

Abstract
Efforts to model and reengineer the putative binding sites of G protein-coupled receptors (GPCRs) have led to an approach to combining small molecule “classical” medicinal chemistry and gene therapy. By this approach, complementary structural changes, for example, based on novel ionic or H bonds, are made in the receptor and ligand for selective enhancement of affinity. Thus, a modified receptor (neoceptor) is designed for activation by tailor-made agonists that do not interact with the native receptor. The neoceptor is no longer activated by the native agonist, but rather acts as scaffold for docking of novel small molecules (neoligands). In theory, the approach could verify the accuracy of GPCR molecular modeling, dissection of signaling, design of small molecules to rescue disease-related mutations, and small-molecule-directed gene therapy. The neoceptor-neoligand pairing may offer spacial specificity by delivering the neoceptor to a target site and temporal specificity by administering neoligand when needed.
Introduction
The use of GPCR agonists for therapy has inherent limitations [1] from desensitization and widespread receptor distribution leading to undesired side effects. We are developing an alternate approach to benefit from GPCR activation in a more spatially and temporally selective manner than the systemic administration of agonists to the native GPCR. This approach of neoceptors [2–4] combines small molecule “classical” medicinal chemistry and gene or cell therapy. By this rational design approach, complementary structural changes are made in the receptor and ligand for selective enhancement of affinity (Figure 1). The activation of a neoceptor in a spatially-selective manner would be achieved by cell- or organ-target delivery of the gene, given the development of an appropriate delivery method.
Figure 1.

Schematic of reengineering of GPCR by mutation of a small region of the putative ligand binding site of the native receptor (A) to recognize a tailored ligand, to achieve the desired orthogonality of activation. Yellow polyhedra symbolize functional groups on the native receptor required for ligand recognition, and at least one of which is modified in the neoceptor. The pair of neoceptor and neoligand (B) is intended for therapeutics via organ-targeted delivery of a reengineered GPCR gene. (C) shows means by which selective affinity enhancement may be achieved.
Molecular modeling based on homology to the best studied GPCR, rhodopsin [5,6], has been used widely to arrive at hypotheses for ligand docking, which are ideally validated using site-directed mutagenesis [7,8]. With this knowledge and the ability to tailor-make new analogues of a native agonist, one may design a matched neoceptor and neoligand, i.e. the binding site of a given GPCR may be engineered to recognize synthetic agonist ligands that do not activate the native receptor. As opposed to de novo receptor design [9], this approach uses the native receptor as a scaffold for docking of novel molecules. This reengineered GPCR (neoceptor) ideally retains its capacity to activate a particular second messenger pathway causing beneficial effects identical to those induced by the native receptor. The uniquely-matched ligands (neoligands) are synthesized based on molecular complementarity with the neoceptor. The structure activity relationship (SAR) profile of such modified receptors need not correspond to that of the parent.
It is envisioned that the neoceptor DNA would be delivered by an appropriate organ-targeted gene or cell therapy. In the absence of the neoligand, the neoceptor would be “silent”, not subject to activation by the native agonist. The side effects normally associated with agonist therapy would not be expected, since the native receptor would not be activated by the tailored ligand. Thus, by design, the interacting pairs of receptor and ligand would have to be orthogonal with respect to the native pair.
The reengineering of enzymes, nuclear receptors, and other proteins is practiced in a variety of contexts [10–16]. Various kinases were reengineered to recognize modified ATP analogues, for example, by the creation of a “bump and hole” [10]. Microscopic complementarity of the β-adrenoceptor and its chemically modified ligands was studied [12]. GPCRs have been engineered for regulation by metal ions leading to insights into the activation mechanism [17]. Reengineered receptors have been proposed for rescue from genetic diseases [4,15].
