

Abstract
A series of 2-imido substituted furans containing tethered unsaturation were prepared by the addition of the lithium carbamate of furan-2-yl carbamic acid tert-butyl ester to a solution of the mixed anhydride of an appropriately substituted 3-butenoic acid. The initially formed imido furans undergo a rapid intramolecular [4+2]-cycloaddition at room temperature to deliver the Diels-Alder cycloadducts in good to excellent yield. Isolation of the highly labile oxabicyclic adduct is believed to be a consequence of the lower reaction temperatures employed as well as the presence of the extra carbonyl group, which diminishes the basicity of the nitrogen atom thereby retarding the ring cleavage/rearrangement reaction generally encountered with related systems. By using a Rh(I)-catalyzed ring opening of the oxabicyclic adduct with various nucleophilic reagents, it was possible to prepare highly functionalized hexahydro-1 H-indol-2(3H)-one derivatives in good yield. The major stereoisomer obtained possesses a cis-relationship between the nucleophile and hydroxyl group in the ring-opened product. The stereochemistry was unequivocally established by X-ray crystallographic analysis. Coordination of Rh(I) to the alkenyl π-bond followed by a nitrogen-assisted cleavage of the carbon-oxygen bond occurs to furnish a π-allyl rhodium(III) species. Addition of the nucleophile then occurs from the least hindered terminus of the resulting π-allyl rhodium(III) complex. Proton exchange followed by rhodium(I) decomplexation ultimately leads to the cis-diastereomer.
Introduction
7-Oxabicyclo[2.2.1]heptenes are valuable intermediates in organic synthesis.1,2 The large number of selective transformations possible with the oxabicyclic system endow this nucleus with impressive versatility.3–9 A crucial synthetic transformation employing these intermediates involves the cleavage of the oxygen bridge to produce functionalized cyclohexene derivatives. Many groups have developed different approaches including β-elimination of suitable derivatives,10 treatment with strong acids,11 reductive elimination of endo functionalities,12 fragmentation,13 hydrolytic ring openings,14 and alkylative bridge cleavage reactions.15 A significant advancement in the ring-opening chemistry of oxabicyclic compounds occurred in the early 1990’s as a result of the elegant work of Lautens and co-workers.16 This research team demonstrated that the ring opening of unsymmetrical oxabicycloheptenes 1 is a highly regioselective process giving rise to products 2 derived from attack of the nucleophile distal to the bridgehead substituent.16 These reactions were carried out with a range of nucleophiles including hydride, stabilized and nonstabilized carbanions, alcohols, amines and carboxylates.1 Highly stereoselective reactions were also observed to occur with organocuprates, silylcuprates, organolithium reagents and organomagnesium compounds1c and these processes were used for the synthesis of acyclic (and cyclic compounds) 3 with multiple stereocenters (Scheme 1).1
Scheme 1.
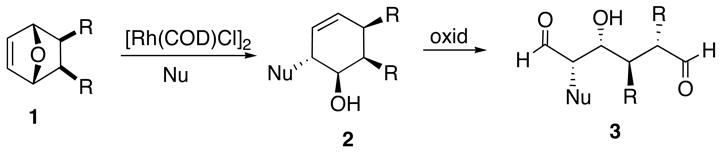
Lautens’ Oxabicyclic Ring Opening Protocol
The most general method for the synthesis of 7-oxabicyclo[2.2.1]heptenes relies on the Diels-Alder reaction of a furan derivative with various dienophiles.17 Furan itself is not very reactive as a diene due to the loss of aromaticity which accompanies the cycloaddition.18 Among the solutions investigated to improve the [4+2]-cycloaddition reaction are catalysis using Lewis acids,2c use of metal salts,19 silica gel,20 zeolites,21 centrifugation,22 ultrasound23 and high-pressure techniques.24 The placement of an electron donor substituent on the furan ring also significantly enhances its reactivity toward dienophiles by raising its HOMO level. The intramolecular version of the Diels-Alder reaction of furans (IMDAF) has been described in several reviews,17,25 including an excellent treatise by Keay and Hunt.26 For the IMDAF reaction to proceed favorably, the aromatic character of the furan ring and the strain associated with the oxabicyclic adduct must be overcome. Steric factors, rather than electronic or solvent effects, appear to have the greatest influence on the outcome of the [4+2]-cycloaddition.27 Electronically disfavored furan cycloadditions can be brought about by creative functional group modifications.
Several years ago we began a synthetic program to provide general access to a variety of alkaloids by [4+2]-cycloaddition chemistry of substituted 2-amidofurans.28 Our synthetic strategy was to take advantage of an intramolecular Diels-Alder reaction of an alkenyl-substituted furanyl carbamate derivative (IMDAF).29,30 Not only does this IMDAF reaction allow for the preparation of aza-polycyclic ring compounds, but they proceed at lower temperatures than their intermolecular counterparts. Even more significantly, unactivated π-bonds are reactive toward the internal cycloaddition reaction. We discovered that the IMDAF reaction of a series of furanyl carbamates (i.e., 4) occurred smoothly to furnish the cyclized indoline 6 as the only isolable product in high yield when a monosubstituted alkenyl tether (R=H) was used (Scheme 2).28 When the alkenyl group possesses a substituent other than hydrogen at the 2-position of the π-bond, the thermal reaction furnished a rearranged hexahydroindolinone (i.e., 8). With this system, the initially formed cycloadduct 5 cannot aromatize. Instead, ring opening of the oxabicyclic intermediate occurs to generate zwitterion 7, which undergoes a subsequent proton elimination by tautomerization to give the rearranged ketone 8.30
Scheme 2.
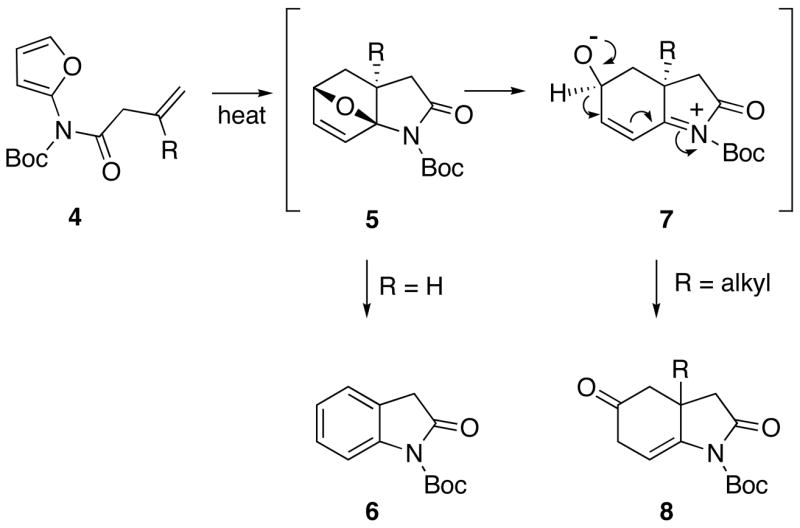
IMDAF Reaction of Furanyl Carbamates
When furanyl carbamates such as 4 were used, the IMDAF reaction required heating at 165 °C for several hours in order to produce the rearranged product (i.e., 6 or 8). The Diels-Alder adduct 5 that was first formed underwent reorganization under the reaction conditions and could not be isolated nor detected in the crude reaction mixture. During the course of investigating the general nature of this reaction, significant rate differences were noticed when the alkenyl tether was incorporated onto the amido side chain. With these systems, the IMDAF cycloaddition occurred at room temperature and in some cases31 it was possible to isolate the initially formed aza-oxabicyclo adduct. For example, the intramolecular [4+2]-cycloaddition of amidofuran 9 afforded cycloadduct 10 in 77% yield when the reaction was carried out at 25 °C (Scheme 3). Further heating of a benzene solution of this cycloadduct at reflux provided the rearranged tricyclic lactam 11 in 75% yield. The notion of further functionalizing an aza-oxabicyclo adduct by a transition metal-catalyzed ring-opening reaction with various nucleophilic reagents was most attractive to us since the resulting product(s) can be envisaged as precursors to a wide assortment of alkaloids. In this paper we detail our recent findings using a series of alkenyl substituted imido-furans where we address the issues of (a) ease of access to the starting aza-oxabicyclo adducts, (b) the efficiency of the IMDAF reaction and (c) regio and stereocontrol in the metal-catalyzed ring opening reaction.
Scheme 3.
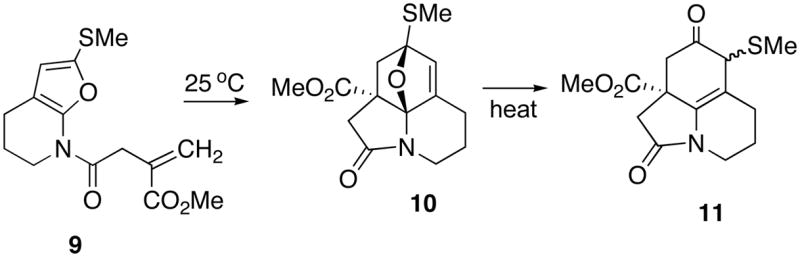
Isolation of the Aza-oxabicyclic Cycloadduct
Results and Discussion
The aza-oxabicyclo[2.2.1] substrates employed in this study were readily prepared from the corresponding imidofurans. Initially, we had envisioned imidofuran 13 arising from the simple acylation of furanyl carbamate 12a with the acid chloride derived from 3-butenoic acid. However, under a variety of conditions, 12a proved to be remarkably resistant toward acylation. After some experimentation, we found that the addition of lithiated carbamate 12b, formed by the action of n-BuLi on 12a, to a solution of the mixed anhydride 14 provided the expected imidofuran 13, which rapidly reacted at room temperature to deliver the Diels-Alder cycloadduct 23 in 55% isolated yield. Intrigued by the ease with which imidofuran 13 underwent the IMDAF cycloaddition, we investigated several other systems containing related tethers as illustrated in Scheme 4. The mixed anhydrides 15–16 were subjected to the acylation protocol and provided the desired imidofurans 19–20 which also underwent cycloaddition at 25 °C (12 h) to furnish the aza-oxabridged cycloadducts 24–25. When the more highly activated anhydride 17 was used, imidofuran 21 could not be observed because the [4+2]-cycloaddition occurred too rapidly to preclude its detection, even at 0 °C. In contrast, the more sterically congested anhydride 18 furnished imidofuran 22, which required heating at 90 °C to give cycloadduct 27.
Scheme 4.
