

Summary
Tumor-associated immune responses assert varied responses toward developing neoplasms that can either act to eradicate malignant cells via engagement of potent cytotoxic programs or alternatively enhance tumor growth through release of multifunctional pro-tumor mediators. Seemingly paradoxical, these disparate activities reflect a continuum of polarization (or activation) states possible for distinct leukocyte subsets that demonstrate tissue, organ, and tumor selectivity. Herein, we review clinical and experimental studies investigating cellular and molecular mechanisms utilized by neoplastic tissues to alternatively polarize immune responses that favor either pro- or anti-tumor immunity.
Keywords: cancer, humoral immunity, inflammation, innate immunity, leukocytes, adaptive immunity
Introduction: cancer and inflammation
The cellular composition of cancers include ‘initiated’ or neoplastic cells harboring genetic mutations, typically referred to as ‘tumor’ or ‘cancer’ cells, as well as diverse populations of genetically stable cell types that are activated and/or recruited to the local microenvironment, e.g. innate and adaptive immune cells, cells composing the hematogenous and lymphatic vasculature, fibroblasts, and other specialized mesenchymal support cells. Reciprocal interactions between responding ‘normal’ cells, their mediators, and structural components of extracellular matrix (ECM) with genetically altered neoplastic cells regulate all aspects of tumorgenicity (1-4). Clinical and experimental studies in the last decade have enhanced our understanding of how activated and/or recruited genetically stable cells contribute to tumorigenesis. For a neoplastic tissue to expand, not only do initiated tumor cells need to acquire unlimited self-renewal capacity (5), but they also need to co-opt cellular programs embedded within the tissue that enhance blood flow, oxygenation, and waste removal, i.e. angiogenesis and lymphangiogenesis, as well as molecular programs that favor tumor cell survival, and enhanced metabolism of soluble and insoluble components of ECM that favor remodeling and expansion of the tissue (4). While some experimental studies have revealed the intrinsic properties of tumor cells that regulate aspects of these programs, also emerging has been the realization that innate (myeloid) and adaptive (lymphoid) immune cells functionally contribute toward all aspects of cancer development (2, 6, 7).
Innate immune cells, including mast cells, macrophages, granulocytes, dendritic cells (DCs), and natural killer (NK) cells, represent the first line of defense against pathogens and foreign agents. In response to disruption of tissue homeostasis, tissue-resident innate immune cells locally secrete soluble factors, such as cytokines and chemokines, matrix-remodeling proteins, and other bioactive mediators, that recruit additional leukocytes from the circulation into the tissue, i.e. inflammation. In response to pathogen infection, some recruited immune cells (also referred to as inflammatory cells) directly eliminate pathogenic agents in situ. Alternatively, professional antigen-presenting cells (APCs) including DCs and macrophages take up foreign antigens (including tumor antigens) and migrate to lymphoid organs where they ‘present’ their antigens to adaptive immune cells. Upon recognition of presented antigens, B lymphocytes, CD8+ cytotoxic T lymphocytes (CTL), and CD4+ T helper (Th) lymphocytes, mount an ‘adaptive’ immune response targeted against the foreign agent. By these mechanisms, acute activation of innate immunity sets the stage for activation of antigenically committed adaptive immune responses. Once foreign agents have been eliminated, inflammation resolves, and tissue homeostasis is restored. Regarding cancer risk and/or cancer development, it is now clear that similar immunological responses required for activating acute inflammation can be co-opted, such that if persistent inflammation is maintained in a tissue, it can instead promote neoplastic programming of that tissue and enhance cancer development (2, 6, 7).
The fact that some leukocytes promote - rather than restrict - tumor growth may be viewed as an apparent paradox (8, 9). Historically, leukocytes found in developing tumors were thought to represent an attempt by the host to eradicate transformed cells. Undeniably, certain leukocytes, such as some T lymphocyte subsets and NK cells, play a vital function in constraining tumor development (10), and it has been postulated that many more tumors arise than those that eventually develop to fully malignant disease owing to such activity. However, it is now clear that tumor-infiltrating leukocytes also play causal roles in cancer development and progression (2, 6-8, 11, 12). Herein, we review clinical and experimental studies investigating cellular and molecular mechanisms utilized by neoplastic tissues to alternatively polarize immune responses to favor either pro- or anti-tumor immunity.
Establishing pro-tumor immunity
In 1863, Virchow first postulated that cancer originates at sites of chronic inflammation, in part based on his hypothesis that some classes of irritants causing inflammation also enhance cell proliferation. When tissues are injured or exposed to chemical irritants, damaged cells are removed by induction of cell death pathways, while cell proliferation is enhanced to facilitate tissue regeneration in an attempt to re-establish tissue homeostasis. Proliferation and acute inflammatory responses are resolved only after insulting agents are removed and/or tissue repair completed. However, when insulting agent persists over time, so too do sustained cycles of cell proliferation/death and localized inflammation, and together these processes generate a microenvironment rich in inflammatory cell bioactive products that can increase neoplastic risk and foster tumor progression. Thus, while sporadic or inherited genetic mutations in critical genes regulating cell cycle, cell death, differentiation, metabolism, and adhesion represent initiating events in tumorigenesis (‘initiation’), chronic inflammatory responses can promote full malignant potential of neoplastic cells (‘promotion’).