Although not intended for therapeutic application, Conklin and colleagues have introduced RASSLs (Receptors Activated Solely by Synthetic Ligands) for mechanistic probing through conditional expression in transgenic mice [13]. RASSLs begin with a GPCR for which a synthetic high affinity agonist probe is known and then mutation reduces the affinity of the endogenous ligands with retention of affinity for the synthetic agonist. The neoceptor approach, however, is based on the rational reengineering of both the putative binding site (genetically) and the ligand. Although the term RASSL would seem to include the neoceptor approach as well, there is an important distinction. In the first reported class of RASSLs, i.e. chimeric κ-opiate receptors containing the second extracellular loop of the δ-opiate receptor, the recognition was not truly orthogonal – the synthetic ligand also activated the native receptor, indicating a reengineered receptor, but not an engineered ligand. A mutant histamine receptor that combines a 200-fold lower potency for the endogenous ligand with improved affinity of the 2-phenylhistamine class of H1 receptor agonists was identified [18]. Random mutagenesis of the A2B AR led to the identification of gain-of-function mutations [19].
This article describes the efforts using the adenosine receptors (ARs) as proof of the neoceptor concept by rationally applying insights from molecular modeling and the envisioned applications as research tools and possibly in a futuristic therapeutic modality.
Neoceptors – development
The neoceptor approach was validated for ARs, which respond to stress-elevated levels of extracellular adenosine and have diverse protective roles against ischemia and tissue damage [20,21]. Much experience has been gained to tailor ligands for the native ARs, and several selective agonists are in advanced clinical trials for inflammation, cancer, arthritis, and cardiac arrhythmias and imaging [20, 22–24]. Because of the widespread distribution of native ARs, agonist therapy has been impeded by side effects. The A2A and A3 ARs have been developed as test cases of the neoceptor approach [2–4] to address the issue of inherent nonselectivity of agonist-based therapies.
In general, hypotheses for receptor docking of nucleosides and the proposed conformational changes of GPCRs that are associated with activation have guided the design of neoligands. Modeling of the putative ligand-binding site of the A3AR receptor [8] led to the identification of a conserved site for mutagenesis, i.e. a His residue (7.43) that has been implicated in agonist recognition [8]. Modeling of A1, A2A and A3 ARs places His(7.43) within a hydrophilic ribose-binding region of the putative agonist binding site. This residue corresponds to the Lys of rhodopsin which forms the Schiff base with retinal, and in the AR has been proposed to be H-bonded to the 3′-hydroxyl group of adenosine [2]. Mutagenesis of A2A and A3 ARs indicates His(7.43) is associated with agonist binding and less important for antagonist binding.
Initially the A3AR was converted into a neoceptor that can recognize uniquely modified nucleosides that are inactive at the native ARs [2]. This concept is also dependent on the retention of the ability of the neoceptor to activate signaling pathways known to be beneficial in cardiac myocyte cultures for antiischemic protection, such as A3AR-activated phospholipase D [4].
His7.43 of the A3AR receptor was mutated to Glu. A complementary functional group was incorporated in a synthetic neoligand, i.e. 3′-amino-3′-deoxyadenosine (MRS1960, Figure 2). A novel electrostatic pair forming between the neoceptor and neoligand was intended for selective recognition of MRS1960 by the carboxylate-modified receptor [2]. The consequences of H272E mutation are: 1) Adenosine (100 μM) no longer binds to the receptor. However, the affinity of a standard agonist radioligand (125I-I-AB-MECA) is fortuitously only 2-fold decreased. Thus, an important synthetic agonist tool may still be used to characterize the mutant receptor. MRS1960 and the mutant H272E A3 AR form a suitable association to achieve a 6-fold enhancement of binding affinity. A novel electrostatic pair would account for the enhanced recognition and subsequent activation of second messenger systems by the tailored ligand.
Figure 2.

