughout the cell cycle and then disappeared in mitosis. In addition, Acm1-5A was degraded earlier than the wild-type protein: not only was it absent from anaphase cells but it was also absent from many cells containing short pre-anaphase spindles, arguing that phosphorylation normally delays Acm1 degradation.
We also found that Acm1 is diffusely localized in nocodazole-arrested cells, in which Cdk activity is very high (Figure 4D). Overexpression of the anti-Cdk1 phosphatase Cdc14 in these cells caused Acm1 to localize primarily inside the nucleus, consistent with the notion that dephosphorylation of Cdk1 sites on Acm1 promotes its accumulation in the nucleus.
Acm1 is a substrate of APCCdc20
The disappearance of Acm1 in anaphase raised the possibility that it is targeted for destruction by APCCdc20. We first investigated this possibility by testing Acm1 as a substrate of APCCdc20 in vitro (Figure 5A). When 35S-methionine-labeled Acm1 was translated in vitro and incubated with APCCdc20, extensive ubiquitination was observed, suggesting that Acm1 is indeed an APCCdc20 substrate.
Figure 5. Acm1 is targeted for destruction by APCCdc20.
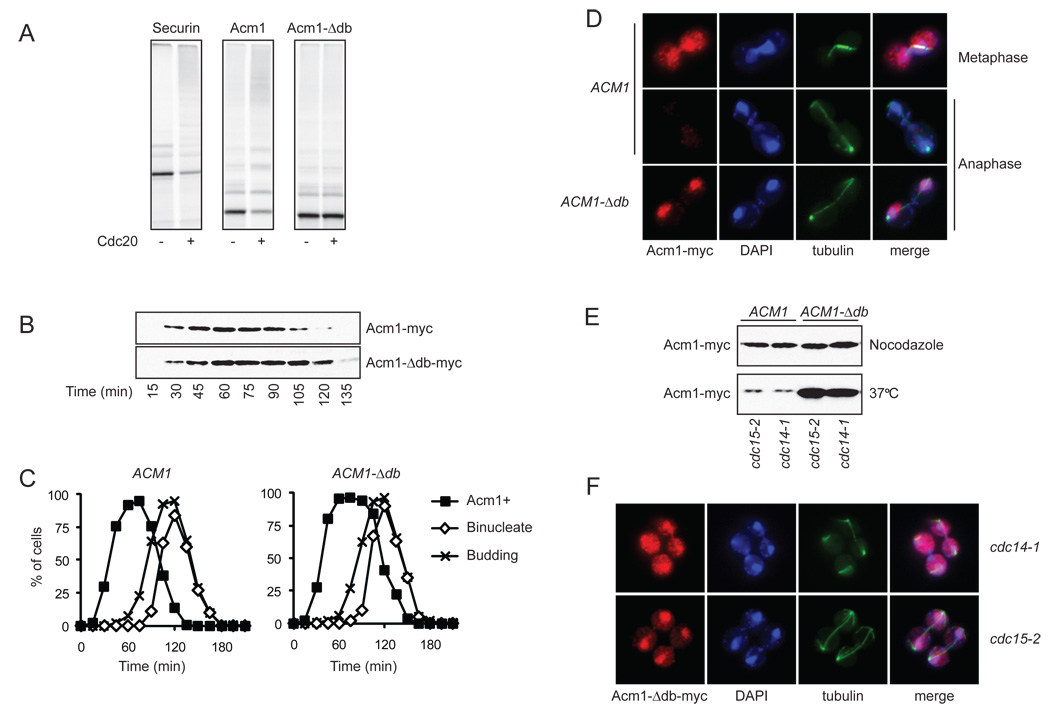
(A) Acm1 and Acm1-Δdb were translated in vitro with 35S-methionine and incubated in an APC reaction with or without Cdc20, as indicated. At left, an APC reaction with securin (Pds1) as substrate was included as a positive control. Reaction products were analyzed by SDS-PAGE and PhosphorImager analysis. Note that the clearest measure of activity is Cdc20-dependent depletion of the unmodified substrate at the bottom of the gel.
(B) ACM1-myc9 and ACM1-Δdb-myc9 yeast were synchronized in G1 with alpha factor and released into the cell cycle. Samples were taken for western blotting analysis of Acm1-myc at the indicated times.
(C) Cells from the experiment in panel B were processed for analysis of budding and binucleate formation, and the presence of Acm1 was followed by immunofluorescence.
(D) Representative images of metaphase and anaphase cells from the experiment in panel B.
(E) ACM1-myc9 and ACM1-Δdb-myc9 cells, carrying either the cdc15-2 or cdc14-1 temperature-sensitive mutation, were arrested either at metaphase by nocodazole treatment or in late anaphase by temperature shift to 37°C. Western blotting was performed to analyze Acm1 levels.