Pre-malignant and malignant tissues are known to be associated with changes in leukocyte profiles as well as their functionality (13, 14), including suppressed CTL responses associated with tumor rejection, in combination with enhanced humoral immunity (HI), that can promote tumor progression (13, 15). Distinctive CD4+ T-cell subsets, e.g. Th1, Th2, or Th17 cells, secrete unique repertoires of cytokines that mediate their responses. Th1 cells produce interleukin-2 (IL-2) and interferon-γ (IFN-γ) for example, and direct CTL responses, whereas Th2 cells produce IL-4, IL-13, and IL-10 and facilitate local HI responses, whereas Th17 polarized cells produce IL-23, IL-6, and tumor necrosis factor-α (TNF-α), factors that promote and/or sustain chronic inflammation. In peripheral blood of patients with bladder and colorectal cancer, proportions of Th1 cells, identified by intracellular production of IFN-γ or IL-2, is decreased, whereas proportions of Th2 cells producing IL-4, IL-6, and/or IL-10 is significantly elevated, as compared with otherwise healthy cohorts (16, 17). A recent study investigating characteristics of leukocytic infiltrations in human cervical carcinomas found that CD3+ tumor-infiltrating T cells display enhanced Th2 cytokine profiles and specifically increased IL-4 and reduced IFN-γ (18). In line with these findings, alterations in immune cell status (suppressed CTL responses and enhanced HI) have also been reported in chronicin flammatory diseases associated with increased cancer risk (reviewed in 13). Taken together, these compelling clinical findings indicate that enhanced pro-tumor immune responses underlie increased risk for neoplastic progression in tissues afflicted with chronic inflammatory disease pathologies and/or tissues that harbor initiated neoplastic cells.
Polarization of Th cell responses
The specific immune response to a tumor is directed by interactions with mature APCs and the nature of the pro-inflammatory milieu (19). In this context, regulatory CD4+ T cells play an important role in orchestrating responses. Naive T cells of the same antigenic specificity can be ‘polarized’ into distinct functional effector cells that depend upon early environmental signals received when antigen is presented (20). These signals are produced by innate leukocytes and cells at the site of injury and are registered by receptors expressed on naive T-cell precursors (21). Following viral infection, for example, infected cells rapidly release type-I interferons (IFN-α and/or β) (22), that activate early viral defense programs, and are also important for polarizing the immune system toward an anti-viral Th1 response (23).
Effector CD4+ T cells of the Th1 lineage evolved to eradicate intracellular pathogens, such as intracellular bacteria and viruses, through activation of CTL responses, as well as induction of immunoglobulin G2a (IgG2a) and IgG3 production (24). In the context of cancer development, it has been postulated that some IFNs mediate anti-tumor immunity, in part through regulating polarized Th1 responses (25). IFN-induced initiation of Th1 cell polarization occurs when T-cell receptor (TCR) activation is accompanied by IFN-induced signaling through the signal transducer and activator of transcription 1 (STAT1) intracellular signaling cascade, the earliest step of Th1 polarization (26). Signaling through STAT1 upregulates expression of the IL-12Rβ2 chain of the IL-12 receptor, thus yielding cells responsive to IL-12, a cytokine important for further differentiation of Th1 cells (27). This process also stimulates IFN-γ production and induces expression of IL-18Rα (28). Mature Th1 effector cells can produce IFN-γ through TCR-dependent pathways but also are capable of producing the cytokine independently of antigen stimulation, if activated by IL-12 and IL-18 (29).
Cells of the Th2 lineage are thought to have evolved to enhance elimination of parasitic infections and are characterized by production of IL-4, IL-5, and IL-13. These cytokines are potent activators of IgE production and recruitment of eosinophils and granulocytes (30). Specification of Th2 cells is initiated by TCR signaling together with IL-4 signaling through STAT6, resulting in induced epigenetic chromatin remodeling in the Th2 cytokine cluster (31) while simultaneously suppressing STAT4 and IL-12Rβ2 expression (32). Together, these events favor expression of Th2-related cytokines, while rendering them insensitive to repolarization toward Th1 lineage commitment.
Recent studies have revealed a greater diversity in the CD4+ T-cell effector repertoire and have linked the cytokines IL-23 and IL-17 to a new arm of the Th cell family, referred to as Th17 cells (33). Development of Th17 cells from naive T cells is induced by transforming growth factor-β (TGF-β) together with IL-6 as an important cofactor (34). TCR stimulation can directly stimulate Th17 cytokine production, while IL-1, IL-18, and IL-23 can potentiate this effect (24). CD4+ Th17 cells have been implicated in clearance of extracellular bacteria and autoimmune disorders, such as experimental autoimmune encephalitis and collagen-induced arthritis, and are associated with recruitment of granulocytes and expression of IgM, IgG, and IgA (24, 35). Th17 responses have also been associated with aspects of cancer development (36). In a mouse model of chemically induced skin carcinogenesis, IL-23 was identified as an important mediator of the inflammatory response associated with promotion (36).
A fourth class of CD4+ cells, namely regulatory T (Treg) cells, suppress effector functions of CTLs and have important physiological roles in preventing autoimmune disease and exacerbated immunity against infections (9, 14). Two main subsets of Treg cells have been identified: natural Treg cells that form in the thymus and adaptive Treg cells that differentiate in peripheral tissues. Natural Treg cells are CD4+CD25+FoxP3+ and form centrally in the thymus by high-affinity TCR with antigens expressed in thymic stroma (14, 37). Natural Treg cells suppress immune responses through cell surface molecules such as cytotoxic T-lymphocyte antigen 4 (CTLA4), membrane-bound TGF-β, and pericellular generation of adenosine. Adaptive Treg cells are CD4+CD25+FoxP3+/low, form in peripheral tissues in the presence of IL-10 or TGF-β, and suppress immune responses mainly through release of soluble factors, such as IL-10 and TGF-β (24). In vivo depletion of Treg cells using neutralizing antibodies directed against CD25 enhance anti-tumor T-cell responses and induce regression of experimental tumors, e.g. sarcomas and melanomas (38, 39). Together with clinical observations revealing that presence of Treg cells in patients with ovarian cancer correlated with reduced survival (40), these findings may indicate their critical role in regulating pro- versus anti-tumor immunity.