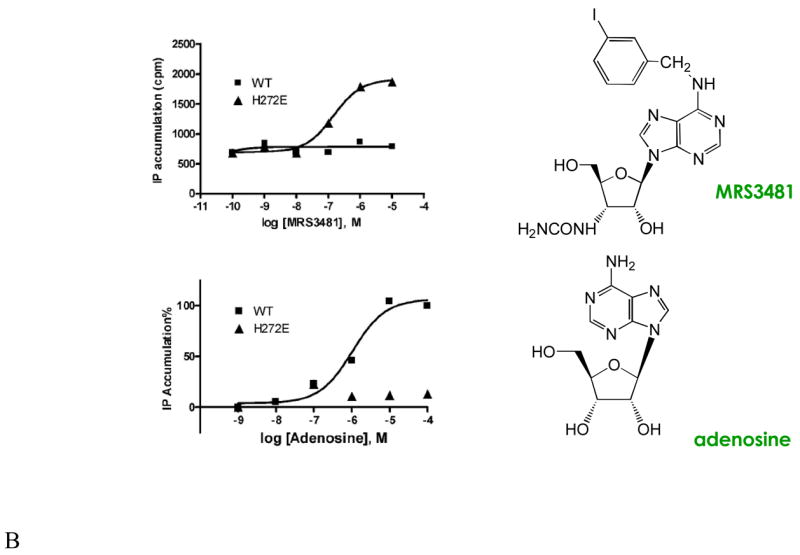
Modifications of the structure of adenosine leading to neoligands for A2A and A3ARs. Known SAR of adenosine derivatives have identified sites of modification for achieving receptor subtype selectivity and in some cases for tuning the efficacy. Cumulative efforts to characterize agonist SARs at ARs have focused on the substituent groups at 2, N6, 5′ positions of the nucleoside and the conformation of the ribose moiety [25]. (A) Amine derivatives, which showed enhancements in binding of 6- (MRS1960) and 20-fold (MRS3176) at the H272E mutant A3AR; and 10-fold (MRS3366) and >300-fold (MRS3417) at mutant A2AARs. The large N6-substituents of MRS3176 and MRS3417 serve to increase the affinity at both wild type and mutant ARs, and the extra methylene group at the ribose 3′ position was included to span a predicted gap in the A3AR docking model. (B) Comparison of the functional effects of adenosine and the 3′-ureido neoligand MRS3481 at wild type and H272E mutant A3ARs. The enhancement of binding was >200-fold.
Since the ratio of selective enhancement of MRS1960 was only modest, we improved the interaction based on prediction from molecular modeling [4,25]. A 3′-aminomethyl neoligand (MRS3176 [26]) achieved a 20-fold enhancement of affinity at the H272E mutant A3 AR. The impetus for this structural change was the prediction from molecular modeling that the 3′-amino group may be at an excessive distance from the His imidazole group to form a direct H bond. This modified nucleoside also contained a substituted N6-benzyl group, which tends to enhance affinity at the wild type and mutant A3 ARs.
Thus, ligands may be tailored with strategically-positioned amino groups for recognition by carboxylate mutants of the native receptor. However, a more dramatic demonstration of selective affinity enhancement and orthogonality was desired. The substituent at the 3′-position of ribose was varied in charge, size, and H bonding ability, and each analogue was examined for binding affinity at several neoceptor variations. The optimal affinity enhancement and orthogonality followed the incorporation of a urea group in place of the 3′-hydroxyl group of adenosine analogues, as in MRS3481 (Figure 2). The urea group is capable of forming multiple H bonds with the neoceptor and appears to preclude binding at the native ARs for steric reasons [27]. A model of this analogue docked in the H272E receptor showed a bidentate coordination of between the carboxylate group and the urea moiety (Figure 3).
Figure 3.

Docking of agonist Cl-IB-MECA in the native human A3AR (left panel) and a neoligand, MRS3481, in the H272E neoceptor (right panel). The homology models were derived from human A3AR model based on rhodopsin and resembling the meta I state [6].