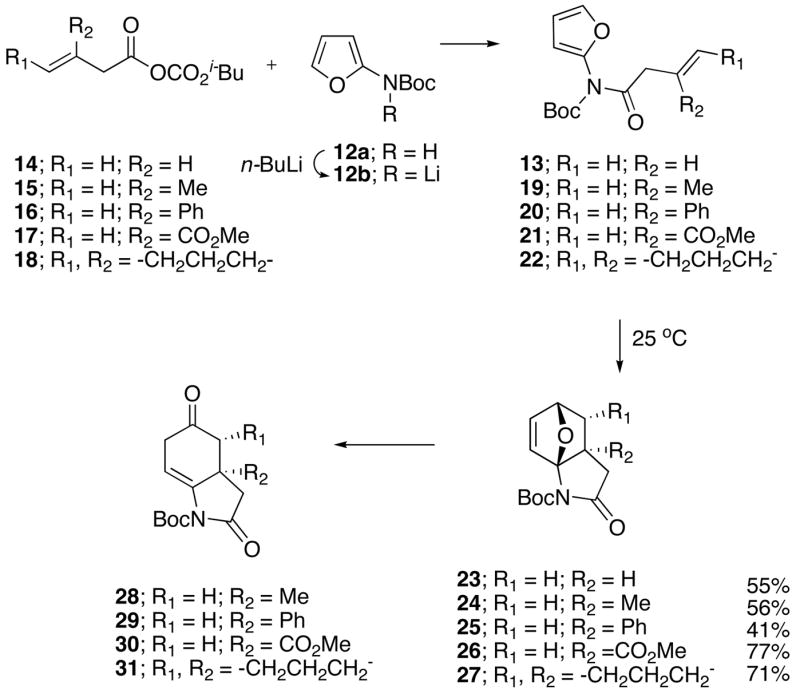
IMDAF Cycloaddition of the 2-Imidofuran System
Several more heavily substituted imidofuran substrates were also found to rapidly undergo the intramolecular [4+2]-cycloaddition at room temperature and gave aza-oxabicyclo adducts in good yield. Thus, the reaction of the mixed anhydride of vinylacetic acid with lithiated carbamates 32 and 33 provided the expected imidofurans as transient species which underwent a subsequent IMDAF reaction at 25 °C to deliver cycloadducts 34 and 35 in 60% yield, respectively (Scheme 5). Interestingly, the IMDAF cycloaddition of the somewhat labile imidofurans 36 and 37 (prepared in a similar manner) occurred with very high diastereoselectivity (> 20:1) producing the cis-substituted cycloadducts 38 and 39 in 96% and 98% yield, respectively.
Scheme 5.
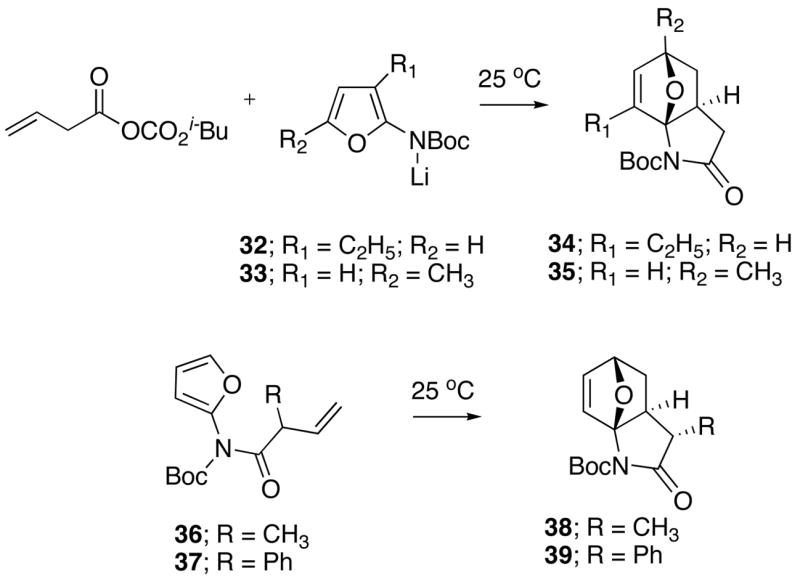
More Heavily Substituted Systems
The increase in reactivity of these 2-imido-substituted furans (0–90 °C) when compared to the related furanyl carbamates32 (> 150 °C) is clearly related to the placement of the carbonyl center within the dienophilic tether. Dramatic effects on the rate of the Diels-Alder reaction were previously noted to occur when an amido group was used to anchor the diene and dienophile.33 The effect of amide-linked tethers on both the rate and diastereoselectivity of intramolecular Diels-Alder reactions were previously attributed to relative transition state stabilities.33 The rate enhancement observed with the imidofuranyl system probably originates from restricted rotation about the C-N bond, producing a lower energy ground state conformer that is more energetically similar to the reactive conformer.34 Our ability to isolate the highly labile aza-oxabicyclic adducts is presumably a result of the lower reaction temperatures employed as well as the presence of the extra carbonyl group, which diminishes the basicity of the nitrogen atom thereby retarding the ring cleavage/rearrangement reaction generally encountered with these systems.32 When exposed to more forcing conditions (i.e., > 100 °C), the oxabridged cycloadducts 24–27 were smoothly transformed (> 90%) into the corresponding hexahydroindolinone systems 28–31. It should also be noted that the IMDAF reaction of these systems proceed by a transition state where the side arm of the tethered alkenyl group is oriented exo with respect to the oxygen bridge. Products resulting from an endo side arm transition state were neither detected nor isolated. This result is quite reasonable since, in these mobile cycloaddition equilibria, the exo adducts are thermodynamically more stable. In fact, the stereochemical results that we have encountered with the IMDAF cycloaddition of these 2-imidofurans is consistent with those reported by others for related systems possessing short tethers.35 As a consequence of this preferred exo orientation, the oxido bridge is located anti to the substituent on the bridgehead carbon. The cis-diastereoselectivity observed in the cycloaddition of imidofurans 36 and 37 can be rationalized as being the result of approach of the furan ring from the less hindered π-face of the double bond.
Regioselective cleavage of the oxygen bridge of the 7-oxabicyclo-[2.2.1]hept-2-ene skeleton to give functionalized cyclohexene derivatives is central to many synthetic strategies.36 Remote electronic effects mediated by an aromatic π-system have been thoroughly investigated for the transformation of substituted oxabenzonorbornadienes to the corresponding naphthols under protic conditions.37 The ring opening reaction of 7-oxanorbornenes with organometallic reagents has also been investigated in some detail by several research groups.38 Lautens and co-workers demonstrated that a variety of oxabicyclo[2.2.1]heptene derivatives readily undergo Rh(I)-catalyzed nucleophilic ring-opening1 and this key reaction was employed for the synthesis of highly functionalized acyclic polypropionate and polyacetate chains using the related oxabicyclo[3.2.1] system.39 Our research plan was to explore the reactivity of the IMDAF-derived aza-oxabicyclo adducts toward several nucleophilic partners with the thought of eventually applying the resulting methodology toward the synthesis of several alkaloid skeletons.
With a representative selection of 10-oxa-2-azatricyclo[5.2.1.01,5]decenes on hand, a standard set of ring-opening conditions was employed. Our first set of experiments were carried out with cycloadducts 23 and 24 using Lauten’s conditions16 ([Rh(COD)Cl]2, DPPF). Phenol and N-methylaniline were employed as the nucleophilic reagents. This led to the ring opened alcohols 40–43 in good yield (Scheme 6) and with cis/trans ratios (Nu to OH) of 5:1 for 40 & 41 and 20:1 for 42 & 43. An X-ray crystal structure of the major diastereomer of 41 unequivocally established the cis relationship between the nucleophile and hydroxyl groups in the ring opened product. Interestingly, the stereochemical outcome of this reaction was exactly opposite to that reported by Lautens for the Rh(I)-catalyzed alcoholysis and aminolysis of oxabenzonorbornadiene.16 When oxabicyclo 24 was subjected to the Rh(I)-catalyzed conditions using 2-bromobenzoic acid and NEt3, a 3:1-mixture of the ring opened product 44 was obtained in 72% yield (Scheme 7). The cis-stereochemical assignment of the major diastereomer 44a was made on the basis of analogy with substrate 41 where X-ray data had been obtained. Subsequent experiments revealed that the reaction of the related oxabicyclo adduct 23 with the Rh(I)-catalyst in the presence of various ammonium carboxylates16c generated the dienyl alcohol 45 in 80% isolated yield as the exclusive product. In the absence of the Rh(I)-catalyst, only starting material was recovered. On the other hand, when the Rh(I)-catalyzed reaction was carried out in the absence of ammonium carboxylate, oxindole 46 was formed in 90% yield.
Scheme 6.
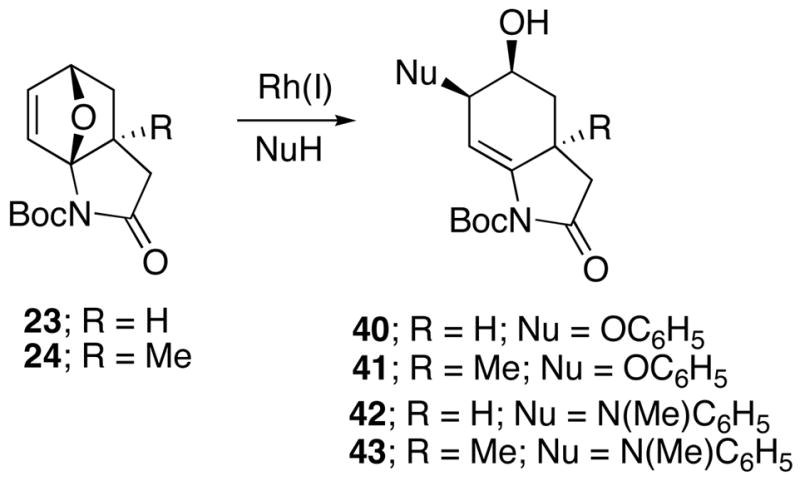
Rh(I) Catalyzed Ring Opening of the 10-Oxa-2-azatricyclodecene System
Scheme 7.
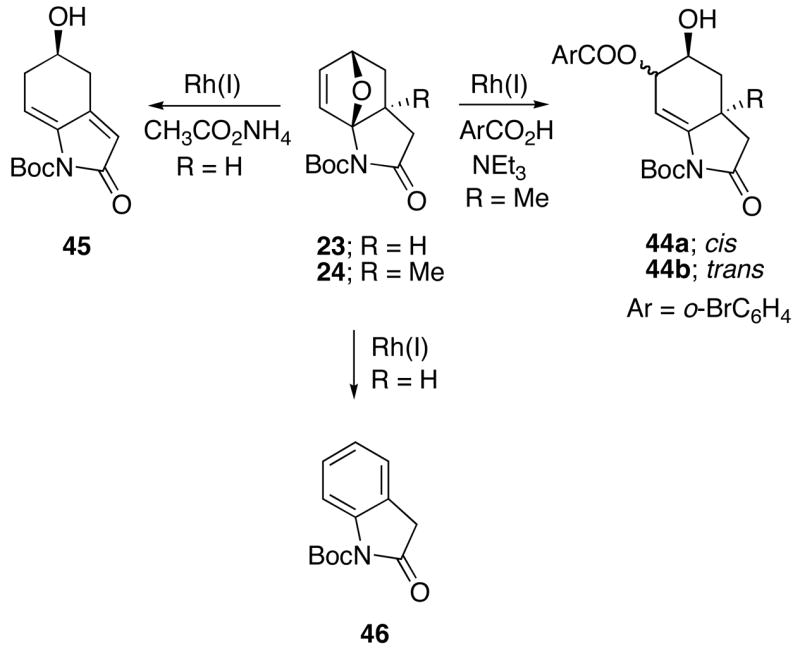
Rh(I)-Catalyzed Ring Opening Reactions Using Carboxylates as the Nucleophile
A reasonable mechanism to account for the Rh(I)-catalyzed reaction of the oxabicyclic adduct is outlined in Scheme 8. Coordination of Rh(I) to the alkenyl π-bond followed by nitrogen-assisted cleavage of the carbon-oxygen bond occurs to furnish the π-allyl rhodium(III) species 47. Nucleophilic addition then occurs from the least hindered terminus of 47 and on the side opposite the rhodium complex.40 Proton exchange of intermediate 48 followed by rhodium decomplexation ultimately leads to the cis diastereromers 40–43. In the presence of the basic ammonium carboxylate, intermediate 47 (R=H) undergoes preferential deprotonation and subsequent loss of Rh(I) to generate a transient diene 49 which then isomerizes to give 45. Without a base to induce double bond isomerization, intermediate 49 undergoes an aromatization reaction to furnish oxindole 46.