Diverse CD4+ T-cell subsets orchestrate a wide range of immune responses depending upon cues received from their microenvironment that can enhance or limit pro- and/or anti-tumor immunity (Fig. 1). Understanding the tissue and organ-specific nuances of these signals may reveal important targets for anti-cancer immune-based therapy whose modulation would enhance anti-tumor immune response and thus eradication or at least stabilization of pre-malignant disease.
Fig. 1. Polarization of immune responses during tumor development.
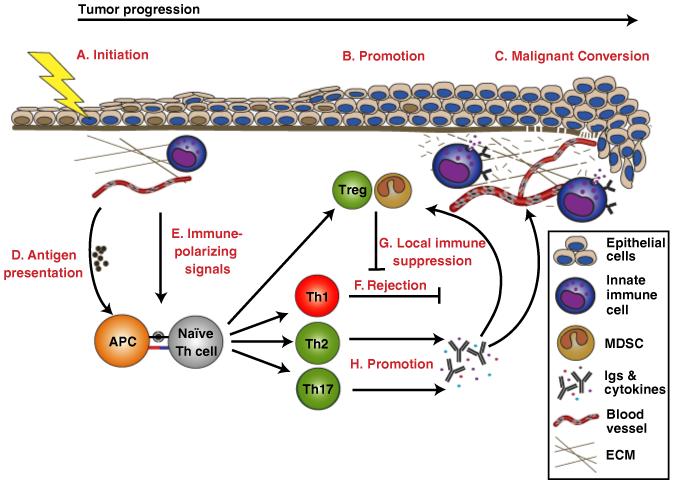
Epithelial cancers develop through expansion of cells harboring mutations in key regulatory genes, i.e. initiated cells (A). Microenvironmental effectors, such as inflammation, promote (B) malignant conversion (C) of initiated cells. When an inflammatory response is triggered during initiation or promotion, antigen-presenting cells (APCs), such as dendritic cells and macrophages, take up tumor-associated antigens and present them (D) to naïve T helper (Th) cells. Depending on the context and the inflammatory microenvironment in which antigen presentation occurs, Th cells can become alternatively polarized. The process of immune polarization integrates a large number of microenvironmental signals (E) into one general outcome. Three discrete and mutually exclusive Th cell lineages, Th1, Th2, and Th17, have been defined. Th1 polarized immune responses (F) are associated with cytotoxic T lymphocytes (CTL)-mediated killing of tumor cells and favor cancer regression. CTL-mediated tumor cell cytotoxicity can be inhibited through recruitment and/or conversion of natural or adaptive regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) (G). Th2 and Th17 polarized immune responses both have been shown to promote tumor progression (H) by establishing chronic inflammation, e.g. through cytokine production and induction of humoral responses. Chronic inflammation can promote tumor progression in multiple ways, such as stimulation of tumor cell proliferation and dedifferentiation, activation of angiogenesis, and recruitment of Tregs and MDSCs, as well as through remodeling of extracellular matrix (ECM) and tissue basement membranes.
Stimulating a polarized Th response
Cell autonomous and cell non-autonomous mechanisms regulate Th polarization. One required signal is presentation of specific peptides by class II major histocompatibility complex molecules to TCRs (41) that effectively determine which tumor antigens immune responses are directed toward (42). Following engagement of TCRs, Th cells require costimulatory signals, such as CD40-CD40L and CD28-CD80/86 receptor-ligand interactions, from APCs to become activated. A well-characterized costimulatory receptor is CD28, which binds CD80 and CD86 on APCs, thereby stimulating survival and proliferation of Th cells (43). Both antigen-specific signals mediated through the TCR and antigen-non-specific costimulatory signals are necessary for T-cell activation. However, an additional and earlier signal is also necessary for APC activation that orientates subsequent Th responses. This signal is induced by ligation of pathogen-recognition receptors (PRRs) that recognize factors associated with infection and tissue damage (20). PRRs have recently been implicated as important for both anti- and pro-tumor activities by the immune system and are discussed further below.
Innate immune responses by resident and recruited leukocytes have historically been viewed as the first line of defense providing rapid non-specific protection to the local tissue/organ and thereby allowing time for adaptive immune response pathways to become engaged. However, it is now clear that adaptive immunity also depends on the level and specificity of the initial ‘danger’ signals perceived by activated innate leukocytes (21, 44). Following infection, innate leukocytes recognize substances produced by microbes through germline-encoded PRRs. The best-studied group of PRRs is the Toll-like receptors (TLRs). TLRs are type I integral membrane glycoproteins with cytoplasmic signaling domains homologous to that of the IL-1R (21). TLRs were initially investigated for their role in defense against microbes, but it is now clear that TLRs play an important role in polarizing immune responses during tumor progression. Twelve TLRs have been described, and each member has a unique pattern of cellular and tissue expression. TLRs are expressed on innate immune cells, including DCs, mast cells, macrophages, neutrophils, and granulocytes, but also on endothelial cells, epithelial cells, and fibroblasts (21). Following engagement, TLRs dimerize and undergo conformal changes required for recruitment of Toll-IL-1 resistance (TIR)-domain-containing adapter molecules to the TIR domain of the TLR. There are four adapter molecules, namely myeloid differentiation factor 88 (MyD88), TIR-associated protein (TIRAP)/MyD88-adapter-like (MAL), TIR-domain-containing molecule 1 (TICAM1), and TIR-domain-containing adapter inducing IFN-β (TRIF)-related adapter molecule (TRAM) (45).
It is now clear that distinct responses are elicited by different TLR ligands that can in part be explained by differential usage of adapter molecules. There are two main outcomes of these signaling pathways: the MyD88-dependent pathway that leads to production of pro-inflammatory cytokines and polarization toward a Th2 phenotype (46) or, alternatively, expression of type I IFNs and polarization toward a cytotoxic Th1 response (46).