It was necessary to probe the coupling pattern of the neoceptor [4]. Although the coupling specificity in GPCRs is principally a function of the second and third intracellular loops [28], and ligand specificity is governed by functionality within the upper third of the TM regions, the preservation of the typical A3AR second messengers could not be assumed. Coupling to one known effector pathway of the A3AR, i.e. stimulation of phospholipase C through the Gβ,γ-subunits, is preserved upon activation of the H272E mutant receptor by MRS3481. Thus, at least part of the downstream signaling of this cytoprotective receptor is maintained. The stimulation of phospholipase C by MRS3481 occurred with an EC50 value of ~100 nM at the neoceptor, while it was inactive at 100 μM at the WT receptor similarly expressed in COS-7 cells. For comparison, the EC50 for adenosine at the WT receptor was ~1 μM and >100,000 at the neoceptor. The effects of MRS3481 acting through the H272E neoceptor on other signaling systems (e.g., cyclic AMP, ion channels, and arrestin) remain to be determined.
In a chick cardiac myocyte culture, which is an established model for cardioprotection [29], the neoligand MRS3481 induced a potent antiischemic protection in cells expressing the H272E neoceptor [4]. Also, this protection correlated with the activation of PLD, as occurs with the native A3 AR transfected in the same cell system. Thus, a neoceptor-neoligand pair has been demonstrated to be beneficial in inducing stages of a response to stress in a tissue known to respond similarly when the native parent receptor is present.
The anti-inflammatory A2A AR was also converted into a neoceptor [3]. The same approach of mutation of the conserved His278 in TM7 could not be used, because mutation of the corresponding His residue to Asp or Glu did not lower the potency of native adenosine. However, a hydrophilic residue, i.e. T88 in TM3, on the other side of the putative subdomain for ribose binding to the ARs was selected for mutation. This residue in the A2A AR is exclusively associated with agonist binding [3]. The T88D mutant receptor was unaffected in the ability to bind the nonselective AR antagonist CGS15943, however it failed to bind the nonselective AR agonist NECA, even at a concentration of 100 μM. The T88D mutant A2A AR recognized a strategically 5′-modified amino derivative (MRS3366, Figure 2B). Moreover, the precise position and spacer length of the amino group was critical to achieving a selective affinity enhancement at the neoceptor. Other mutant A2A ARs displayed even greater degrees of enhancement for neoligands. For example, MRS3417 was functionally enhanced at the N181D mutant receptor by 110-fold, however, this combination was not truly orthogonal since this modified receptor was still capable of being activated by known AR agonists.
In addition to mutating the ligand binding pocket, it is also possible to mutate sites involved in phosphorylation and desensitization to retard these processes in neoceptors. This is particularly important with regard to A3 receptors since these are known to undergo exceptionally rapid desensitization. It should also be possible to mutate promoter regions of the neoceptor transcript. This could be used to enable induction of the neoreceptor mRNA - thus adding an additional layer of control in the response to the neoligand.
Neoceptors – potential applications
Ligand docking in rhodopsin-based molecular models of GPCRs has been controversial. Neoceptors provide a means of verifying the accuracy of predictions based on molecular modeling of GPCRs, which have been subject to discrepancies, especially for agonist docking [6]. A gain-of-function mutation and a complementary ligand provide powerful evidence that the predicted binding pocket is correct. Moreover, neoceptors can be used for mechanistic probing of the role of a specific GPCR in cells or tissues.
Therapeutic applications are also envisioned for gene therapy, dependent on site specific gene delivery, e.g. in the cardiovascular system [30] (Table 1). Novel proposed applications include donor stem cells, which hold great promise in repairing or regenerating diseased tissues such as heart or other organs. Methods to enhance the survival of exogenous stem cells when they are implanted in the recipient subjects may greatly increase their ability to achieve repair. Although the native A3 AR has a potent cytoprotective effect, its ubiquitous presence will cause significant side effects. The use of a tailor-made neoligand is proposed for selectively activating, as needed, a cytoprotective neoceptor to be expressed in the donor stem cells. Another potential therapeutic application of the orthogonal neoceptor-neoligand pair is in skeletal muscle, based on cytoprotection by adenosine acting at the A1AR [32].