Scheme 8.
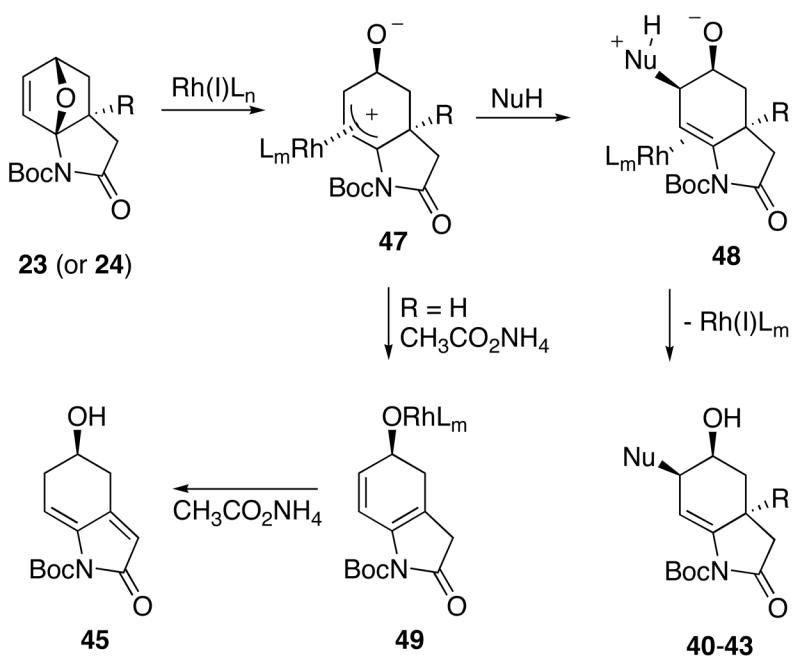
Proposed Mechanism for the Rh(I)-Catalyzed Ring-Opening Reactions
In recent years, the Rh(I)-catalyzed addition of arylboronic acids to olefins has become an active research area in organic synthesis.41 Conjugate addition generally occurs with electron deficient olefins such as enone,42 alkenylphosphonates43 and nitroalkenes.44 The facile addition of boronic acids to oxabenzonorbornenes has also been achieved using a catalytic amount of a rhodium(I) complex.45 A common step in these reactions is the carborhodation of the carbon-carbon double bond followed by hydrolysis of the organorhodium intermediate. These earlier findings prompted us to examine whether a related transformation might occur upon treating the IMDAF-derived cycloadducts 23/24 with organoboronic acids in the presence of a Rh(I)-catalyst. With this in mind, the reaction of 5,5-dimethyl-2-phenyl-[1,3,2]-dioxaborinane (50) with oxabicyclics 23 and 24, using 5 mol% of the Rh(I) catalyst and 2.0 equiv of Cs2CO3 (5 M in H2O) in THF at 65 °C, led to the ring-opened alcohols 51 and 52 in 70–80% yield (Scheme 9). The cis isomer was formed exclusively (X-ray structure of 51 was obtained) and parallels the results observed with the alcoholysis and aminolysis experiments. When the Rh(I)-catalyzed reaction was carried out with phenylboronic acid and without added base, the ring opened boronates 53 and 54 were obtained in excellent yield (>95%). The cis-stereochemistry of 53 was unequivocally established by an X-ray crystal structure. Both boronates were cleaved to the corresponding diols46 which were subsequently transformed into dioxolanes 55 and 56 by reaction with 2,2-dimethoxypropane. It was also possible to prepare the same 1,3-dioxolanes (>90%) by treating oxabicyclic adducts 23 and 24 with catalytic anhydrous SnCl2 in acetone.47
Scheme 9.
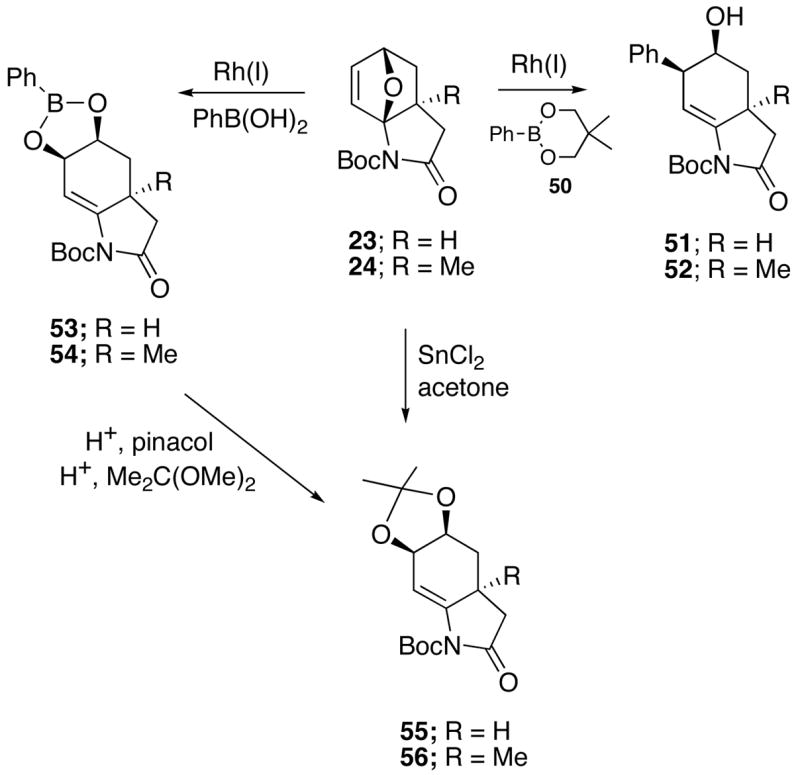
Rh(I)-Catalyzed Addition of Arylboronic Acids to the IMDAF Cycloadducts
The following mechanistic scheme is suggested to account for the formation of the ring opened alcohol 51 (or 52). Initially, phenylrhodium(I) 57 is generated by transmetallation of a rhodium(I) chloride or hydroxide with dioxaborinane 50. This process might well be promoted by the presence of a base. The resulting rhodium species 57 then undergoes exo-selective carborhodation at the oxabicycle olefin to generate intermediate 58. Chelation of the rhodium metal with the olefin and oxygen atom of the oxabicycle probably contributes to the high exo-selectivity. β-Oxygen elimination, perhaps assisted by the nitrogen lone pair, furnishes the ring opened intermediate 59 which undergoes eventual protonolysis with water to produce the final product (i.e., 51) (Scheme 10). The rhodium(I) species that is regenerated in this step is available to promote the next catalytic cycle.
Scheme 10.
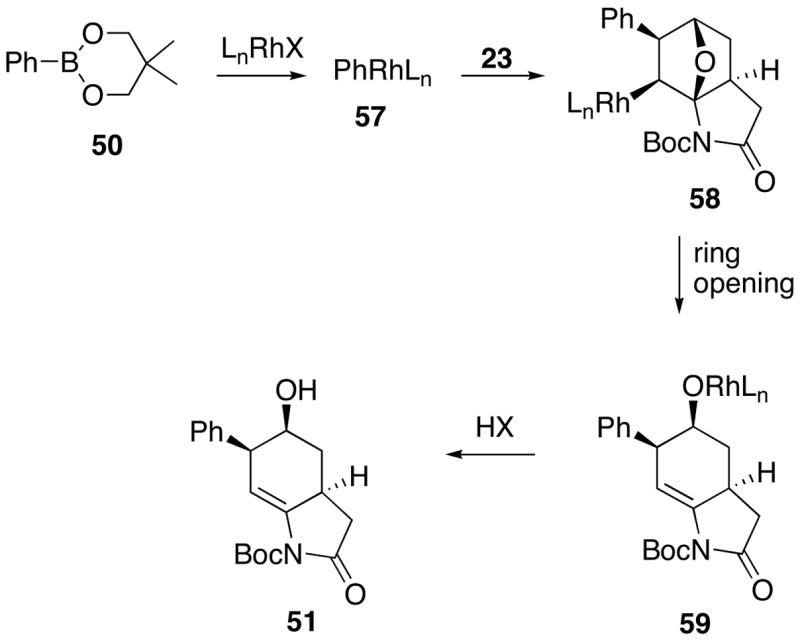
Rh(I)-Catalyzed Reaction of the Oxabicyclic System Using 5,5-Dimethyl-2-phenyl-[1,3,2]-dioxaborinane (50)
When phenylboronic acid is used without added base, the oxabicyclic adduct 23 seemingly prefers to undergo a Rh(I) catalyzed oxygen elimination reaction as the first step, perhaps as a consequence of the higher Lewis acidity of PhB(OH)2 vs dioxaborinane 50, and gives the ring-opened intermediate 60.
This intermediate then undergoes reaction with the boronic acid from the side opposite the rhodium metal to produce 61 which eventually affords 53 (or 54) by cyclization and liberation of rhodium(I) hydroxide (Scheme 11).
Scheme 11.
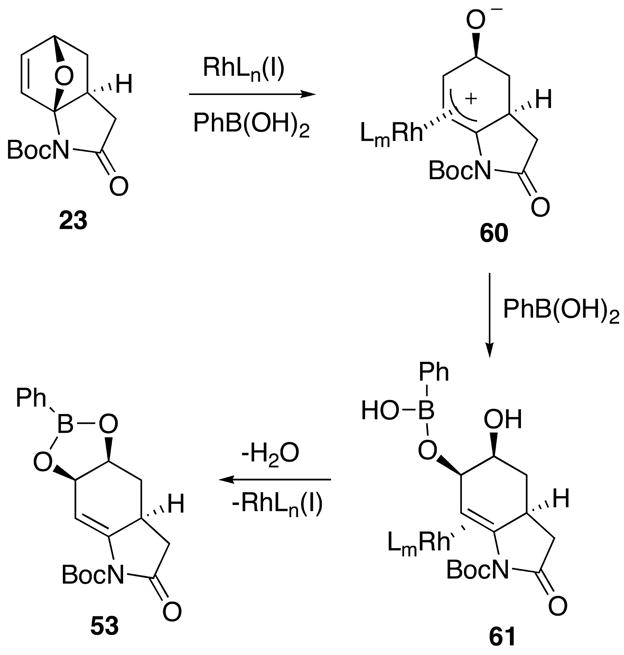
Rh(I) Catalyzed Reaction using PhB(OH)2
In summary, a highly convergent synthesis of hexahydro-1H-indol-2-(3H)-one derivatives has been devised using an IMDAF cycloaddition reaction of 2-imido substituted furans. The increase in reactivity of these imidofurans when compared to the related furanyl carbamates is related to the placement of the carbonyl center within the dienophilic tether. Isolation of the highly labile aza-oxabicyclo adducts is a result of the lower reaction temperatures used (ca 25 °C) as well as the presence of the extra carbonyl group which diminishes the basicity of the nitrogen atom thereby retarding the thermal cleavage/rearrangement reaction generally encountered with these systems. The Rh(I)-catalyzed ring-opening reaction of the IMDAF cycloadducts were investigated using a variety of nucleophiles. The catalyzed reactions proceed in high yield under very mild conditions and occur with excellent diasteroselectivity. Application of this methodology to various alkaloid skeletons is currently under investigation, the results of which will be disclosed in due course.