Stimulation with TLR3, TLR4, TLR7, and TLR9 agonists, but not TLR2 agonists, leads to type I IFN production, and cytokines with potent anti-tumor effects including DC maturation, upregulation of the activation markers CD40, CD80, and CD86 and expression of IFN-α and IFN-β (33), giving rise primarily to anti-viral and Th1-polarized cytotoxic responses. This polarization can be either MyD88-dependent or independent for different TLRs. Stimulation of TLR9, for example by its endogenous ligand, free DNA, elicits strong Th1 responses that have demonstrated anti-tumor activity and are currently being tested in phase II and phase III clinical trials (47). Recently, Apetoh et al. (48) reported that following chemotherapy or radiation, dead tumor cells release endogenous TLR ligands, such as high-mobility group box 1 protein (HMGB1). HMGB1 activates TLR4, which activates DCs through a MyD88-dependent pathway and promotes an anti-tumor CTL response (48). The authors further demonstrated that breast cancer patients with a mutation in TLR4, rendering it unable to bind HMGB1, harbor increased risk for development of metastatic disease (48).
Other TLRs can elicit pro-inflammatory responses that are largely mediated through MyD88. MyD88 participates in signaling downstream of IL-1R and all TLRs except TLR3 (49). Stimulation of this pathway recruits MyD88 and TIRAP to the receptor and the subsequent activation of the nuclear factor κB (NF-κB) transcription factor, resulting in production of pro-inflammatory cytokines such as IL-6 and IL-12 (21). Two recent papers demonstrated that MyD88-dependent pathways are important for promotion of tumor progression in inflammation-associated carcinomas (50, 51). Karin and colleagues (51) found that MyD88-dependent upregulation of IL-6 following administration of a hepatic carcinogen and ablation of MyD88 prevented IL-6 upregulation and protected mice from tumor development. Rakoff-Nahoum and Medzhitov (50) investigated intestinal tumorigenesis and found that MyD88 was required for progression from micro- to macro-adenomas, while tumor initiation was unaffected. Signaling through TLR4 and MyD88 is also important for induction of HI responses toward human papilloma virus (HPV)-16 capsid proteins, a virus that is associated with cervical cancer in humans (52). Interestingly, TLRs not only respond to pathogen-derived substances but also to endogenous ligands often associated with tissue damage or tumor development (53). TLR4, for example, responds to extravasated fibrin, heparan sulfate, fibronectin, and hyaluronate (54); these ligands are commonly found in tumor stroma (55, 56). Taken together, tumor antigens are well known to trigger immune responses, which depending upon context and vigor, can either foster or inhibit pro-tumor immunity
Pro-tumor HI
B lymphocytes are the central component of HI and exert their effector function through antibody production, antigen presentation, and secretion of pro-inflammatory cytokines. In the context of cancer development, B cells have also been found to inhibit Th1-mediated anti-tumor immune responses (13). In experimental mouse models of lung adenocarcinoma, B-cell deficiency significantly enhanced therapeutic efficacy when treated with combinations of chemotherapy and IL-15, a Th-1 cytokine with IL-2-like anti-tumor bioactivities (57). In a syngeneic mouse xenograft model of colorectal cancer, partial B-cell depletion resulted in significantly reduced tumor burden (58). Moreover, in a mouse model of melanoma, the absence of B cells was associated with increased therapeutic efficacy of a melanoma vaccine, with enhanced tumor protection in B-cell-deficient mice characterized by increased magnitude and long-evity of specific cellular immune responses provoked by vaccination (59). In addition, B-cell-deficient mice exhibit resistance to several histologically diverse primary syngeneic tumors, associated with enhanced anti-tumor Th1 cytokines and CTL responses (60). In these studies, tumor growth was restored by adoptive transfer of B lymphocytes but not serum from wildtype mice to B-cell-deficient mice, accompanied by reduced Th1 cytokine levels and CTL response, indicating antibody-independent mechanisms mediating the response (60). Together, these experimental studies support the concept that B cells limit anti-tumor immunity through inhibition of Th1 and CTL responses while simultaneously bolstering Th2-effector cell pro-tumor functions.
Antibodies directed toward tumor-associated antigens are commonly found in cancer patients, e.g. c-myc, HER-2/neu, and p53 (61). However, production of these autoantibodies does not confer protection but paradoxically, correlates with poor prognosis and decreased survival for several human cancer types (62). Antibodies directed toward tumor antigens are thought to enhance tumor growth by promoting pro-tumor inflammatory responses and in general protecting tumor cells from CTL-mediated killing (63). In addition, extravasation of antigen-specific Ig into tumor stroma results in formation of immune complexes (ICs) that engender tumor-promoting inflammatory responses (8, 64). Ig-IC formation is indeed a common feature of cancer development. High levels of circulating ICs are associated with increased tumor burden and poor prognosis in patients with breast, genitourinary, and head and neck malignancies (65-67), and Ig deposition in neoplastic stroma has been reported in pre-malignant and malignant human breast and prostate tissues (8).