Table 1.
Future Therapeutic Applications of AR-Derived Neoceptors.
| Receptor | Target tissue | Effect | Referencea |
|---|---|---|---|
| A1 | A-V node | Antiarrhythmic | 31 |
| A1 | Skeletal myocytes | Antiischemic protection | 32 |
| A1 | Kidneys | Reduced hyperfiltration | 33 |
| A1, A3 | Cardiac myocytes | Antiischemic protection | 4 |
| A1, A3 | Hematopoietic stem cells | Myeloprotection, enhanced survival | 34 |
| A2A | Neutrophils, T cells | Antiinflammatory | 35 |
| A2A | Platelets | Antithrombotic | 36 |
| A2A | Liver, bowel | Cytoprotection | 37,38 |
| A2A, A2B | Endothelial cells/Vascular smooth muscle cells | Vasodilatation, angiogenesis | 39,40 |
| A3 | Cancer cells/tumors | Cytostatic, anticancer effect | 22 |
| A3 | Synoviocytes | Antiarthritic | 23 |
| A3 | Lungs | Antiischemic protection | 24 |
The basis for these projected applications in the physiological effects of the native ARs is described in reference 25 and as indicated.
Additionally, increasing evidence suggests that mutations in genes encoding GPCRs are an important cause of human disease. The neoceptor approach and binding site modeling could be used to design small molecules to specifically rescue disease-related mutations.
Conclusions
Neoceptors represent a rational design approach for new pharmacological tools and possible therapies. By iterative steps of modeling and ligand design, one may identify and refine a neoceptor/neoligand pair. The success of the neoceptor strategy for the ARs validates the use of GPCR homology modeling, and provides a means for dissection of signaling, design of small molecules to rescue disease-related mutations, and small-molecule-directed gene therapy. The neoceptor-neoligand pairing may offer spacial specificity by delivering the neoceptor to a target site and temporal specificity by administering neoligand when needed. The success of this approach depends on orthogonality of the interaction, which is not required by the RASSL approach, to avoid undesirable, nonselective activation of the native receptor. This process may now be applied to other GPCRs, even in the absence of X-ray crystallographic structures.
Acknowledgments
We thank the NIDDK Intramural Research Program, NIH for support and Dr. Soo-Kyung Kim for helpful discussions.
Glossary
- SAR
structure activity relationships
- Ligand docking
process of computational identification of an energetically favorable binding mode of a small molecule in its receptor site
- Orthogonal
multiple systems in which individual elements interact only within a system and do not cross-react, from the Greek “ortho”, meaning “right” and “gonia”, meaning “angle”
References
- 1.Anderson SD, Brannan JD. Long-acting beta 2-adrenoceptor agonists and exercise-induced asthma: lessons to guide us in the future. Paediatr Drugs. 2004;6:161–75. doi: 10.2165/00148581-200406030-00003. [DOI] [PubMed] [Google Scholar]
- 2.Jacobson KA, et al. Neoceptor concept based on molecular complementarity in GPCRs: A mutant adenosine A3 receptor with selectively enhanced affinity for amine-modified nucleosides. J Med Chem. 2001;44:4125–4136. doi: 10.1021/jm010232o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacobson KA, et al. A neoceptor approach to unraveling microscopic interactions between the human A2A adenosine receptor and its agonists. Chem Biol. 2005;12:237–247. doi: 10.1016/j.chembiol.2004.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao ZG, et al. Orthogonal activation of the reengineered A3 adenosine receptor (neoceptor) using tailored nucleoside agonists. J Med Chem. 2006;49:2689–2702. doi: 10.1021/jm050968b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visiers I, et al. Three-dimensional representations of G protein-coupled receptor structures and mechanisms. Methods Enzymol. 2002;343:329–371. doi: 10.1016/s0076-6879(02)43145-x. [DOI] [PubMed] [Google Scholar]