(F) Immunofluorescence analysis of cells arrested at high temperature in the experiment in panel E, showing the localization of Acm1-Δdb in late anaphase.
We next identified the sequence in Acm1 that is required for targeting by Cdc20. We found that the central inhibitory region of Acm1, which contains multiple destruction motifs as described earlier (residues 98–122), is not required for Cdc20-dependent ubiquitination in vitro (Figure 2). However, deletion of the highly conserved D-box at the N terminus of Acm1 (residues 8–11, termed the Acm1-Δdb mutant) greatly reduced ubiquitination by APCCdc20 in vitro (Figure 5A), suggesting that this is the key determinant of Acm1 destruction. Acm1-Δdb was still fully capable of inhibiting APCCdh1 both in vitro and in vivo (data not shown).
We constructed a yeast strain expressing an epitope-tagged version of the mutant ACM1-Δdb gene under the control of its own promoter. We analyzed Acm1 protein levels in cells released from a G1 arrest and then arrested again in the following G1 with mating pheromone. Deletion of the N-terminal D-box resulted in a significant (~20-min) delay in the destruction of Acm1 (Figure 5B, C). Most importantly, analysis of Acm1 localization in these cells revealed that Acm1-Δdb was present in anaphase cells, whereas wild-type Acm1 was not (Figure 5D). These results are consistent with the possibility that Acm1 is destroyed in early anaphase as a result of ubiquitination by APCCdc20. If Cdc20-dependent degradation is prevented by mutation of the N-terminal D-box, then Acm1 is destroyed after anaphase by some other mechanism (see below).
Acm1-Δdb is found in the nucleus of anaphase cells (Figure 5D). Considering our evidence that Cdk1-dependent phosphorylation reduces the nuclear accumulation of Acm1 (Figure 4), this observation suggests that Acm1 is dephosphorylated in early anaphase, perhaps as a result of cyclin destruction and the separase-dependent activation of Cdc14 (Queralt et al., 2006; Stegmeier et al., 2002).
We further addressed the timing and mechanism of Acm1 destruction by determining its levels and localization in cdc14-1 and cdc15-2 mutant cells, which arrest at the end of anaphase with high APCCdc20 activity but no APCCdh1 activity. After 2.5 hours incubation of mutant cells at the restrictive temperature, wild-type Acm1 was barely detectable in both strains, but the Acm1-Δdb mutant protein was present at high levels (Figure 5E), further supporting the notion that Acm1 is normally degraded in anaphase by a mechanism that depends on an interaction between the N-terminal D-box and APCCdc20. Furthermore, Acm1-Δdb was dispersed throughout the cell in the cdc14-1 arrest, as expected if Cdc14-dependent dephosphorylation normally promotes Acm1 import into the nucleus (Figure 5F). In the cdc15-2 arrest, where separase-dependent Cdc14 activation has occurred (Stegmeier et al., 2002), most Acm1-Δdb was in the nucleus.
Acm1 is both an inhibitor and substrate of APCCdh1
We next addressed the mechanism underlying the disappearance of the Acm1-Δdb mutant protein after anaphase. The timing of Acm1-Δdb destruction, coupled with the evidence that it is not degraded in a cdc15-2 arrest, raised the possibility that Acm1-Δdb, despite its Cdh1-inhibitory activity, is ubiquitinated in vivo by APCCdh1 at a rate that is sufficient to promote its destruction. To test this idea, we analyzed Acm1-Δdb protein levels during the cell cycle of cdh1Δ cells. It is difficult to arrest these cells with mating pheromone, and so we analyzed Acm1-Δdb levels after release from a cdc15-2 arrest in late anaphase. In the presence of wild-type CDH1, Acm1-Δdb levels dropped abruptly 45–60 min after release from the arrest and then rose again as the cells entered the next cell cycle (Figure 6A, B). In contrast, levels of Acm1-Δdb remained constant after release from the arrest in the cdh1Δ background, clearly demonstrating that destruction of the protein depends on APCCdh1.
Figure 6. APCCdh1 promotes the destruction of Acm1-Δdb.