ICs have long been suspected as acting as initiators of inflammatory cascades associated with tissue destruction in autoimmune disease; however, underlying molecular mechanisms have remained elusive (68). These mechanisms have recently been investigated using an experimental mouse model of airway remodeling following Mycoplasma pulmonis infection. This study demonstrated that peripheral B-cell responses and local Ig-IC deposition are critical triggers for recruiting innate leucocytes into infected airways, that subsequently activate pro-angiogenic and tissue remodeling pathways necessary for resolving infection (69). As potentiators of inflammation associated with cancer development, B-cell activation is significant. Using a spontaneous transgenic mouse model of squamous carcinogenesis, i.e. K14-HPV16 mice (70), we reported that combined B- and T-lymphocyte deficiency eliminated IC deposition and attenuated innate immune cell infiltration into pre-malignant skin (71). As a consequence of deficient adaptive immunity and abated innate immune cell responses, there was an overall decrease in tissue remodeling activity, failure to activate angiogenic vasculature, retention of terminal differentiation programs in skin keratinocytes, and a 43% reduction in squamous cell carcinoma incidence (71). Adoptive transfer of B-lymphocytes or serum isolated from HPV16 mice (but not from wildtype naive mice) into B- and T-lymphocyte-deficient/HPV16 mice restored IC deposition, chronic innate immune cell infiltration in pre-malignant skin, and reinstated parameters for full malignancy, i.e. inflammation, tissue remodeling, angiogenesis, and keratinocyte hyperproliferation (71). These data indicate that B-lymphocyte-derived factors in serum, possibly Ig, are essential for establishing chronic inflammatory pathways that promote tumor development. In support of this concept, antibodies directed toward tumor antigens are known to enhance outgrowth and invasion of murine and human tumor-cell xenografts by recruiting and activating granulocytes and macrophages (64), important sources of vascular endothelial growth factor (72) that stimulates angiogenesis. Thus, serum proteins (presumably antibodies) produced by B cells are, at least in some scenarios, crucially involved in initiation of chronic inflammation that in addition to resolving acute tissue damage also are essential for promoting tumor development in neoplastic tissues.
Mechanisms by which ICs promote inflammation are still not fully understood; however, IgG Fc receptors (FcγRs), especially FcγRIII, and complement factors, in particular C5a, are recognized as co-dominant effectors in this process (73). Different types of FcγRs either activate or inhibit immune responses. In humans, three types of FcγRs exist: FcγRI/CD64, FcγRII/CD32, and FcγRIII/CD16. FcγRII is further subdivided into FcγRIIA, FcγRIIB, and FcγRIIC. Mice express a fourth FcγR, FcγRIV (13). Both mice and humans express a single inhibitory FcγR, FcγRIIB. Upon ligation of FcγRs with IgG that are complexed with antigen, different cellular responses are triggered depending on which receptors are engaged (13). FcγRI and FcγRIII, predominantly FcγRIII, mediate immune cell activation via their FcRγ chain that contains an immunoreceptor tyrosine-based activating motif (ITAM). ITAM-mediated activation triggers oxidative bursts, cytokine release, and phagocytosis by macrophages, antibody-dependent cell-mediated cytotoxicity by NK cells, and mast cell degranulation (68). Conversely, engagement of FcγRIIB, which instead contains immunoreceptor tyrosine-based inhibitory motifs, inhibits these same inflammatory responses (68). A further complication is that immune cells frequently co-express both activating and inhibitory receptors, and the net cellular response is determined by a balance between activating and inhibitory signals. Regulatory functions of FcγRs have been studied using genetically engineered mouse models. Mice deficient in the FcRγ chain or activating type FcγRs are resistant to a wide range of hypersensitive reactions, such as vasculitis, glomerulonephritis, and skin Arthus reaction (68). In contrast, mice deficient in FcγRIIB exhibit enhanced IC-mediated inflammatory responses (68).
Nimmerjahn and Ravetch (74, 75) utilized in vivo activities of various IgG isotypes, in combination with determining their affinity for different FcγR, as a predictor of cellular response. In this scheme, affinities of IgGs to FcγRs are determined in the context of an activation-to-inhibitory ratio (A/I ratio), calculated by dividing affinity of an IgG subclass for the relevant activating receptor (FcγRIII or FcγRIV, depending on IgG isotype) by the affinity for the inhibitory FcγRIIB (74). IgG2a subclasses demonstrate the highest A/I ratio, followed by IgG2b and IgG1 having the lowest, whereas IgG3 demonstrates no detectable binding. The functional significance of these ratios were assessed in vivo, where it was found that IgG2a, the IgG with the highest A/I ratio, enhanced clearing of lung metastases in mice when injected with tumor cells; thus, the response correlated with the ratio. In additional experiments, A/I ratios were modified by altering IgG glycosylation profiles, and once again, antibodies with higher A/I ratios exhibited enhanced in vivo efficacy - effects that were attenuated in mice deficient in FcγRs but unchanged in mice deficient in components of the complement system, demonstrating that in vivo activity of IgGs, at least in the experimental setting, depends on FcγRs and not on complement activation (74). When investigated further, it was found that glycolytic modifications through sialylation of Fc regions of IgG reduce binding affinities toward FcγRs and thereby inhibit pro-inflammatory activities of IgG, and in contrast, reduction of sialylation, upon antigen challenge, switches the immune response pathway from anti-inflammatory to pro-inflammatory via differential engagement with FcγRs on effector cells (76). Together, these studies demonstrate functional importance of FcγRs in inflammation and autoimmune diseases; their role(s) during tumor development remains to be determined.
Inhibiting anti-tumor immunity
The predominant role of the mammalian immune system is to protect the body against infectious agents and to facilitate healing after injury; thus, the dogma has been that immune cells would similarly offer protection against primary tumor development and/or metastases. In support of this are clinical data revealing that patients taking immunosuppressive drugs or suffering from various types of immune deficiency disorders exhibit increased risk for some viral- and/or carcinogen-associated cancers (8, 77), thus indicating that absence of anti-viral immunity, presumably CTLs, affects relative cancer risk. However, for tumors not commonly associated with carcinogen exposure or viral infection (e.g. prostate, breast, ovarian, and uterine cancer), relative risk for cancer development is decreased in similar cohorts of immunocompromised individuals (8). Thus, an apparent paradox exists, where both anti- and pro-tumor immune programs can be engaged depending on organ specificity and cancer etiology.