- 6.Kim SK, et al. Docking studies of agonists and antagonists suggest an activation pathway of the A3 adenosine receptor. J Mol Graphics and Modelling. 2006;25:562–577. doi: 10.1016/j.jmgm.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kristiansen K. Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol Ther. 2004;103:21–80. doi: 10.1016/j.pharmthera.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 8.Kim J, et al. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J Biol Chem. 1995;270:13987–13997. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuhlmann B, et al. Design of a novel globular protein fold with atomic-level accuracy. Science. 2005;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 10.Kenski DM, et al. Chemical genetic engineering of G protein-coupled receptor kinase 2. J Biol Chem. 2005;280:35051–35061. doi: 10.1074/jbc.M507594200. [DOI] [PubMed] [Google Scholar]
- 11.Tedesco R, et al. The estrogen receptor: a structure-based approach to the design of new specific hormone-receptor combinations. Chem Biol. 2001;8:277–287. doi: 10.1016/s1074-5521(01)00006-0. [DOI] [PubMed] [Google Scholar]
- 12.Strader CD, et al. Allele-specific activation of genetically engineered receptors. J Biol Chem. 1991;266:5–8. [PubMed] [Google Scholar]
- 13.Redfern CH, et al. Conditional expression and signaling of a specifically designed Gi-coupled receptor in transgenic mice. Nat Biotechnol. 1999;17:165–169. doi: 10.1038/6165. [DOI] [PubMed] [Google Scholar]
- 14.Baker D. Proteins by design. The Scientist. 2006 July;:26–32. [Google Scholar]
- 15.Koh JT, Biggins JB. Ligand-receptor engineering and its application towards the complementation of genetic disease and target identification. Curr Top Med Chem. 2005;5:413–420. doi: 10.2174/1568026053828420. [DOI] [PubMed] [Google Scholar]
- 16.Doyle DF, et al. Engineering orthogonal ligand-receptor pairs from “near drugs”. J Am Chem Soc. 2001;123:11367–11371. doi: 10.1021/ja0164632. [DOI] [PubMed] [Google Scholar]
- 17.Elling CE, et al. Metal ion site engineering indicates a global toggle switch model for seven-transmembrane receptor activation. J Biol Chem. 2006;281:17337–17346. doi: 10.1074/jbc.M512510200. [DOI] [PubMed] [Google Scholar]
- 18.Bruysters M, et al. A Gq/11-coupled mutant histamine H1 receptor F435A activated solely by synthetic ligands (RASSL) J Biol Chem. 2005;280:34741–34746. doi: 10.1074/jbc.M504165200. [DOI] [PubMed] [Google Scholar]
- 19.Beukers MW, et al. Random mutagenesis of the human adenosine A2B receptor followed by growth selection in yeast. Identification of constitutively active and gain of function mutations. Mol Pharmacol. 2004;65:702–710. doi: 10.1124/mol.65.3.702. [DOI] [PubMed] [Google Scholar]
- 20.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 21.Linden J. Adenosine in tissue protection and tissue regeneration. Mol Pharmacol. 2005;67:1385–1387. doi: 10.1124/mol.105.011783. [DOI] [PubMed] [Google Scholar]
- 22.Madi L, et al. The A3 adenosine receptor is highly expressed in tumor versus normal cells: potential target for tumor growth inhibition. Clin Cancer Res. 2004;10:4472–9. doi: 10.1158/1078-0432.CCR-03-0651. [DOI] [PubMed] [Google Scholar]
- 23.Fishman P, et al. The PI3K–NF- B signal transduction pathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Research & Therapy. 2006;8:R33. doi: 10.1186/ar1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rivo J, et al. Activation of A3 adenosine receptors attenuates lung injury after in vivo reperfusion. Anesthesiology. 2004;101:1153–1159. doi: 10.1097/00000542-200411000-00015. [DOI] [PubMed] [Google Scholar]