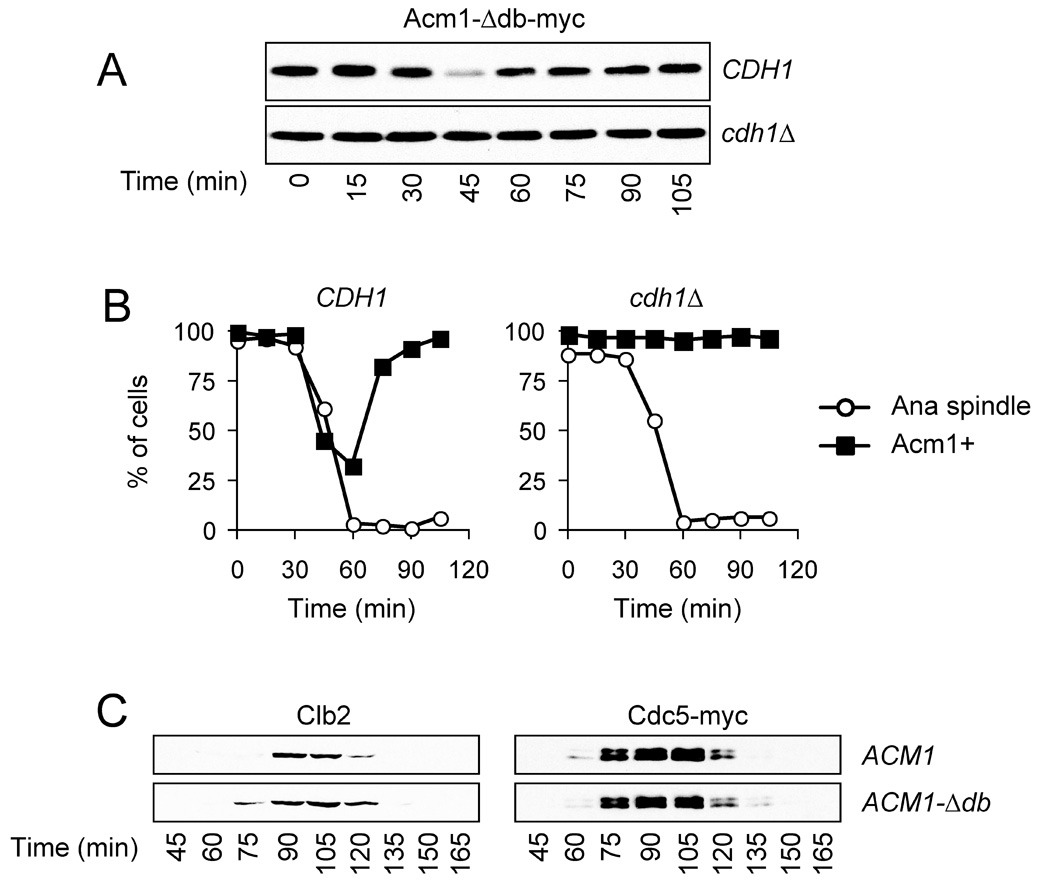
(A) ACM1-Δdb-myc9 cells carrying the cdc15-2 mutation, either with or without CDH1, were synchronized in late anaphase by growth at 37°C for 2.5 h. Cells were released back into the cell cycle by temperature downshift, and samples were taken every 15 min to measure Acm1 levels.
(B) Immunofluorescence analysis of Acm1 and tubulin was performed on cells from the experiment in panel A, and the number of cells containing Acm1 or anaphase spindles was determined.
(C) ACM1 and ACM1-Δdb cells carrying Cdc5-myc9 were synchronized in G1 with alpha factor and released into a new cell cycle. At the indicated times, samples were taken for western blotting analysis of the levels of Clb2 and Cdc5-myc9.
Acm1 appears to occupy the substrate-binding site on Cdh1 with high affinity, thereby competitively inhibiting the binding of other proteins at that site. However, some low rate of Acm1 ubiquitination might occur while it occupies that site, and inside the cell this might lead to proteasome-dependent stripping of Acm1 from Cdh1. Indeed, we found that wild-type Acm1 and Acm1-Δdb were ubiquitinated at low but significant rates by APCCdh1 in vitro, even under conditions where Cdh1 was fully occupied by Acm1 (Figure S4; summarized in Figure 2). The rate of Acm1 ubiquitination by APCCdh1 increased substantially after removal of various regions of Acm1, including the central inhibitory region (residues 98–122) and other regions at the N or C termini (Figure 2). As many of these deletions reduce Acm1-Cdh1 binding affinity, we suspect that increasing the rate of dissociation of Acm1 from Cdh1 allows more rapid Acm1 turnover by the enzyme, thereby transforming Acm1 from a high-affinity inhibitor to a relatively lowaffinity substrate. APCCdh1 is therefore capable of catalyzing the ubiquitination and thus destruction of its own inhibitor, which has interesting implications for the behavior of the late mitotic regulatory system (see Discussion).
If Acm1 destruction in anaphase is an important determinant of the timing of APCCdh1 activation, then delaying Acm1 destruction might result in delays in the destruction of APCCdh1 targets. However, we found that when ACM1-Δdb cells were released from a G1 arrest, degradation of the Cdh1 target Clb2 was only slightly delayed (Figure 6C). There were no significant effects on the timing of destruction of other APCCdh1 targets, including Cdc5. We suspect that Acm1-Δdb causes only minor delays in APCCdh1 activation because the dephosphorylation of Cdh1 in anaphase results in sufficient APCCdh1 activity to trigger Acm1-Δdb destruction.