Lymphocytes and some innate immune cells possess potent anti-cancer activities that can affect growth and/or metastatic spread. Recently, CD3+ T-cell densities within colorectal cancer biopsies were found to be a better predictor of patient survival than standard histopathological staging methods (78). Similarly, infiltration of NK cells in human gastric or colorectal carcinoma is associated with a favorable prognosis (79). NK cells can play a role in protection against experimental tumor growth by directly killing tumor cells and indirectly by producing mediators with anti-angiogenic properties (80, 81).
The idea that neoplasms can be recognized and attacked by the adaptive immune system has encouraged several groups to attempt to activate adaptive immune cells to achieve anti-tumor immune responses (82). CD8+ T cells were found to be particularly important and required for anti-tumor effects in several experimental mouse models (77). Furthermore, CTLs were able to eliminate only tumor cells expressing their cognate antigen, indicating a specific immune response (82). A subset of innate immune cells may contribute to CTL suppression, namely myeloid-derived suppressor cells (MDSCs), which are CD11b+Gr1+ cells and accumulate in peripheral blood of cancer patients (83, 84) as well as in tumors and lymphoid organs (9, 84-86). MDSCs are known to induce T-lymphocyte dysfunction by direct cell-cell contact and by production of immunosuppressive mediators, e.g. arginase and TGF-β production (84, 86, 87). TGF-β also converts naive CD4+ T cells into adaptive Treg cells (14), indicating that MDSCs can inhibit anti-tumor adaptive immunity directly, as well as through polarization of local immune responses. In addition, MDSCs can promote tumor growth by contributing to tumor-associated angiogenesis (88). Together, these studies support the concept that B and T lymphocytes can contribute to inhibition of Th1 and CTL responses and thereby aid tumor development.
Conclusion
B cells are crucial in defense against extracellular bacteria and parasites. They are also known to initiate autoimmune diseases through several mechanistic pathways, including production of autoantibodies, formation of ICs, activation of DCs and T cells, as well as cytokine production (89). Cancer patients contain elevated levels of serum Igs, some with anti-tumor specificity, but the role of these antibodies have been debated. Initially, they were assumed to represent an anti-tumor response; however, their presence often instead correlates with poor prognosis. This observation in combination with experimental studies demonstrating that treatment with tumor-specific B cells promote tumor progression, while also suppressing antitumor CTL responses has fostered a shift in thinking regarding the role of B cells and HI during cancer development (13, 60, 71, 90). Are there clinical implications for these more recent points of view?
In autoimmune disease, therapeutic depletion of B cells using a chimeric monoclonal antibody specific for human CD20, i.e. rituximab, in patients with rheumatoid arthritis, systemic lupus erythematosus, and others, have demonstrated clinical efficacy (91, 92). Rituximab has also proven to be clinically effective in adult acute lymphoblastic leukemia in combination with chemotherapy (93). Could rituximab then potentially be used therapeutically for treatment of solid tumors? A limited clinical study has been performed where advanced colon cancer patients were treated with rituximab (58). In these individuals, numbers of CD21-hyperpositive lymphocytes were reduced in parallel with a 50% reduction in tumor burden with no side effects due to therapy (58). Even though rituximab effectively deletes the vast majority of circulating B cells, no increased susceptibility to infection has been reported in treated patients (91), likely due to the fact that circulating concentrations of Igs are unchanged by rituximab, because memory B cells and/or plasma cells are not targets for rituximab. The efficacy of rituximab in safely manipulating HI as a therapeutic strategy is encouraging but is as of yet untested.
Clinical data as well as experimental animal studies support the notion that a sustained humoral response has the ability to elicit significant pro-tumor effects in developing neoplasms. Established tumors represent formidable opponents that often develop resistance toward available drug options. To develop new treatment strategies, one promising approach is to combine drugs that act on neoplastic cells together with compounds that disassemble microenvironmental support programs utilized by tumors for survival and spread. In this context, a more thorough understanding of immune-based modulation of tumor development will be instrumental for development of new treatment strategies for cancer.
Acknowledgements
The authors were supported by grants from the National Institutes of Health (CA72006, CA94168, CA098075), Sandler Program in Basic Sciences, National Technology Center for Networks and Pathways (U54 RR020843) and a Department of Defense Era of Hope Scholar Award (BC051640); as well as two postdoctoral training grants (VR P2-M & ACS PF-07-264-01-LIB).