Experimental Section
tert-Butyl 3-oxo-10-oxa-2-azatricyclo[5.2.1.01,5]dec-8-ene-2-carboxylate (23)
To a solution of 0.2 g (1.2 mmol) of tert-butyl furan-2-yl-carbamate (12a) in 4 mL of THF at 0 °C was added dropwise 0.8 mL (1.3 mmol) of n-BuLi (1.5 M in hexane). The reaction mixture was stirred at 0 °C for 20 min. In a separate flask, 0.1 mL (1.2 mmol) of 3-butenoic acid was dissolved in 5 mL of THF at 0 °C and 0.13 mL (1.2 mmol) of 4-methylmorpholine, and 0.15 mL (1.2 mmol) of isobutyl chloroformate were added dropwise. After stirring for 5 min, the white precipitate was removed via filtration and washed with 2 mL of THF. The filtrate was cooled to 0 °C and the preformed lithiate was added dropwise via syringe to the above solution. After stirring at 0 °C for an additional 5 min, the reaction mixture was quenched with H2O and extracted with EtOAc. The organic layer was washed with a saturated aqueous NaHCO3 solution, dried over MgSO4, and concentrated under reduced pressure. The residue was subjected to flash silica gel chromatography to afford a colorless oil which underwent spontaneous cycloaddition while standing at room temperature for 12 h. The crude yellow solid was subjected to flash silica gel chromatography to provide 0.09 g (55%) of 23 as a white solid: mp 126–127 °C; IR (neat) 1792, 1767, 1726, 1357, 1296, and 1157 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.52 (s, 9H), 1.65 (dd, 1 H, J= 11.6 and 7.6 Hz), 1.80 (dt, 1 H, J= 11.6 and 4.4 Hz), 2.05–2.13 (m, 1 H), 2.41 (dd, 1 H, J= 17.2 and 10.0 Hz), 2.75 (dd, 1H, J= 17.2 and 8.8 Hz), 5.02 (dd, 1H, J= 4.4 and 2.0 Hz), 6.30 (dd, 1H, J= 6.0 and 2.0 Hz), and 6.52 (d, 1H, J= 6.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 28.2, 32.6, 35.9, 38.4, 77.6, 84.1, 102.0, 133.6, 134.7, 149.4, and 174.6; Anal. Calcd. for C13H17NO4: C, 62.14; H, 6.82; N, 5.57. Found: C, 62.01; H, 6.80; N, 5.52.
tert-Butyl 5-methyl-3-oxo-10-oxa-2-azatricyclo[5.2.1.01,5]dec-8-ene-2-carboxylate (24)
To a solution of 0.6 g (3.2 mmol) of carbamate 12a in 10 mL of THF at 0 °C was added dropwise 1.6 mL (3.4 mmol) of n-BuLi (2.1 M in hexane). The reaction mixture was stirred at 0 °C for 20 min. In a separate flask, 0.35 g (3.5 mmol) of 3-methylbut-3-enoic acid48 was dissolved in 10 mL of THF at 0 °C and 0.38 mL (3.5 mmol) of 4-methylmorpholine and 0.45 mL (3.5 mmol) of isobutyl chloroformate were added dropwise to the above solution. After stirring for 5 min, the white precipitate was removed via filtration and washed with 4 mL of THF. The filtrate was cooled to 0 °C and the preformed lithiate was added dropwise via syringe. After stirring at 0 °C for an additional 5 min, the reaction was quenched with H2O and extracted with EtOAc. The organic layer was washed with a saturated aqueous NaHCO3 solution, dried over MgSO4, and concentrated under reduced pressure. The residue was subjected to flash silica gel chromatography to afford a clear oil which underwent spontaneous cycloaddition while standing at room temperature for 12 h. The crude yellow oil was subjected to flash silica gel chromatography to provide 0.5 g (56%) of 24 as a white solid: mp 96–98 °C; IR (neat) 1793, 1767, 1730, 1353, 1306, and 1157 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 0.97 (s, 3H), 1.25 (d, 1 H, J= 11.6 Hz), 1.50 (s, 9H), 2.13 (dd, 1H, J= 11.6 and 4.4 Hz), 2.39 (d, 1H, J= 16.8 Hz), 2.67 (d, 1H, J= 16.8 Hz), 4.93 (dd, 1 H, J= 4.4 and 2.0 Hz), 6.32 (dd, 1 H, J= 6.0 and 2.0 Hz), and 6.48 (d, 1H, J= 6.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 24.3, 28.1, 39.9, 41.8, 46.5, 77.1, 84.0, 104.0, 132.8, 133.3, 149.7, and 174.5; Anal. Calcd. for C14H19NO4: C, 63.38; H, 7.22; N, 5.28. Found: C, 63.33; H, 7.13; N, 5.24.
tert-Butyl 3-oxo-5-phenyl-10-oxa-2-azatricyclo[5.2.1.01,5]dec-8-ene-2-carboxylate (25)
To a solution of 0.41 g (2.2 mmol) of carbamate 12a in 8 mL of THF at 0 °C was added dropwise 1.7 mL (2.5 mmol) of n-BuLi (1.5 M in hexane). The reaction mixture was stirred at 0°C for 20 min. In a separate flask, 0.40 g (2.5 mmol) of 3-phenylbut-3-enoic acid49 was dissolved in 10 mL of THF at 0 °C and 0.27 mL (2.5 mmol) of 4-methylmorpholine and 0.32 mL (2.5 mmol) of isobutyl chloroformate were added dropwise. After stirring for 5 min, the white precipitate was removed via filtration and washed with 4 mL of THF. The filtrate was cooled to 0 °C and the preformed lithiate was added dropwise via syringe to the above solution. After stirring at 0 °C for an additional 5 min, the reaction was quenched with H2O and extracted with EtOAc. The organic layer was washed with saturated aqueous NaHCO3, dried over MgSO4, and concentrated under reduced pressure. The residue was subjected to flash silica gel chromatography to afford a pale yellow oil which underwent spontaneous cycloaddition while standing at room temperature for 12 h. The crude yellow oil was subjected to flash silica gel chromatography to provide 0.3 g (41%) of 25 as a clear oil: IR (neat) 1798, 1762, 1726, 1357, and 1296 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.53 (s, 9H), 2.17 (d, 1H, J= 12.0 Hz), 2.52 (dd, 1H, J= 12.0 and 4.8 Hz), 2.84 (d, 1H, J= 16.8 Hz), 3.02 (d, 1H, J= 16.8 Hz), 5.10 (dd, 1H, J= 4.8 and 2.0 Hz), 6.31 (dd, 1 H, J= 6.0 and 2.0 Hz), 6.36 (d, 1 H, J= 6.0 Hz), 7.11–7.14 (m, 2H), and 7.17–7.28 (m, 3H); 13C-NMR (CDCl3, 100 MHz) δ 28.2, 40.4, 48.4, 49.9, 77.0, 84.2, 104.5, 126.9, 127.1, 128.8, 133.0, 134.4, 143.2, 149.3, and 174.4; Anal. Calcd. for C19H21NO4: C, 69.71; H, 6.47; N, 4.28. Found: C, 69.52; H, 6.37; N, 4.38.
tert-Butyl 3-oxo-5-carbomethoxy-10-oxa-2-azatricyclo[5.2.1.01,5]dec-8-ene-2-carboxylate (26)
To a solution of 0.36 g (2.0 mmol) of tert-butyl furan-2-yl-carbamate (12a) in 8 mL of THF at 0 °C was added dropwise 1.3 mL (2.2 mmol) of n-BuLi (1.6 M in hexane). The reaction mixture was stirred at 0 °C for 20 min. In a separate flask, 0.37 g (2.6 mmol) of 3-carbomethoxy-but-3-enoic acid was dissolved in 10 mL of THF at 0 °C and 0.28 mL (2.6 mmol) of 4-methylmorpholine, and 0.33 mL (2.6 mmol) of isobutyl chloroformate were added dropwise. After stirring for 5 min, the white precipitate was removed via filtration and washed with 4 mL of THF. The filtrate was cooled to 0 °C and the preformed lithiate was added dropwise via syringe to the above soltuion. After stirring at 0°C for an additional 10 min, the reaction was quenched with H2O and extracted with EtOAc. The organic layer was washed with saturated aqueous NaHCO3, dried over MgSO4, and concentrated under reduced pressure. The residue was subjected to flash silica gel chromatography to afford 0.47 g (77%) of 26 as a yellow oil: IR (neat) 2971, 2955, 1801, and 1728 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.50 (s, 9H), 2.08 (dd, 1H, J= 12.0 and 4.4 Hz), 2.28 (d, 1H, J= 12.0 Hz), 2.67 (d, 1H, J= 17.0 Hz), 2.89 (d, 1H, J= 17.0 Hz), 3.63 (s, 3H), 5.03 (dd, 1H, J= 4.4 and 2.0 Hz), 6.38 (dd, 1H, J= 6.0 and 2.0 Hz), and 6.41 (d, 1H, J= 6.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 28.1, 36.7, 42.4, 51.7, 52.9, 77.4, 84.2, 103.2, 132.7, 134.8, 149.0, 172.3 and 172.9; FAB HRMS Calcd for [(C15H19NO6)+Li]+: 316.1372. Found: 316.1372.
tert-Butyl 2-(cyclopent-1-enylacetyl)furan-2-yl)carbamate (22)
To a solution of 0.5 g (2.6 mmol) of furanyl carbamate 12a in 8 mL of THF at 0 °C was added dropwise 1.9 mL (2.8 mmol) of n-BuLi (1.5 M in hexane). The reaction mixture was stirred at 0 °C for 20 min. In a separate flask, 0.36 g (2.8 mmol) of 2-cyclopent-1-enylacetic acid50 was dissolved in 10 mL of THF at 0 °C and 0.3 mL (2.8 mmol) of 4-methylmorpholine, and 0.36 mL (2.8 mmol) of isobutyl chloroformate were added dropwise. After stirring for 5 min, the white precipitate was removed via filtration and washed with 4 mL of THF. The filtrate was cooled to 0 °C and the preformed lithiate was added dropwise via syringe to the above solution. After stirring at 0 °C for an additional 5 min, the reaction was quenched with H2O and extracted with EtOAc. The organic layer was washed with saturated aqueous NaHCO3 solution, dried over MgSO4, and concentrated under reduced pressure. The residue was subjected to flash silica gel chromatography to afford 0.43 g (58%) of 22 as a white solid: mp 55–56 °C; IR (neat) 1792, 1746, 1608, 1265, and 1147 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.42 (s, 9H), 1.85–1.92 (m, 4H), 2.33 (t, 2H, J= 7.4 Hz), 3.57 (s, 2H), 5.52 (s, 1H), 6.14 (dd, 1H, J= 3.2 and 0.8 Hz), 6.41 (dd, 1H, J= 3.2 and 2.4 Hz), and 7.32 (dd, 1H, J= 2.4 and 0.8 Hz); 13C-NMR (CDCl3, 100 MHz) δ 23.7, 27.9, 32.8, 35.4, 39.8, 84.0, 106.1, 111.4, 128.4, 137.1, 140.7, 144.1, 151.6, and 173.0; Anal. Calcd. for C16H21NO4: C, 65.96; H, 7.27; N, 4.81. Found: C, 65.83; H, 7.14; N, 4.88.