Discussion
Acm1 is a high-affinity pseudosubstrate inhibitor of Cdh1
Our work suggests that Acm1 is a Cdh1-specific inhibitor with a central role in the control of APCCdh1 activation in late mitosis. Considerable evidence suggests that Acm1 inhibits Cdh1 function by competitively inhibiting its interaction with APC targets. Previous work showed that deletion of ACM1 increases the binding of Cdh1 to its targets in vivo (Martinez et al., 2006) and that Acm1 reduces Cdh1-Clb2 binding (but not Cdh1-APC binding) in vitro (Dial et al., 2007). Our work suggests that the inhibitory function of Acm1 depends on a central inhibitory region (residues 98–122) that contains sequence motifs that are very similar to known destruction motifs on APC targets. We speculate that these motifs interact with KEN and D-box binding sites on Cdh1, thereby blocking the binding of other substrates. Thus, inhibition by Acm1 involves mechanisms that are much like those used by other APC inhibitors, such as Mad3 and Emi1 (Burton and Solomon, 2007; Miller et al., 2006).
The binding of Acm1 to Cdh1 depends not only on the central inhibitory region at residues 98–122 but also on another region in the vicinity of residues 60–70. This region includes a sequence (FMLYEETAEER) that is rich in aromatic and acidic residues and has been well conserved in evolution (Figure S1). It seems likely that this region interacts with a conserved surface of Cdh1 that is adjacent to the KEN- and D-box-binding sites. In the context of wild-type Acm1, this region enhances Acm1-Cdh1 affinity and thereby makes Acm1 a more effective, high-affinity inhibitor.
As discussed earlier (see Results), we found that when Acm1-Cdh1 affinity is reduced by partial deletion of Cdh1-binding sites on Acm1, Acm1 is changed from a Cdh1 inhibitor to a more efficient APCCdh1 substrate. For example, an Acm1 fragment (residues 1–97) lacking the central inhibitory region is a poor Cdh1 inhibitor but an efficient substrate of APCCdh1. Thus, in the context of some Acm1 fragments, the Cdh1-binding region around residues 60–70 might serve as a ‘destruction’ sequence that drives a low-affinity Cdh1 interaction that promotes ubiquitination. This finding has interesting implications for mechanisms of APC-substrate recognition. It is well known that D- and KEN-boxes are required but often not sufficient for APC recognition, suggesting that the APC also recognizes other, as yet unidentified, sequence motifs in its targets. Perhaps the region around residues 60–70 of Acm1 represents a version of such a motif.
The Acm1-Cdh1 binding interaction is not just high in affinity but also very specific: Acm1 binds tightly to Cdh1 but not to the related activator Cdc20, suggesting that the pseudosubstrate and other binding motifs on Acm1 are highly specialized for Cdh1 binding. On the other hand, the N-terminal D-box of Acm1 is remarkably specific for Cdc20 and seems to have no impact on Acm1 recognition by Cdh1. There are very few other known APCCdc20 targets, and in most cases it is thought that a single destruction sequence targets these proteins to both Cdc20 and Cdh1. Detailed analyses of the Cdh1- and Cdc20-specific binding regions of Acm1 might therefore provide important insights into the nature of activator-substrate interactions (Yu, 2007).
Acm1 localization and destruction are governed by phosphorylation
Our results reveal that Acm1 function in the cell is modulated by a remarkable array of regulatory mechanisms. These mechanisms include Cdk1-dependent phosphorylation and Cdc14-dependent dephosphorylation, which appear to influence the subcellular localization of Acm1. Acm1 contains five highly conserved Cdk1 consensus sites and is an excellent substrate for Cdk1 in vitro (Ubersax et al., 2003). It localizes to the nucleus in late G1, when Cdk1 activity is low, and the nonphosphorylatable Acm1-5A mutant localizes constitutively to the nucleus. CDC14 overexpression drives Acm1 into the nucleus, and nuclear accumulation is reduced in the cdc14-1 mutant. Finally, the Cdc20-resistant Acm1-Δdb mutant is imported into the nucleus at the onset of anaphase, as might be expected if separase-dependent Cdc14 activation, together with some cyclin destruction, triggers Acm1 dephosphorylation. We speculate that phosphorylation alters the relative rates of nuclear import and export of Acm1; for example, phosphorylation may block the function of some nuclear localization sequence on Acm1. It is known that phosphorylated Acm1 interacts with the 14-3-3 proteins Bmh1 and Bmh2 (Dial et al., 2007; Martinez et al., 2006), and the binding of these proteins may interfere with nuclear import – much like phosphorylation-dependent 14-3-3 binding interferes with the nuclear import of the phosphatase Cdc25C in vertebrate cells (Yang et al., 1999).