References
- 1.Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 4.Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol Mech Dis. 2006;1:119–150. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 6.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 7.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 8.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 9.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 10.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 11.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 12.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 13.Tan T, Coussens LM. Humoral immunity, inflammation and cancer. Curr Opin Immunol. 2007;19:209–216. doi: 10.1016/j.coi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Colombo MP, Piconese S. Regulatory T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer. 2007;7:880–887. doi: 10.1038/nrc2250. [DOI] [PubMed] [Google Scholar]
- 15.Dalgleish AG, O’Byrne KJ. Chronic immune activation and inflammation in the pathogenesis of AIDS and cancer. Adv Cancer Res. 2002;84:231–276. doi: 10.1016/s0065-230x(02)84008-8. [DOI] [PubMed] [Google Scholar]
- 16.Agarwal A, Verma S, Burra U, Murthy NS, Mohanty NK, Saxena S. Flow cytometric analysis of Th1 and Th2 cytokines in PBMCs as a parameter of immunological dysfunction in patients of superficial transitional cell carcinoma of bladder. Cancer Immunol Immunother. 2006;55:734–743. doi: 10.1007/s00262-005-0045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanazawa M, et al. Effects of PSK on T and dendritic cells differentiation in gastric or colorectal cancer patients. Anticancer Res. 2005;25:443–449. [PubMed] [Google Scholar]
- 18.Sheu BC, Lin RH, Lien HC, Ho HN, Hsu SM, Huang SC. Predominant Th2/Tc2 polarity of tumor-infiltrating lymphocytes in human cervical cancer. J Immunol. 2001;167:2972–2978. doi: 10.4049/jimmunol.167.5.2972. [DOI] [PubMed] [Google Scholar]
- 19.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 20.Guy B. The perfect mix: recent progress in adjuvant research. Nat Rev Microbiol. 2007;5:505–517. doi: 10.1038/nrmicro1681. [DOI] [PubMed] [Google Scholar]
- 21.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 23.Proietti E, et al. Type I IFN as a natural adjuvant for a protective immune response: lessons from the influenza vaccine model. J Immunol. 2002;169:375–383. doi: 10.4049/jimmunol.169.1.375. [DOI] [PubMed] [Google Scholar]
- 24.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Dunn GP, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6:722–729. doi: 10.1038/ni1213. [DOI] [PubMed] [Google Scholar]
- 26.Lighvani AA, et al. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci USA. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szabo SJ, Jacobson NG, Dighe AS, Gubler U, Murphy KM. Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity. 1995;2:665–675. doi: 10.1016/1074-7613(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 28.Dinarello CA. IL-18: a TH1-inducing, proinflammatory cytokine and new member of the IL-1 family. J Allergy Clin Immunol. 1999;103:11–24. doi: 10.1016/s0091-6749(99)70518-x. [DOI] [PubMed] [Google Scholar]
- 29.Yang J, Murphy TL, Ouyang W, Murphy KM. Induction of interferon-gamma production in Th1 CD4+T cells: evidence for two distinct pathways for promoter activation. Eur J Immunol. 1999;29:548–555. doi: 10.1002/(SICI)1521-4141(199902)29:02<548::AID-IMMU548>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 30.Lee NA, Gelfand EW, Lee JJ. Pulmonary T cells and eosinophils: coconspirators or independent triggers of allergic respiratory pathology? J Allergy Clin Immunol. 2001;107:945–957. doi: 10.1067/mai.2001.116002. [DOI] [PubMed] [Google Scholar]
- 31.Reiner SL. Epigenetic control in the immune response. Hum Mol Genet. 2005;14:R41–R46. doi: 10.1093/hmg/ddi115. [DOI] [PubMed] [Google Scholar]
- 32.Ouyang W, et al. Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity. 2000;12:27–37. doi: 10.1016/s1074-7613(00)80156-9. [DOI] [PubMed] [Google Scholar]
- 33.Cua DJ, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 34.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 35.Annunziato F, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langowski JL, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 37.Fehervari Z, Sakaguchi S. CD4+Tregs and immune control. J Clin Invest. 2004;114:1209–1217. doi: 10.1172/JCI23395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999;59:3128–3133. [PubMed] [Google Scholar]
- 39.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–5218. [PubMed] [Google Scholar]
- 40.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 41.Delves PJ, Roitt IM. The immune system. Second of two parts. N Engl J Med. 2000;343:108–117. doi: 10.1056/NEJM200007133430207. [DOI] [PubMed] [Google Scholar]
- 42.Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721–728. doi: 10.1007/s00262-004-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1:220–228. doi: 10.1038/35105024. [DOI] [PubMed] [Google Scholar]
- 44.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 45.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 46.Pulendran B, Ahmed R. Translating innate immunity into immunological memory: implications for vaccine development. Cell. 2006;124:849–863. doi: 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 47.Krieg AM. Development of TLR9 agonists for cancer therapy. J Clin Invest. 2007;117:1184–1194. doi: 10.1172/JCI31414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Apetoh L, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 49.Janssens S, Beyaert R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci. 2002;27:474–482. doi: 10.1016/s0968-0004(02)02145-x. [DOI] [PubMed] [Google Scholar]
- 50.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- 51.Naugler WE, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317:121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 52.Yang R, et al. B lymphocyte activation by human papillomavirus-like particles directly induces Ig class switch recombination via TLR4-MyD88. J Immunol. 2005;174:7912–7919. doi: 10.4049/jimmunol.174.12.7912. [DOI] [PubMed] [Google Scholar]
- 53.Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 54.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mignatti P, Rifkin DB. Biology and biochemistry of proteinases in tumor invasion. Physiol Rev. 1993;73:161–195. doi: 10.1152/physrev.1993.73.1.161. [DOI] [PubMed] [Google Scholar]
- 56.Hayen W, Goebeler M, Kumar S, Riessen R, Nehls V. Hyaluronan stimulates tumor cell migration by modulating the fibrin fiber architecture. J Cell Sci. 1999;112:2241–2251. doi: 10.1242/jcs.112.13.2241. [DOI] [PubMed] [Google Scholar]