tert-Butyl 5,6-cyclopentyl-3-oxo-10-oxa-2-azatricyclo[5.2.1.01,5]dec-8-ene-2-carboxylate (27)
A solution of 0.2 g (0.7 mmol) of imidofuran 22 in 4 mL of toluene was heated at 90 °C for 4 h. The solution was cooled to room temperature and the solvent was removed under reduced pressure. The residue was subjected to flash silica gel chromatography to afford 0.15 g (71%) of 27 as a white solid: mp 154–155 °C; IR (neat) 1792,1357, 1255, and 1157 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.20–1.26 (m, 1 H), 1.32–1.40 (m, 1 H), 1.53 (s, 9H), 1.55–1.79 (m, 4H), 2.51 (d, 1H, J= 17.0 Hz), 2.76–2.81 (m, 1H), 2.96 (d, 1H, J = 17.0 Hz), 4.88 (dd, 1H, J= 5.2 and 2.0 Hz), 6.41 (dd, 1H, J= 6.0 and 2.0 Hz), and 6.59 (d, 1H, J= 6.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 27.2, 28.2, 29.2, 35.1, 46.9, 55.2, 56.7, 80.1, 84.1, 104.4, 133.9, 134.8, 149.6, and 174.8; Anal. Calcd. for C16H21NO4: C, 65.96; H, 7.27; N, 4.81. Found: C, 65.68; H, 7.20; N, 4.90.
tert-Butyl 2,3,3a,4,5,6-hexahydro-3a-methyl-2,5-dioxoindole-1-carboxylate (28)
A solution containing 0.05 g (0.19 mmol) of oxabicycle 24 in 2 mL of toluene in a sealed tube was heated at 140 °C for 4 h. Concentration under reduced pressure and purification by silica gel chromatography provided 0.046 g (92%) of 28 as a yellow oil: IR (neat) 1763, 1724, 1681,1301, 1150 and 845 cm−1; 1H-NMR (CDCl3, 400 MHz) 51.18 (s, 3H), 1.57 (s, 9H), 2.43 (d, 1 H, J= 17.2 Hz), 2.56 (d, 1H, J= 14.6 Hz), 2.57 (d, 1H, J= 17.2 Hz), 2.63 (d, 1H, J= 14.6 Hz), 3.03 (d, 2H, J= 3.8 Hz), and 6.00 (t, 1H, J= 3.8 Hz); 13C-NMR (CDCl3, 100 MHz) δ 25.8, 28.2, 37.8, 37.9, 46.0, 51.4, 84.7, 102.6, 142.0, 149.5, 171.8, and 207.6; HRMS Calcd. for C14H19NO4: 265.1314. Found: 265.1312.
tert-Butyl 2,5-dioxo-3a-phenyl-2,3,3a,4,5,6-hexahydroindole-1-carboxylate (29)
A solution containing 0.02 g (0.06 mmol) of hexahydroindolinone 25 in 1 mL of toluene was heated at 135 °C for 14 h and then cooled to rt. Concentration of the solution under reduced pressure followed by purification using silica gel chromatography afforded 0.018 g (91%) of 29 as clear oil: IR (neat) 2986, 1766, 1725, 1298, 1149 and 707 cm−1; 1H-NMR (CDCl3, 600 MHz) δ 1.59 (s, 9H), 2.82 (dd, 1H, J= 23.4 and 2.4 Hz), 2.84 (d, 1H, J= 14.4 Hz), 2.94 (s, 2H), 3.01 (dd, 1 H, J= 23.4 and 5.4 Hz), 3.17 (d, 1 H, J= 14.4 Hz), 6.35 (dd, 1 H, J= 5.4 and 2.4 Hz), 7.23–7.26 (m, 3H), and 7.32 (t, 2H, J= 7.8 Hz); 13C-NMR (CDCl3, 150 MHz) δ 28.2, 38.2, 46.1, 47.8, 53.0, 84.9, 106.0, 126.0, 128.2, 129.6, 140.1, 141.0, 149.4, 171.2, and 206.6; HRMS Calcd. for C19H21NO4: 327.1471. Found: 327.1470.
1 -tert-Butyl 3a-methyl-2,5-dioxo-2,3,3a,4,5,6-tetrahydro-1H-indole-1,3a(4H)-dicarboxylate (30)
A solution containing 0.04 g of oxabicycle 26 in 2 mL of toluene in a sealed tube was heated at 140 °C for 14 h. Concentration under reduced pressure and purification by silica gel chromatography provided 0.035 g (90%) of 30 as a yellow solid: mp 101–103 °C; IR (neat) 2980, 1774, 1736, 1302, 1153, and 842 cm−1; 1H-NMR (CDCl3, 600 MHz) δ 1.58 (s, 9H), 2.42 (d, 1H, J = 16.2 Hz), 2.54 (d, 1H, J= 17.4 Hz), 3.01 (d, 1H, J= 16.2 Hz), 3.02 (dd, 1H, J = 22.8 and 6.0 Hz), 3.11 (d, 1H, J= 17.4 Hz), 3.15 (dd, 1H, J= 22.8 and 2.4 Hz), 3.73 (s, 3H), and 6.27 (dd, 1H, J= 6.0 and 2.4 Hz); 13C-NMR (CDCl3, 100 MHz) δ 28.2, 37.2, 41.0, 46.7, 47.0, 53.7, 85.0, 106.4, 135.5, 149.2, 170.7, 171.3, and 204.7; HRMS Calcd. for C15H19NO6: 309.1212. Found: 309.1214.
tert-Butyl 2,6-dioxo-1,2,5,6,6a,7,8,9-octahydro-3-aza-cyclopenta[d]indene-3-carboxylate (31)
A solution containing 0.03 g (0.1 mmol) of oxabicycle 27 in 2 mL of toluene in a sealed tube was heated at 140 °C for 3 h. Concentration under reduced pressure and purification by silica gel chromatography provided 0.027 g (90%) of 31 as a yellow oil: IR (neat) 1767, 1728, 1683, 1295, and 1151 cm−1; 1H-NMR (CDCl3, 600 MHz) δ 1.47–1.57 (m, 1 H), 1.58 (s, 9H), 1.65–1.73 (m,1H), 1.84–2.75 (m, 2H), 2.40–3.45 (m, 1H), 2.45 (d, 1H, J= 16.8 Hz), 2.49 (dd, 1H, J= 16.8 and 1.6 Hz), 2.54 (d, 1H, J=7.2 Hz), 3.05 (dd, 1H, J= 21.6 and 4.8 Hz), 3.10 (dd, 1H, J= 21.6 and 3.6 Hz), and 5.99 (dd, 1 H, J= 4.8 and 3.6 Hz); 13C-NMR (CDCl3, 150 MHz) δ 21.6, 24.8, 28.2, 37.5, 37.7, 45.0, 49.0, 56.7, 84.6, 102.6, 139.7, 149.5, 171.9, and 208.6; HRMS Calcd. for C16H21NO4: 291.1471. Found: 291.1470.
tert-Butyl (3-ethyl-furan-2-yl)carbamate (32)
A modification of the procedure of Burness51 was used to prepare methyl 3-ethylfuran-2-carboxylate. To a mixture containing 23 g (156 mmol) of 1,1-dimethoxy-3-pentanone52 22 mL (250 mmol) of methyl chloroacetate, and 125 mL of dry Et2O at −10 °C was added 13.5 g (250 mmol) of freshly prepared powdered NaOMe in small portions over 30 min maintaining an internal temperature below −5 °C. The reaction mixture was stirred an additional 2 h at −10 °C and then warmed to room temperature overnight. The slurry was cooled to 0 °C, quenched by the slow addition of a solution of 3 mL of AcOH in 50 mL of H2O, and then the aqueous layer was extracted with Et2O. The combined organic layers were washed with a dilute NaHCO3 solution and brine, then dried over anhydrous MgSO4 and concentrated under reduced pressure. The crude residue was transferred to a flask equipped with a 5 inch vigreaux column and distillation head and was heated at 170 °C until MeOH distillation ceased. The mixture was cooled to room temperature and distilled under reduced pressure to afford 11.7 g (49%) of 3-ethyl-furan-2-carboxylic acid methyl ester as a colorless oil: bp 80–95 °C (15 mm); IR (neat) 1716, 1598, 1490, 1434, 1301 and 1198 cm−1; 1H-NMR (CDCl3, 400 MHz) 51.18 (t, 3H, J= 7.6 Hz), 2.81 (q, 2H, J= 7.6 Hz), 3.87 (s, 3H), 6.40 (d, 1H, J= 1.6 Hz) and 7.43 (d, 1H, J= 1.6 Hz); 13C-NMR (CDCl3, 100 MHz) δ 14.3, 19.1, 51.7, 113.5, 137.8, 139.5, 145.2 and 160.1; HRMS Calcd. for C8H10O3: 154.0630. Found: 154.0631.
A suspension of 4.0 g (26 mmol) of the above 3-ethylfuran-2-carboxylic acid methyl ester in 30 mL of a 10% aqueous NaOH solution was heated at reflux for 2 h. The homogeneous reaction mixture was cooled to 0 °C and was slowly acidified with cone. HCI. The resultant white precipitate was collected by filtration, washed with several portions of cold H2O, and dried under high-vacuum to afford 3.4 g (94%) of 3-ethylfuran-2-carboxylic acid as a white solid: mp 106–107 °C; IR (neat) 3139, 1675, 1593, 1490, and 1285 cm−1; 1H-NMR (CDCl3, 400 MHz) 5 1.22 (t, 3H, J= 7.6 Hz), 2.85 (q, 2H, J= 7.6 Hz), 6.46 (d, 1 H, J= 1.6 Hz), 7.52 (d, 1H, J= 1.6 Hz) and 12.31 (brs, 1H); 13C-NMR (CDCl3, 100 MHz) δ 14.2, 19.4, 113.9, 138.9, 140.3, 146.5 and 165.0; Anal. Calcd. for C7H8O3: C, 59.99; H, 5.75. Found: C, 59.82; H, 5.74.