What function might the import of dephosphorylated Acm1 serve? The APC activator Cdc20 is found primarily inside the nucleus (Jaquenoud et al., 2002), and so the Cdc14-dependent dephosphorylation and thus nuclear import of Acm1 may promote its ubiquitination by nuclear APCCdc20 (Figure 7). Consistent with this possibility, we found that the nonphosphorylatable Acm1-5A mutant was degraded earlier than the wild-type protein. Phosphorylation may also delay Acm1 destruction more directly: one of its Cdk1 phosphorylation sites lies next to its N-terminal D-box, where it may interfere with Acm1-Cdc20 binding. It is known, for example, that phosphorylation of Cdk sites near the D-box of human Cdc6 blocks its ubiquitination by APCCdh1 (Mailand and Diffley, 2005).
Figure 7. Model of the regulatory system governing APCCdh1 activation.
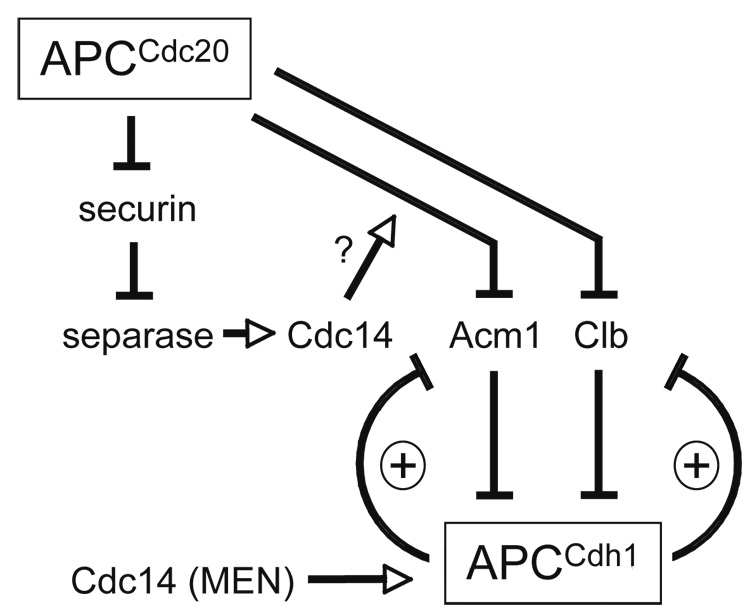
APCCdc20 promotes APCCdh1 activation by triggering the destruction of two Cdh1 inhibitors: Acm1 and cyclins (Clb). These inhibitors are also targets of APCCdh1, resulting in two double-negative feedback loops that are likely to generate bistability in APCCdh1 activity. Cdc14, following activation by the mitotic exit network (MEN), also promotes APCCdh1 activation by dephosphorylating Cdh1. Our results suggest that separase-dependent Cdc14 activation also acts on Cdh1 by promoting the APCCdc20-dependent destruction of Acm1. For simplicity, this diagram does not include the double-negative feedback loop between Clb and the Cdk1 inhibitor Sic1, whose destruction is stimulated by Clb-Cdk1 activity.
Acm1 is a key component of the late mitotic regulatory system
Acm1 serves as an important node in the complex regulatory network governing Cdh1 activation (Figure 7). Prior to anaphase, Acm1 acts together with Cdh1 phosphorylation to restrain APCCdh1 activity and thus prevent the premature destruction of Cdh1-specific targets. As phosphorylated Cdh1 is thought to reside primarily in the cytoplasm (Jaquenoud et al., 2002), an additional function of cytoplasmic Acm1 may be to simply prevent Cdh1 from binding and thereby disrupting the function of its cytoplasmic and bud neck targets, such as Clb2 and Hsl1 (Martinez et al., 2006).
APCCdc20 catalyzes Acm1 ubiquitination, resulting in an abrupt decline in Acm1 levels at anaphase onset. This provides a clear mechanism by which APCCdc20 promotes the activation of its successor, APCCdh1. It is well known that Cdc20 also promotes Cdh1 activation through the destruction of mitotic cyclins (which activate the Cdks that phosphorylate Cdh1) and less directly through the activation of Cdc14 (which dephosphorylates Cdh1) (Figure 7). APCCdc20-dependent Acm1 destruction thus joins Cdh1 dephosphorylation as the second major mechanism underlying APCCdh1 activation. Although Acm1 is not essential for cell viability, and its destruction alone may not be a key determinant of the timing of Cdh1 activation, it is likely that the existence of two distinct Cdh1-activating mechanisms results in a more robust activation process.