- 57.Chapoval AI, Fuller JA, Kremlev SG, Kamdar SJ, Evans R. Combination chemotherapy and IL-15 administration induce permanent tumor regression in a mouse lung tumor model: NK and T cell-mediated effects antagonized by B cells. J Immunol. 1998;161:6977–6984. [PubMed] [Google Scholar]
- 58.Barbera-Guillem E, et al. B lymphocyte pathology in human colorectal cancer. Experimental and clinical therapeutic effects of partial B cell depletion. Cancer Immunol Immunother. 2000;48:541–549. doi: 10.1007/PL00006672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perricone MA, et al. Enhanced efficacy of melanoma vaccines in the absence of B lymphocytes. J Immunother. 2004;27:273–281. doi: 10.1097/00002371-200407000-00003. [DOI] [PubMed] [Google Scholar]
- 60.Shah S, et al. Increased rejection of primary tumors in mice lacking B cells: inhibition of anti-tumor CTL and TH1 cytokine responses by B cells. Int J Cancer. 2005;117:574–586. doi: 10.1002/ijc.21177. [DOI] [PubMed] [Google Scholar]
- 61.Abu-Shakra M, Buskila D, Ehrenfeld M, Conrad K, Shoenfeld Y. Cancer and autoimmunity: autoimmune and rheumatic features in patients with malignancies. Ann Rheum Dis. 2001;60:433–441. doi: 10.1136/ard.60.5.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gumus E, Erdamar S, Demirel G, Horasanli K, Kendirci M, Miroglu C. Association of positive serum anti-p53 antibodies with poor prognosis in bladder cancer patients. Int J Urol. 2004;11:1070–1077. doi: 10.1111/j.1442-2042.2004.00948.x. [DOI] [PubMed] [Google Scholar]
- 63.Schreiber H, Wu TH, Nachman J, Rowley DA. Immunological enhancement of primary tumor development and its prevention. Semin Cancer Biol. 2000;10:351–357. doi: 10.1006/scbi.2000.0331. [DOI] [PubMed] [Google Scholar]
- 64.Barbera-Guillem E, May KF, Jr, Nyhus JK, Nelson MB. Promotion of tumor invasion by cooperation of granulocytes and macrophages activated by anti-tumor antibodies. Neoplasia. 1999;1:453–460. doi: 10.1038/sj.neo.7900054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aziz M, Das TK, Rattan A. Role of circulating immune complexes in prognostic evaluation and management of genitourinary cancer patients. Indian J Cancer. 1997;34:111–120. [PubMed] [Google Scholar]
- 66.Das TK, Aziz M, Rattan A, Sherwani R. Prognostic significance of circulating immune complexes in malignant tumours of head and neck. J Indian Med Assoc. 1995;93:3–7. [PubMed] [Google Scholar]
- 67.Dass TK, Aziz M, Rattan A, Tyagi SP. Clinical utility and monitoring of breast cancer by circulating immune complexes. Indian J Pathol Microbiol. 1992;35:298–307. [PubMed] [Google Scholar]
- 68.Takai T. Fc receptors and their role in immune regulation and autoimmunity. J Clin Immunol. 2005;25:1–18. doi: 10.1007/s10875-005-0353-8. [DOI] [PubMed] [Google Scholar]
- 69.Aurora AB, et al. Immune complex-dependent remodeling of the airway vasculature in response to a chronic bacterial infection. J Immunol. 2005;175:6319–6326. doi: 10.4049/jimmunol.175.10.6319. [DOI] [PubMed] [Google Scholar]
- 70.Coussens LM, Hanahan D, Arbeit JM. Genetic predisposition and parameters of malignant progression in K14-HPV16 transgenic mice. Am J Path. 1996;149:1899–1917. [PMC free article] [PubMed] [Google Scholar]
- 71.de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7:411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 72.Barbera-Guillem E, Nyhus JK, Wolford CC, Friece CR, Sampsel JW. Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res. 2002;62:7042–7049. [PubMed] [Google Scholar]
- 73.Schmidt RE, Gessner JE. Fc receptors and their interaction with complement in autoimmunity. Immunol Lett. 2005;100:56–67. doi: 10.1016/j.imlet.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 74.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310:1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 75.Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. Curr Opin Immunol. 2007;19:239–245. doi: 10.1016/j.coi.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 76.Kaneko Y, Nimmerjahn F, Ravetch JV. Antiinflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 77.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–727. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- 78.Galon J, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 79.Coca S, et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. 1997;79:2320–2328. doi: 10.1002/(sici)1097-0142(19970615)79:12<2320::aid-cncr5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 80.Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol. 2001;13:459–463. doi: 10.1093/intimm/13.4.459. [DOI] [PubMed] [Google Scholar]
- 81.Hayakawa Y, et al. IFN-gamma-mediated inhibition of tumor angiogenesis by natural killer T-cell ligand, alpha-galactosylceramide. Blood. 2002;100:1728–1733. [PubMed] [Google Scholar]
- 82.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–675. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Almand B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6:1755–1766. [PubMed] [Google Scholar]
- 84.Serafini P, et al. Derangement of immune responses by myeloid suppressor cells. Cancer Immunol Immunother. 2004;53:64–72. doi: 10.1007/s00262-003-0443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zea AH, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005;65:3044–3048. doi: 10.1158/0008-5472.CAN-04-4505. [DOI] [PubMed] [Google Scholar]
- 86.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+myeloid cells. J Immunol. 2001;166:5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 87.Chen ML, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci USA. 2005;102:419–424. doi: 10.1073/pnas.0408197102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang L, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6:409–421. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 89.Martin F, Chan AC. B cell immunobiology in disease: evolving concepts from the clinic. Annu Rev Immunol. 2006;24:467–496. doi: 10.1146/annurev.immunol.24.021605.090517. [DOI] [PubMed] [Google Scholar]
- 90.Johansson M, Tan T, de Visser KE, Coussens LM. Immune cells as anti-cancer therapeutic targets and tools. J Cell Biochem. 2007;101:918–926. doi: 10.1002/jcb.21230. [DOI] [PubMed] [Google Scholar]
- 91.Silverman GJ. Therapeutic B cell depletion and regeneration in rheumatoid arthritis: emerging patterns and paradigms. Arthritis Rheum. 2006;54:2356–2367. doi: 10.1002/art.22020. [DOI] [PubMed] [Google Scholar]
- 92.Edwards JC, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. 2006;6:394–403. doi: 10.1038/nri1838. [DOI] [PubMed] [Google Scholar]
- 93.Gokbuget N, Hoelzer D. Novel antibody-based therapy for acute lymphoblastic leukaemia. Best Pract Res Clin Haematol. 2006;19:701–713. doi: 10.1016/j.beha.2006.06.008. [DOI] [PubMed] [Google Scholar]