- 25.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Rev Drug Disc. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Rompaey P, et al. Exploring human adenosine A3 receptor complementarity and activity for adenosine analogues modified in the ribose and purine moiety. Bioorg Med Chem. 2005;13:973–983. doi: 10.1016/j.bmc.2004.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeong LS, et al. Design and synthesis of 3′-ureidoadenosine-5′-uronamides: Effects of the 3′-ureido group on binding to the A3 adenosine receptor. Bioorg Med Chem Lett. 2004;14:4851–4854. doi: 10.1016/j.bmcl.2004.07.042. [DOI] [PubMed] [Google Scholar]
- 28.Swaminath G, et al. Probing the β2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. J Biol Chem. 2005;280:22165–22171. doi: 10.1074/jbc.M502352200. [DOI] [PubMed] [Google Scholar]
- 29.Liang BT. Direct preconditioning of cardiac ventricular myocytes via adenosine A1 receptor and KATP channel. Am J Physiol. 1996;271:H1769–H1777. doi: 10.1152/ajpheart.1996.271.5.H1769. [DOI] [PubMed] [Google Scholar]
- 30.Fishbein I, et al. Site specific gene delivery in the cardiovascular system. J Control Release. 2005;109:37–48. doi: 10.1016/j.jconrel.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 31.Belardinelli L, et al. Ionic basis of the electrophysiological actions of adenosine on cardiomyocytes. FASEB J. 1995;9:359–365. doi: 10.1096/fasebj.9.5.7896004. [DOI] [PubMed] [Google Scholar]
- 32.Zheng J, et al. Adenosine A1 receptors mediate potent anti-ischemic skeletal muscle protection in a mouse hindlimb model. J Mol Cell Cardiol. 2005;38:64. [Google Scholar]
- 33.Lee HT, et al. A1 adenosine receptor knockout mice exhibit increased renal injury following ischemia and reperfusion. Am J Physiol Renal Physiol. 2004;286:F298–F306. doi: 10.1152/ajprenal.00185.2003. [DOI] [PubMed] [Google Scholar]
- 34.Budak-Alpdogan T, et al. Hematopoietic stem cell gene therapy with drug resistance genes: an update. Cancer Gene Ther. 2005;12:849–863. doi: 10.1038/sj.cgt.7700866. [DOI] [PubMed] [Google Scholar]
- 35.Lappas CM, et al. Adenosine A2A agonists in development for the treatment of inflammation. Expert Opin Investig Drugs. 2005;14:797–806. doi: 10.1517/13543784.14.7.797. [DOI] [PubMed] [Google Scholar]
- 36.Camaioni E, et al. Adenosine receptor agonists: synthesis and biological evaluation of the diastereoisomers of 2-(3-hydroxy-3-phenyl-1-propyn-1-yl)NECA. Bioorg Med Chem. 1997;5:2267–2275. doi: 10.1016/s0968-0896(97)00172-7. [DOI] [PubMed] [Google Scholar]
- 37.Day YJ, et al. Protection from ischemic liver injury by activation of A2A adenosine receptors during reperfusion: inhibition of chemokine induction. Am J Physiol Gastrointest Liver Physiol. 2004;286:G285–G293. doi: 10.1152/ajpgi.00348.2003. [DOI] [PubMed] [Google Scholar]
- 38.Odashima M, et al. Activation of A2A adenosine receptor attenuates intestinal inflammation in animal models of inflammatory bowel disease. Gastroenterology. 2005;129:26–33. doi: 10.1053/j.gastro.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 39.Feoktistov I, et al. Hypoxia modulates adenosine receptors in human endothelial and smooth muscle cells toward an A2B angiogenic phenotype. Hypertension. 2004;44:649–654. doi: 10.1161/01.HYP.0000144800.21037.a5. [DOI] [PubMed] [Google Scholar]
- 40.Yang D, et al. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest. 2006;116:1913–1923. doi: 10.1172/JCI27933. [DOI] [PMC free article] [PubMed] [Google Scholar]