Interestingly, our studies of the Acm1-Δdb mutant revealed that when Cdc20-dependent Acm1 destruction is prevented, Cdh1 dephosphorylation triggers sufficient APCCdh1 activation to promote Acm1 ubiquitination and destruction. We suspect, therefore, that Acm1, although present in large excess over Cdh1 in the cell (Martinez et al., 2006), cannot completely restrain APCCdh1 once Cdh1 dephosphorylation occurs (except when Acm1 is overexpressed, in which case it suppresses the ill effects of the unphosphorylated Cdh1-m11 mutant; (Martinez et al., 2006)). More importantly, the ability of APCCdh1 to promote the destruction of its own inhibitor has major implications for the behavior of the regulatory circuits governing APCCdh1 activity. The mutually antagonistic relationship between Acm1 and Cdh1 generates a double-negative feedback loop – which is essentially the equivalent of a positive feedback loop (Figure 7). These regulatory loops have the potential to promote bistability in regulatory systems (Ferrell, 2002; Tyson et al., 2003): in this case, for example, the Acm1-Cdh1 loop is predicted to generate a system in which Cdh1 switches completely between a stable state of low activity and a stable state of high activity.
In fact, this system is already known to contain double-negative feedback because mitotic cyclins and Cdh1 are also mutual antagonists (Figure 7). We therefore propose that the Cdh1 regulatory system includes a pair of interlinked double-negative feedback loops, which together produce a bistable system ensuring that APCCdh1 activation is switch-like, robust, and irreversible (Brandman et al., 2005). According to this scheme, Cdh1 is held during most of the cell cycle in a stable inactive state by Acm1 and Cdk1-dependent phosphorylation. When APCCdc20 begins to remove both of these inhibitory mechanisms in anaphase, the partial activation of APCCdh1 then allows it to gain the upper hand over its inhibitors, flipping the switch to a stable state of full APCCdh1 activity. As is often the case in bistable systems with strong feedback, APCCdh1 is predicted to remain in the on state even after the original trigger mechanism (APCCdc20 activity) is removed in late mitosis.
Acm1 thus lies at the center of a complex web of regulatory interactions (Figure 7). As a target of both APCCdc20 and APCCdh1, Acm1 helps ensure that APCCdh1 is activated after APCCdc20, in a switch-like and robust manner. In addition, Acm1 integrates inputs from Cdk1 and Cdc14, and presumably from transcriptional regulators as well, to refine the spatial and temporal features of its regulation and behavior. The detailed dissection of these mechanisms promises to unveil interesting new features of late mitotic regulation, and is also likely to provide insights into the operation of complex regulatory systems in general.
Experimental Procedures
Strains and Plasmids
All experiments were performed in the W303 budding yeast strain. Epitope tagging of endogenous genes and gene deletions was performed by gene targeting using polymerase chain reaction (PCR) products. The ACM1 gene, along with 9 C-terminal Myc epitopes and 323 bp of the ACM1 promoter, was amplified from budding yeast and cloned to allow integration of ACM1-myc9 at either the LEU2 or URA3 locus. This plasmid was modified to generate ACM1-Δdb by removal of the coding sequence for amino acids 8 to 11 (RTIL). This plasmid was also modified to generate the Cdk resistant form acm1-5A by mutagenesis of serines 3, 31, 48 and 102, plus threonine 161, to alanine. For in vitro APC assays, ACM1 or ACM1-Δdb was cloned into vector pME34, which contains a T7 promoter and a C-terminal ZZ tag. All other ACM1 fragments were generated by PCR without the ZZ tag.
Strains with galactose-inducible expression of CDH1-m11 were generated by transformation of W303 with plasmid pRS303-CDH1-m11, which integrates at the HIS3 locus. The toxicity of CDH1-m11 expression can be suppressed by galactose-induced expression of ACM1-TAP from the centromeric plasmid pRS314-1234 (created by in vivo recombination). Mutant versions of Acm1 were generated by similar methods and comparable expression was confirmed by Western blotting.
APC assays and inhibition assays
E1, E2 and APC were expressed and purified as described previously (Carroll et al., 2005; Carroll and Morgan, 2005). APC activators were transcribed and translated in vitro using TnT Quick Coupled Transcription/Translation Systems (Promega). Substrates (Pds1/securin and Acm1) were transcribed and translated in vitro from plasmids with 35S-Methionine and treated with 10 mM NEM (10 min) followed by 20 mM DTT (10 min) to inactivate ubiquitin chain-extending activities in the reticulocyte lysate. For APC inhibition assays, unlabeled Acm1 was pre-incubated with APC activator for 10 min before addition to the APC assay (5 µl of TnT Acm1 with 2 µl TnT activator). APC assays were performed as described (Carroll et al., 2005; Carroll and Morgan, 2005). Briefly, E1 (Uba1, 300 nM), E2 (Ubc4, 50 µM), ubiquitin (150 µM), and ATP (1 mM) were incubated for 15 min. APC (0.1–1 nM), substrate (2 µl of TnT mix into 15 µl reaction), and activator (2 µl of TnT mix into 15 µl reaction) were added. Reactions were incubated for the indicated times at room temperature, stopped by the addition of SDS sample buffer, separated by SDS-PAGE, and visualized and quantified with a Molecular Dynamics PhosphorImager (Amersham Biosciences).