To a solution of 2.0 g (14.3 mmol) of the above acid in 60 mL of CH2Cl2 were added 1.5 mL (17 mmol) of oxalyl chloride and 1 drop of DMF. The reaction mixture was stirred at rt for 1 h and then concentrated under reduced pressure. The crude residue was dissolved in 40 mL of Et2O and a solution of 1.1 g (17 mmol) of NaN3 in 20 mL of H2O was added. The biphasic reaction mixture was stirred vigorously for 3 h and then the organic layer was separated, dried over MgSO4, and concentrated under reduced pressure. The crude residue was dissolved in 40 mL of t-BuOH and heated at reflux for 12 h. The reaction mixture was cooled to rt and concentrated under reduced pressure. The residue was subjected to flash silica gel chromatography to afford 1.6 g (52 %) of tert-butyl (3-ethyl-furan-2-yl)carbamate (32) as a pale yellow oil: IR (neat) 3313, 1716, 1644, 1511, 1244 and 1157 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.13 (t, 3H, J= 7.6 Hz), 1.46 (s, 9H), 2.34 (q, 2H, J= 7.6 Hz), 6.09 (brs, 1H), 6.25 (d, 1H, J= 2.0 Hz), and 7.13 (d, 1H, J= 2.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 14.2, 17.7, 28.3, 81.2, 111.8, 119.2, 138.9, 139.3, and 154.0; HRMS Calcd. for C11H17NO3:211.1208. Found: 211.1215.
tert-Butyl 9-ethyl-3-oxo-10-oxa-2-azatricyclo[5.2.1.01,5]dec-8-ene-2-carboxylate (34)
To a solution of 0.27 g (1.2 mmol) of the above furan in 4 mL of THF at 0 °C was added dropwise 1.0 mL (1.4 mmol) of n-BuLi (1.4 M in hexane). The reaction mixture was stirred at 0 °C for 20 min so as to generate the lithiated carbamate 32. In a separate flask, 0.12 mL (1.4 mmol) of vinylacetic acid was dissolved in 5 mL of THF at 0 °C and 0.15 mL (1.4 mmol) of 4-methyl-morpholine and 0.18 mL (1.4 mmol) of isobutyl chloroformate were added dropwise. After stirring for 5 min, the white precipitate was removed by filtration and was washed with 2 mL of THF. The filtrate was cooled to 0 °C and the preformed lithiate was added dropwise via syringe. After stirring at 0 °C for an additional 5 min, the reaction was quenched with H2O and extracted with EtOAc. The organic layer was washed with a saturated aqueous NaHCO3 solution, dried over MgSO4 and concentrated under reduced pressure. The residue was subjected to flash silica gel chromatography to afford the expected imidofuran as a labile oil which underwent spontaneous cycloaddition while standing at room temperature for 12 h. The crude colorless oil was subjected to flash silica gel chromatography to afford 0.21 g (60%) of 34 as a colorless oil: IR (neat) 1798, 1762, 1731, 1296 and 1157 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.09 (t, 3H, J = 7.2 Hz), 1.53 (s, 9H), 1.68 (dd, 1 H, J= 11.6 and 8.0 Hz), 1.79 (dt, 1 H, J= 11.6 and 4.0 Hz), 1.92–2.06 (m, 2H), 2.29–2.37 (m, 1H), 2.40 (dd, 1H, J= 17.2 and 10.4 Hz), 2.74 (dd, 1H, J= 17.2 and 8.8 Hz), 4.99 (d, 1H, J= 4.0 Hz) and 5.89 (q, 1H, J=2.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 11.4, 18.8, 28.0, 35.0, 35.4, 38.3, 77.5, 84.0, 103.3, 126.5, 149.6, 150.0 and 175.1; Anal Calcd. for C15H21NO4: C, 64.48; H, 7.58; N, 5.02. Found: C, 64.36; H, 7.35; N, 5.37.
tert-Butyl 7-methyl-3-oxo-10-oxa-2-azatricyclo[5.2.1.01,5]dec-8-ene-2-carboxylate (35)
To a solution of 0.3 g (1.5 mmol) of the known tort-butyl (5-methyl-furan-2-yl) carbamate53 in 5 mL of THF at 0 °C was added dropwise 1.2 mL (1.7 mmol) of n-BuLi (1.4 M in hexane). The reaction mixture was stirred at 0 °C for 20 min so as to generate the lithiated carbamate 33. In a separate flask, 0.14 mL (1.7 mmol) of vinylacetic acid was dissolved in 5 mL of THF at 0 °C and 0.18 mL (1.7 mmol) of 4-methylmorpholine and 0.22 mL (1.7 mmol) of isobutyl chloroformate were added dropwise. After stirring for 5 min, the white precipitate that formed was removed by filtration and washed with 2 mL of THF. The filtrate was cooled to 0 °C and the preformed lithiate was added dropwise via syringe. After stirring at 0 °C for an additional 5 min, the reaction was quenched with H2O and extracted with EtOAc. The organic layer was washed with a saturated aqueous NaHCO3 solution, dried over MgSO4 and concentrated under reduced pressure. The residue was subjected to flash silica gel chromatography to afford the expected imidofuran as a labile oil which underwent spontaneous cycloaddition while standing at room temperature for 12 h. The crude yellow solid was subjected to flash silica gel chromatography to give 0.3 g (68%) of 35 as a white solid: mp 86–87 °C; IR (neat) 1796,1763, 1731, 1295 and 1160 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.46 (dd, 1 H, J= 7.6 and 4.4 Hz), 1.49 (s, 9H), 1.60 (s, 3H), 1.71 (dd, 1H, J= 11.6 and 7.6 Hz), 2.12–2.19 (m, 1H), 2.40 (dd, 1H, J = 17.0 and 10.2 Hz), 2.70 (dd, 1H, J= 17.0 and 8.6 Hz), 6.10 (d, 1H, J= 5.8 Hz) and 6.49 (d, 1H, J= 5.8 Hz); 13C-NMR (CDCl3, 100 MHz) δ 19.5, 28.1, 38.5, 38.9, 39.0, 83.8, 85.2, 101.2, 135.2, 136.6, 149.4 and 174.6; Anal Calcd. for C14H19NO4: C, 63.38; H, 7.22; N, 5.28. Found: C, 63.45; H, 7.32; N, 5.33.
tert-Butyl 4-methyl-3-oxo-10-oxa-2-aza-tricyclo[5.2.1.01,5]dec-8-ene-2-carboxylate (38)
To a solution of 0.7 g (4.1 mmol) of tert-butyl furan-2-yl carbamate (12a) in 12 mL of THF at 0 °C was added dropwise 2.1 mL (4.3 mmol) of n-BuLi (2.1 M in hexane). The reaction mixture was stirred at 0 °C for 20 min. In a separate flask, 0.47 mL (4.5 mmol) of 2-methyl-but-3-enoic acid was dissolved in 15 mL of THF at 0 °C and 0.5 mL (4.5 mmol) of 4-methyl-morpholine and 0.58 mL (4.5 mmol) of isobutyl chloroformate were added dropwise. After stirring for 5 min, the white precipitate that formed was removed by filtration and washed with 2 mL of THF. The filtrate was cooled to 0 °C and the preformed lithiate was added dropwise via syringe to the above solution. After stirring at 0 °C for an additional 10 min, the reaction mixture was quenched with H2O and extracted with EtOAc. The organic layer was washed with a saturated aqueous NaHCO3 solution, dried over MgSO4, and concentrated under reduced pressure. The residue was subjected to flash silica gel chromatography to afford 0.69 g (65%) of furan 36 as a colorless oil which underwent spontaneous cycloaddition while standing at room temperature for 12 h. The 1H-NMR spectra revealed the presence of a 20:1-mixture of two diastereomers. The crude residue was subjected to flash silica gel chromatography to provide 0.66 g (96%) of the major cis diastereomer 38 as a white solid: mp 102–104 °C; IR (neat) 2979,1794, 1766, 1731 and 1159 cm−1; 1H NMR (CDCl3, 400 MHz) δ 1.24 (d, 3H, J= 6.8 Hz), 1.52 (s, 9H), 1.64 (dd, 1 H, J= 11.2 and 7.2 Hz), 1.73 (ddd, 1 H, J= 10.0, 7.2 and 3.2 Hz), 1.81 (ddd, 1H, J= 11.2, 4.4 and 3.2 Hz), 2.43 (dq, 1H, J= 10.0 and 6.8 Hz), 5.03 (dd, 1H, J= 4.4 and 2.0 Hz), 6.31 (dd, 1H, J= 6.0 and 2.0 Hz) and 6.50 (d, 1H, J=6.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 14.3, 28.2, 31.7, 44.0, 44.5, 77.7, 83.9, 100.0, 133.8, 134.6, 149.6 and 177.0; Anal. Calcd. for C14H19NO4: C, 63.38; H, 7.22, N, 5.28. Found: C, 63.44; H, 7.32, N, 5.30.
tert-Butyl 3-oxo-4-phenyl-10-oxa-2-aza-tricyclo[5.2.1.01,5]dec-8-ene-2-carboxylate (39)
To a solution of 0.35 g (1.9 mmol) of carbamate 12a in 10 mL of THF at 0 °C was added dropwise 1.1 mL (2.2 mmol) of n-BuLi (2.1 M in hexane). The reaction mixture was stirred at 0 °C for 20 min. In a separate flask, 0.37 g (2.3 mmol) of 2-phenyl-but-3-enoic acid54 was dissolved in 15 mL of THF at 0 °C and 0.25 mL (2.3 mmol) of 4-methyl-morpholine and 0.3 mL (2.3 mmol) of isobutyl chloroformate were added dropwise. After stirring for 5 min, the white precipitate that formed was removed by filtration and was washed with 2 mL of THF. The filtrate was cooled to 0 °C and the preformed lithiate was added dropwise via syringe to the above solution. After stirring at 0 °C for an additional 10 min, the reaction mixture was quenched with H2O and extracted with EtOAc. The organic layer was washed with a saturated aqueous NaHCO3 solution, dried over MgSO4, and concentrated under reduced pressure. The residue was subjected to flash silica gel chromatography to afford 0.34 g (55%) of furan 37 as a colorless oil, which underwent spontaneous cycloaddition while standing at room temperature for 12 h. The 1H-NMR spectrum of the solid showed a 30:1-mixture of two diastereomers. The crude residue was subjected to flash silica gel chromatography to provide 0.34 g (98%) of the major cis isomer 39 as a white solid: mp 124–125 °C; IR(neat) 1793, 1732, 1296 and 1157 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.54 (s, 9H), 1.63 (dd, 1H, J= 12.0 and 8.0 Hz), 1.94 (dt, 1H, J= 12.0 and 3.6 Hz), 2.27 (ddd, 1H, J= 10.8, 8.0 and 3.6 Hz), 3.63 (d, 1H, J = 10.8 Hz), 5.09 (dd, 1H, J= 3.6 and 2.0 Hz), 6.36 (dd, 1H, J= 6.0 and 2.0 Hz), 6.59 (d, 1H, J= 6.0 Hz) and 7.22–7.36 (m, 5H); 13C-NMR (CDCl3, 100 MHz) 5 28.1, 31.9, 44.9, 55.6, 77.8, 84.2, 99.7, 127.7, 128.6, 128.9, 134.1, 134.4, 136.5, 149.5 and 174.4; Anal. Calcd. for C19H21NO4: C, 69.71; H, 6.47, N, 4.28. Found: C, 69.76; H, 6.42, N, 4.22.
tert-Butyl 2,3,3a,4,5,6-hexahydro-5-hydroxy-2-oxo-6-phenoxyindole-1-carboxylate (40)
To a solution containing 0.05 g (0.2 mmol) of oxabicycle 23 in 0.5 mL THF was added 3 mg (0.006 mmol) of [Rh(COD)Cl]2, 6 mg (0.012 mmol) of 1,1′-bis(diphenylphosphino)ferrocene (DPPF) and 0.19 g (2.0 mmol) of phenol. The mixture was heated at 80 °C for 12 h, cooled to rt and diluted with ether. The organic layer was washed with a 4% aqueous NaOH solution and dried over MgSO4. The solvent was removed under reduced pressure and a 1H-NMR spectrum of the crude mixture showed the presence of a 5:1-mixture of isomeric products. Purification by flash silica gel chromatography afforded 0.02 g (62%) of the major isomer 40 as a clear oil; IR (neat) 3448, 1771, 1732, 1300, and 1152 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.50 (s, 9H), 1.82 (d, 1H, J= 11.6 Hz), 2.18 (dt, 1 H, J= 11.6 and 4.4 Hz), 2.43 (dd, 1 H, J= 16.8 and 11.6 Hz), 2.57 (d, 1 H, J = 11.6 Hz), 2.66 (dd, 1H, J= 16.8 and 8.4 Hz), 2.76–2.88 (m,1H), 3.96 (tt, 1 H, J = 11.6 and 4.4 Hz), 4.94 (t, 1 H, J= 4.4 Hz), 6.21 (dd, 1 H, J= 4.4 and 2.4 Hz), 6.97–7.00 (m, 3H), and 7.29 (t, 2H, J= 8.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 28.1, 32.5, 34.9, 37.7, 68.7, 72.6, 84.7, 104.4, 116.2, 122.0, 130.0, 143.6, 149.2, 157.8, and 172.3; HRMS Calcd. for C19H23NO5: 345.1576. Found: 345.1574.
tert-Butyl 5-hydroxy-3a-methyl-2-oxo-6-phenoxy-2,3,3a,4,5,6-hexahydro-indole-1-carboxylate (41)
To a solution containing 0.06 g (0.22 mmol) of oxabicycle 24 in 0.5 mL THF was added 3 mg (0.007 mmol) of [Rh(COD)Cl]2, 7 mg (0.014 mmol) of DPPF and 0.22 g (2.3 mmol) of phenol. The mixture was heated at 80 °C for 2 h, cooled to rt and diluted with ether. The organic layer was washed with a 4 % aqueous NaOH solution and dried over MgSO4. The solvent was removed under reduced pressure and a NMR spectrum of the crude reaction mixture showed the presence of a 5:1-mixture of isomeric products. Purification by flash silica gel chromatography afforded 0.06 g (75%) of the major isomer 41 as a white solid: mp 126–128 °C; IR (neat) 3508, 1774, 1732, 1302, and 1153 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.29 (s, 3H), 1.52 (s, 9H), 1.95 (t, 1H, J = 12.1 Hz), 2.02 (dd, 1H, J= 12.1 and 4.1 Hz), 2.36 (d, 1H, J= 16.2 Hz), 2.47 (d, 1H, J= 16.2 Hz), 2.57 (d, 1H, J=10.5 Hz), 4.14–4.23 (m, 1H), 4.94 (t, 1H, J= 4.7 Hz), 6.15 (d, 1H, J= 4.7 Hz), 6.97–7.01 (m, 3H), and 7.28–7.32 (m, 2H); 13C-NMR (CDCl3, 100 MHz) δ 25.9, 28.1, 38.8, 38.9, 47.4, 66.1, 72.3, 84.5, 104.6, 116.2, 122.0, 129.9, 147.2, 149.5, 157.8, and 171.8; HRMS Calcd. for C20H25NO5: 359.1733. Found: 359.1733.
The minor isomer (0.01 g (15%)) was obtained as a clear oil; IR (neat) 1786, 1733, 1301, and 1148 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.37 (s, 3H), 1.44(s, 9H), 1.86 (t, 1H, J= 16.8 Hz), 2.15 (dd, 1H, J= 16.8 and 5.2 Hz), 2.35–2.46 (m, 3H), 4.28–4.38 (m, 1H), 4.91 (dd, 1H, J= 9.6 and 4.0 Hz), 5.98 (d, 1H, J = 4.0 Hz), 6.96–7.00 (m, 3H), and 7.26–7.32 (m, 2H); 13C-NMR (CDCl3, 100 MHz) δ 26.5, 28.1, 28.2, 38.3, 40.8, 47.6, 68.8, 81.0, 84.5, 105.1, 116.6, 121.8, 129.9, 145.1, 157.8, and 173.4; HRMS Calcd. for C20H25NO5: 359.1733. Found: 359.1730.
tert-Butyl 6-(N-methyl-N-phenylamino)-5-hydroxy-2-oxo 2,3,3a,4,5,6-hexahydroindole-1-carboxylate (42)
To a solution containing of 0.06 g (0.24 mmol) of oxabicycle 23 in 1 mL THF was added 3 mg (0.007 mmol) of [Rh(COD)Cl]2, 8 mg (0.014 mmol) of DPPF, 0.13 mL (1.2 mmol) of N-methylaniline and 0.44 g (1.2 mmol) of tetrabutylammonium iodide. The mixture was heated at reflux for 12 h, cooled to rt, diluted with water and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated under reduced pressure. A 1H-NMR spectrum of the crude reaction mixture indicated a 20:1 -mixture of two isomeric products. Purification by silica gel chromatography afforded 0.03 g (33%) of the major isomer 42 as a clear oil: IR (neat) 3451, 1786, 1732, 1306, and 1150 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.51 (s, 9H), 1.67 (q, 1H, J= 12.0 Hz), 2.29 (dd, 1H, J= 16.8 and 12.0 Hz), 2.36 (dt, 1H, J= 10.0 and 4.0 Hz), 2.51 (brs, 1H), 2.64 (dd, 1H, J= 16.8 and 8.8 Hz), 2.78 (s, 3H), 2.88–2.97 (m, 1H), 4.01 (ddd, 1H, J= 12.0, 8.8 and 4.0 Hz), 4.49 (dt, 1H, J= 8.8 and 2.8 Hz), 5.64 (t, 1H, J= 2.8 Hz), 6.80 (t, 1H, J= 7.6 Hz), 6.95 (d, 2H, J= 7.6 Hz), and 7.25 (t, 2H, J=7.6 Hz); 13C-NMR (CDCl3, 100 MHz) δ 28.2, 32.8, 34.1, 38.3, 66.1, 68.4, 84.73, 107.5, 115.2, 118.8, 129.5, 140.1, 148.9, 151.1, and 172.8; HRMS Calcd. for C20H26N2O4: 358.1892. Found: 358.1892.
tert-Butyl 6-(N-methyl-N-phenylamino)-5-hydroxy-3a-methyl-2-oxo 2,3,3a,4,5,6-hexahydroindole-1-carboxylate (43)
To a solution containing 0.05 g (0.19 mmol) of oxabicycle 24 in 1 mL THF was added 3 mg (0.006 mmol) of [Rh(COD)Cl2]2, 6 mg (0.012 mmol) of DPPF, 0.13 g (0.94 mmol) of N-methyl aniline and 0.13 g (0.94 mmol) of Et3NHCl. The mixture was heated at 80 °C for 10 h, cooled to rt, diluted with water and extracted with CHCl3. The organic layer was dried over MgSO4 and concentrated under reduced pressure. A 1H-NMR spectrum of the crude mixture showed the presence of two isomeric products in a ratio of 10:1. Purification by silica gel chromatography afforded 0.04 g (58%) of the major isomer 43 as a clear oil; IR (neat) 3462, 1786, 1733, 1301, and 1149 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.34 (s, 3H), 1.51 (s, 9H), 1.82 (t, 1 H, J = 12.4 Hz), 2.17 (dd, 1H, J= 12.4 and 4.0 Hz), 2.36 (d, 1H, J= 16.4 Hz), 2.36 (brs, 1H), 2.43 (d, 1H, J= 16.4 Hz), 2.80 (s, 3H), 4.19 (ddd, 1H, J= 12.0, 8.8 and 4.0 Hz), 4.47 (dd,1 H, J= 8.8 and 2.8 Hz), 5.60 (d, 1 H, J= 2.8 Hz), 6.80 (t, 1 H, J = 7.6 Hz), 6.95 (d, 2H, J= 7.6 Hz), and 7.25 (t, 2H, J= 7.6 Hz); 13C-NMR (CDCl3, 100 MHz) δ 26.5, 28.2, 32.9, 38.2, 41.1, 47.9, 66.0, 66.4, 84.2, 107.9, 115.1, 118.7, 129.5, 143.7, 149.2, 151.0 and 172.4; HRMS Calcd. for C21H28N2O4: 372.2049. Found: 372.2048.
tert-Butyl 6-(2-bromo-benzoyloxy)-5-hydroxy-3a-methyl-2-oxo-2,3,3a,4,5,6-hexahydroindole-1-carboxylate (44a)
To a solution containing 0.05 g (0.19 mmol) of oxabicycle 24 in 1 mL of THF was added 3 mg (0.006 mmol) of [Rh(COD)Cl]2, 6 mg (0.012 mmol) of DPPF, 0.38 g (1.9 mmol) of 2-bromo-benzoic acid and 0.26 mL (1.9 mmol) of Et3N. The mixture was heated at 80 °C for 10 h, cooled to room temperature, diluted with water and extracted with ether. The organic layer was dried over MgSO4 and concentrated under reduced pressure. A NMR spectrum of the crude mixture showed the presence of two isomeric products in a ratio of 10:3. Purification by silica gel chromatography afforded 0.06 g (72%) of the major cis-isomer 44a as a clear oil: IR (neat) 3487, 1786, 1732 and 1148 cm−1; 1H-NMR (CDCl3, 600 MHz) δ 1.33 (s, 3H), 1.54 (s, 9H), 1.85 (t, 1H, J= 12.0 Hz), 2.19 (dd, 1H, J= 12.0 and 4.8 Hz), 2.37 (d, 1H, J = 16.2 Hz), 2.45 (d, 1H, J= 16.2 Hz), 3.26 (brs, 1H), 4.32 (ddd, 1H, J= 12.0, 7.2 and 4.8 Hz), 5.58 (dd, 1 H, J= 7.2 and 3.6 Hz), 5.96 (d, 1 H, J= 3.6 Hz), 7.34 (td, 1 H, J= 7.8 and 1.8 Hz), 7.37 (td, 1 H, J= 7.8 and 1.8 Hz), 7.66 (dd, 1 H, J= 7.8 and 1.8 Hz) and 7.80 (dd, 1H, J= 7.8 and 1.8 Hz); 13C-NMR (CDCl3, 150 MHz) δ 26.1, 28.2, 37.6, 41.6, 47.2, 68.8, 80.6, 84.7, 104.4, 121.9, 127.5, 131.7, 132.2, 133.1, 134.6, 146.8, 149.2, 167.9 and 172.0; HRMS Calcd. for C21H24NO6Br: 465.0787. Found: 465.0784.