Binding assays
ZZ-tagged activator was transcribed and translated in vitro from plasmids. Acm1 was transcribed and translated in vitro from PCR products in the presence of 35S-Methionine. Translated products were incubated with IgG-magnetic beads (IgG coupled to Epoxy Magnetic Beads (Dynal)) for 2 h at 4°C. After 3 washes with binding buffer (20 mM Hepes pH 8.0, 150 mM NaCl, 0.1% NP-40), beads were boiled in SDS Sample Buffer and the supernatants analyzed by SDS-PAGE. For the time course in Figure 3C, an additional wash was performed at each time point. Reaction products were separated by SDS-PAGE and visualized and quantified with a Molecular Dynamics PhosphorImager (Amersham Biosciences).
Immunofluorescence, growth and antibodies
Conditions for growth and release of synchronous cultures from arrest in G1 by alphafactor were performed as described previously (Loog and Morgan, 2005). Western blot, budding counts and immunofluorescence were performed as described previously (Sullivan et al., 2004). The antibodies used were anti-tubulin YOL1/34 (Serotec), anti-HA clone 16B12 (Babco), anti-Clb2 (a kind gift of D. Kellogg) and anti-Myc clone 9E10 (Babco).
Supplementary Material
Acknowledgements
We thank members of the Morgan laboratory for helpful suggestions and support. This work was supported by funding from the National Institute of General Medical Sciences (GM53270).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Brandman O, Ferrell JE, Jr, Li R, Meyer T. Interlinked fast and slow positive feedback loops drive reliable cell decisions. Science. 2005;310:496–498. doi: 10.1126/science.1113834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JL, Solomon MJ. Mad3p, a pseudosubstrate inhibitor of APCCdc20 in the spindle assembly checkpoint. Genes Dev. 2007;21:655–667. doi: 10.1101/gad.1511107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CW, Enquist-Newman M, Morgan DO. The APC subunit Doc1 promotes recognition of the substrate destruction box. Curr Biol. 2005;15:11–18. doi: 10.1016/j.cub.2004.12.066. [DOI] [PubMed] [Google Scholar]
- Carroll CW, Morgan DO. Enzymology of the Anaphase-Promoting Complex. Meth. Enzymol. 2005;398:219–230. doi: 10.1016/S0076-6879(05)98018-X. [DOI] [PubMed] [Google Scholar]
- D'Amours D, Amon A. At the interface between signaling and executing anaphase--Cdc14 and the FEAR network. Genes Dev. 2004;18:2581–2595. doi: 10.1101/gad.1247304. [DOI] [PubMed] [Google Scholar]
- Dial JM, Petrotchenko EV, Borchers CH. Inhibition of APCCdh1 activity by Cdh1/Acm1/Bmh1 ternary complex formation. J Biol Chem. 2007;282:5237–5248. doi: 10.1074/jbc.M606589200. [DOI] [PubMed] [Google Scholar]
- Ferrell JE., Jr Self-perpetuating states in signal transduction: positive feedback, double-negative feedback and bistability. Curr Opin Cell Biol. 2002;14:140–148. doi: 10.1016/s0955-0674(02)00314-9. [DOI] [PubMed] [Google Scholar]
- Grosskortenhaus R, Sprenger F. Rca1 inhibits APC-Cdh1(Fzr) and is required to prevent cyclin degradation in G2. Dev Cell. 2002;2:29–40. doi: 10.1016/s1534-5807(01)00104-6. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Loktev AV, Ban KH, Jackson PK. Plk1 regulates activation of the anaphase promoting complex by phosphorylating and triggering SCFbetaTrCP-dependent destruction of the APC Inhibitor Emi1. Mol Biol Cell. 2004;15:5623–5634. doi: 10.1091/mbc.E04-07-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JY, Reimann JD, Sorensen CS, Lukas J, Jackson PK. E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC(Cdh1) Nat Cell Biol. 2002;4:358–366. doi: 10.1038/ncb785. [DOI] [PubMed] [Google Scholar]
- Jaquenoud M, van Drogen F, Peter M. Cell cycle-dependent nuclear export of Cdh1p may contribute to the inactivation of APC/C(Cdh1) Embo J. 2002;21:6515–6526. doi: 10.1093/emboj/cdf634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Morgan DO. Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr. Biol. 1999;9:227–236. doi: 10.1016/s0960-9822(99)80111-0. [DOI] [PubMed] [Google Scholar]
- King RW, Glotzer M, Kirschner MW. Mutagenic analysis of the destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Mol. Biol. Cell. 1996;7:1343–1357. doi: 10.1091/mbc.7.9.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C, Herzog F, Gieffers C, Mechtler K, Hagting A, Pines J, Peters JM. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 2003;22:6598–6609. doi: 10.1093/emboj/cdg627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loog M, Morgan DO. Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature. 2005;434:104–108. doi: 10.1038/nature03329. [DOI] [PubMed] [Google Scholar]
- Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 2005;122:915–926. doi: 10.1016/j.cell.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Martinez JS, Jeong DE, Choi E, Billings BM, Hall MC. Acm1 is a negative regulator of the CDH1-dependent anaphase-promoting complex/cyclosome in budding yeast. Mol Cell Biol. 2006;26:9162–9176. doi: 10.1128/MCB.00603-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JJ, Summers MK, Hansen DV, Nachury MV, Lehman NL, Loktev A, Jackson PK. Emi1 stably binds and inhibits the anaphase-promoting complex/cyclosome as a pseudosubstrate inhibitor. Genes Dev. 2006;20:2410–2420. doi: 10.1101/gad.1454006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. Segregating sister genomes: the molecular biology of chromosome separation. Science. 2002;297:559–565. doi: 10.1126/science.1074757. [DOI] [PubMed] [Google Scholar]
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Pfleger CM, Kirschner MW. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 2000;14:655–665. [PMC free article] [PubMed] [Google Scholar]
- Queralt E, Lehane C, Novak B, Uhlmann F. Downregulation of PP2A(Cdc55) phosphatase by separase initiates mitotic exit in budding yeast. Cell. 2006;125:719–732. doi: 10.1016/j.cell.2006.03.038. [DOI] [PubMed] [Google Scholar]
- Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK. Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell. 2001;105:645–655. doi: 10.1016/s0092-8674(01)00361-0. [DOI] [PubMed] [Google Scholar]
- Rudner AD, Murray AW. Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol. 2000;149:1377–1390. doi: 10.1083/jcb.149.7.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteinberg M, Protopopov Y, Listovsky T, Brandeis M, Hershko A. Phosphorylation of the cyclosome is required for its stimulation by Fizzy/cdc20. Biochem Biophys Res Commun. 1999;260:193–198. doi: 10.1006/bbrc.1999.0884. [DOI] [PubMed] [Google Scholar]
- Stegmeier F, Amon A. Closing mitosis: the functions of the Cdc14 phosphatase and its regulation. Annu Rev Genet. 2004;38:203–232. doi: 10.1146/annurev.genet.38.072902.093051. [DOI] [PubMed] [Google Scholar]
- Stegmeier F, Visintin R, Amon A. Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell. 2002;108:207–220. doi: 10.1016/s0092-8674(02)00618-9. [DOI] [PubMed] [Google Scholar]
- Sullivan M, Higuchi T, Katis VL, Uhlmann F. Cdc14 phosphatase induces rDNA condensation and resolves cohesin-independent cohesion during budding yeast anaphase. Cell. 2004;117:471–482. doi: 10.1016/s0092-8674(04)00415-5. [DOI] [PubMed] [Google Scholar]
- Sullivan M, Morgan DO. Finishing mitosis, one step at a time. Nat Rev Mol Cell Biol. 2007;8:894–903. doi: 10.1038/nrm2276. [DOI] [PubMed] [Google Scholar]
- Thornton BR, Toczyski DP. Precise destruction: an emerging picture of the APC. Genes Dev. 2006;20:3069–3078. doi: 10.1101/gad.1478306. [DOI] [PubMed] [Google Scholar]
- Tyson JJ, Chen KC, Novak B. Sniffers, buzzers, toggles and blinkers: dynamics of regulatory and signaling pathways in the cell. Curr Opin Cell Biol. 2003;15:221–231. doi: 10.1016/s0955-0674(03)00017-6. [DOI] [PubMed] [Google Scholar]
- Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425:859–864. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]
- Yang J, Winkler K, Yoshida M, Kornbluth S. Maintenance of G2 arrest in the Xenopus oocyte: a role for 14-3-3-mediated inhibition of Cdc25 nuclear import. EMBO J. 1999;18:2174–2183. doi: 10.1093/emboj/18.8.2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell. 2007;27:3–16. doi: 10.1016/j.molcel.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, Seufert W. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the Anaphase Promoting Complex. Science. 1998;282:1721–1724. doi: 10.1126/science.282.5394.1721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.