tert-Butyl 5-hydroxy-2-oxo-2,4,5,6-tetrahydroindole-1-carboxylate (45)
To the mixture of 0.18 g (0.72 mmol) of oxabicycle 23, 7 mg (0.014 mmol) of [Rh(COD)Cl]2 and 4.9 piL (0.029 mmol) P(OEt)3 in 2 mL THF was added 0.28 g (3.6 mmol) of ammonium acetate. The reaction mixture was heated at reflux for 12 h and cooled to rt. The mixture was poured into a saturated NaHCO3 aqueous solution and was extracted with EtOAc. The combined organic layer was washed with brine and dried over MgSO4. The solvent was removed under reduced pressure and the residue was subjected to flash column chromatography to give 0.14 g (80%) of 45 as a white solid: mp 167–168 °C; IR (neat) 3452, 1733, 1326, and 818 cm−1; 1 H-NMR (400 MHz, CDCl3) δ 1.56 (s, 9H), 2.34 (brs, 1H), 2.44 (ddd, 1H, J= 17.6, 8.4 and 4.0 Hz), 265–2.71 (m, 2H), 2.92 (dd, 1H, J = 16.8 and 4.0 Hz), 4.15 (m, 1H), 5.77 (s, 1H), and 6.54 (ddd, 1H, J =5.6, 4.0 and 1.6 Hz); 13C-NMR (100 MHz, CDCl3) δ 28.3, 33.7, 33.8, 66.2, 83.9, 115.2, 117.1, 136.0, 149.4, 149.7, and 168.2; Anal. Calcd for C13H17NO4: C, 62.14; H, 6.82; N, 5.57. Found: C, 61.73; H, 6.79; N, 5.41.
tert-Butyl 2-oxo-2,3-dihydro-indole-1-carboxylate (46)
To a solution of 0.18 g (0.72 mmol) of oxabicycle 23 in 2 ml of THF was added 7 mg (0.014 mmol) of [Rh(COD)Cl]2 and 4.9 μL (0.029 mmol) of P(OEt)3. The reaction mixture was heated at reflux for 2 h and cooled to room temperature. The solvent was removed under reduced pressure. The residue was subjected to flash silica gel chromatography to give 0.16 g (90%) of 46 as a colorless oil: 1H-NMR (CDCl3, 400 MHz) δ1.59 (s, 9H), 3.61 (s, 2H), 7.12 (t, 1H, J = 7.6 Hz), 7.23 (d, 1H, J = 7.6 Hz), 7.28 (t, 1 H, J = 7.6 Hz), and 7.77 (d, 1 H, J = 7.6 Hz); 13C-NMR (CDCl3, 100 MHz) δ 28.3, 33.7, 84.6, 115.3, 123.4, 124.4, 128.3, 141.2, 149.4 and 173.3; HRMS Calcd. for C13H15NO3: 233.1052. Found: 233.1050.
tert-Butyl 5-hydroxy-2-oxo-6-phenyl-2,3,3a,4,5,6-hexahydroindole-1-carboxylate (51)
To a solution containing 0.06 g (0.24 mmol) of oxabicycle 23, 3 mg (0.007 mmol) of [Rh(COD)Cl]2,2.5 μL (0.014 mmol) of P(OEt)3 in 1 ml THF was added 0.09 g (0.48 mmol) of 5,5-dimethyl-2-phenyl-[1,3,2]-dioxaborinane (50) and 0.1 ml (0.48 mmol) of CS2CO3 (5 M in H2O). The reaction mixture was heated at 65 °C for 2 h, cooled to rt and quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layer was dried over MgSO4. The solvent was removed under reduced pressure and the residue was subjected to the flash silica gel chromatography to afford 0.06 g (72%) of 51 as a white solid: mp 175–176 °C; IR (neat) 3437, 1777, 1724, 1292, and 705 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.05 (d, 1 H, J = 10.2 Hz), 1.51 – 1.55 (m, 1 H), 1.55 (s, 9H), 1.99 (ddd, 1H, J = 12.0, 5.2 and 3.2 Hz), 2.37 (d, 1H, J = 17.2 Hz), 2.71 (dd, 1H, J = 17.2 and 8.8 Hz), 2.92–2.95 (m, 1H), 3.91–3.95 (m,1H), 4.12–4.20 (m, 1H), 5.91 (dd, 1H, J = 4.4 and 2.4 Hz), 7.25–7.28 (m, 2H), and 7.30–7.40 (m, 3H); 13C-NMR (CDCl3, 100 MHz) δ 28.2, 32.6, 38.6, 46.0, 68.7, 84.3, 109.1, 127.9, 128.7, 130.7, 138.2, 138.8, 149.4, and 172.8; Anal. Calcd. for C19H23NO4: C, 69.28; H, 7.04; N, 4.25. Found: C, 69.19; H, 7.07; N, 4.21.
tert-Butyl 5-hydroxy-3a-methyl-2-oxo-6-phenyl-2,3,3a,4,5,6-hexahydro-indole-1-carboxylate (52)
To a solution containing 0.05 g (0.19 mmol) of oxabicycle 24, 3.0 mg (0.006 mmol) of [Rh(COD)Cl]2, 2.0 μL (0.012 mmol) of P(OEt)3 in 1 mL THF was added 0.07 g (0.4 mmol) of 5,5-dimethyl-2-phenyl-[1,3,2]-dioxaborinane (50) and 0.08 ml (0.38 mmol) of Cs2CO3 (5 M in H2O). The reaction mixture was heated at 65 °C for 2 h, cooled to rt and quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layer was dried over MgSO4. The solvent was removed under reduced pressure and the residue was subjected to flash silica gel chromatography to afford 0.05 g (80%) of 52 as a white solid: mp 170–173 °C; IR (neat) 3491, 1785, 1729, 1302, and 714 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.34 (s, 3H), 1.54 (s, 9H), 1.61 (t, 1 H, J = 12.4 Hz), 1.82 (dd, 1 H, J = 12.4 and 3.6 Hz), 2.42 (d, 1 H, J = 16.0 Hz), 2.49 (d, 1 H, J = 16.0 Hz), 3.93 (dd, 1 H, J = 6.4 and 4.4 Hz), 4.27–4.37 (m,1H), 5.85 (d, 1H, J = 4.4 Hz), 7.22 (d, 2H, J = 7.2 Hz), 7.31 (t, 1 H, J = 7.2 Hz), and 7.36 (t, 2 H, J = 7.2 Hz); 13C-NMR (CDCl3, 100 MHz) δ 26.4, 28.2, 38.9, 39.0, 46.1, 48.2, 66.2, 84.2, 109.5, 127.9, 128.7, 130.6, 137.9, 142.4, 149.7, and 172.4;Anal. Calcd. for C20H25NO4: C, 69.95; H, 7.34; N, 4.08. Found: C, 69.67; H, 7.26; N, 4.01.
tert-Butyl 2,3,3a,4,5,6-hexahydro-2-oxo-5,6-phenylboronate-indole-1-carboxylate (53)
To a solution containing 0.1 g (0.4 mmol) of oxabicycle 23, 6 mg (0.01 mmol) of [Rh(COD)Cl]2,4.0 μL (0.02 mmol) of P(OEt)3 in 2 ml THF was added 0.07 g (0.6 mmol) of phenyl boronic acid. The reaction mixture was heated at 65°C for 2 h, cooled to rt and quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layer was washed with a saturated aqueous NaHCO3 solution and dried over MgSO4. The solvent was removed under reduced pressure and the residue was subjected to the flash silica gel chromatography to afford 0.14 g (98%) of 53 as a white solid: mp 139–141°C; IR (neat) 1798, 1736, 1340, 1150, and 702 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.42 (q, 1H, J = 12.0 Hz), 1.58, (s, 9H), 2.28 (dd, 1H, J =20.0 and 13.6 Hz), 2.45 (ddd, 1H, J = 12.0, 5.6 and 4.0 Hz), 2.61–2.68 (m, 1H), 2.68 (dd, 1H, J = 20.0 and 9.2 Hz), 4.67 (ddd, 1H, J = 12.0, 7.6 and 5.6 Hz), 5.07 (ddd, 1H, J = 7.6, 3.2 and 2.0 Hz), 6.24 (dd, 1H, J= 4.0 and 2.0 Hz), 7.37 (t, 2H, J = 7.2 Hz), 7.47 (tt, 1 H, J = 7.2 and 1.6 Hz), and 7.80 (dd, 2H, J = 7.2 and 1.6 Hz); 13C-NMR (CDCl3, 100 MHz) δ 28.1, 31.1, 33.8, 36.9, 73.7, 74.6, 84.8, 104.9, 128.0, 131.8, 135.0, 142.5, 148.9, and 172.6; HRMS Calcd. for C19H22BNO5: 355.1591. Found: 355.1593.
tert-Butyl 2,3,3a,4,5,6-hexahydro-3a-methyl-2-oxo-5,6-phenylboronate-indole-1-carboxylate (54)
To a solution containing 0.05 g (0.19 mmol) of oxabicycle 24, 2 mg (0.003 mmol) of [Rh(COD)Cl]2,1.0.μL (0.02 mmol) of P(OEt)3 in 1 mL THF was added 0.035 g (0.28 mmol) of phenyl boronic acid. The reaction mixture was heated at 65 °C for 2 h, cooled to rt and quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layer was washed with a saturated aqueous NaHCO3 solution and dried over MgSO4. The solvent was removed under reduced pressure and the residue was subjected to the flash silica gel chromatography to afford 0.06 g (95%) of 54 as a white solid: mp 136–137 °C; IR (neat) 1771, 1735, 1369, 1153, and 692 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.23 (s, 3H), 1.56 (t, 1H, J = 12.0 Hz), 1.59, (s, 9H), 2.37 (d, 1H, J = 16.4 Hz), 2.39 (dd, 1H, J = 12.0 and 6.0 Hz), 2.45 (d, 1H, J = 16.4 Hz), 4.89 (ddd, 1H, J = 12.0, 7.6 and 6.0 Hz), 5.13 (dd, 1H, J = 7.6 and 3.2 Hz), 6.21 (d, 1 H, J = 3.2 Hz), 7.38 (t, 2H, J = 7.6 Hz), 7.48 (tt, 1 H, J = 7.6 and 1.6 Hz), and 7.80 (dd, 2H, J = 7.6 and 1.6 Hz); 13C-NMR (CDCl3, 100 MHz) δ 25.3, 28.2, 36.6, 40.0, 46.3, 73.3, 73.6, 84.8, 104.8, 128.0, 131.9, 135.0, 146.1, 149.3, and172.0;Anal. Calcd. forC20H24BNO5: C, 65.06; H, 6.55; N, 3.79. Found: C, 64.80; H, 6.57; N, 3.73.
tert-Butyl 2,3,3a,4,5,6-hexahydro-5,6-acetonide-2-oxoindole-1-carboxylate (55)
To a solution containing 0.1 g (0.39 mmol) of oxabicycle 23 in acetone was added 5 mg (0.03 mmol) of anhydrous stannous chloride. After stirring at rt for 15 min, the solvent was removed under reduced pressure and the residue was subjected to the flash silica gel chromatography to afford 0.11 g (91%) of 55 as a white solid: mp 103–105 °C; IR (neat) 1794, 1731, 1370, and 727 cm−1; 1H-NMR (CDCl3, 400 MHz) δ 1.39 (s, 3H), 1.45 (q, 1 H, J = 11.6 Hz), 1.48 (s, 3H), 1.55 (s, 9H), 2.17 (ddd, 1 H, J = 11.6, 5.6 and 4.0 Hz), 2.25–2.33 (m, 1 H), 2.62–2.69 (m, 2H), 4.22 (dt, 1H, J = 11.6 and 5.6 Hz), 4.68–4.71 (m, 1H), and 6.09 (dd, 1H, J = 4.0 and 2.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 25.9, 28.1, 28.5, 32.1, 32.8, 37.1, 71.5,73.1,84.6, 103.4, 109.2, 143.1, 148.9, and 172.9; Anal. Calcd. for C16H23NO5: C, 62.12; H, 7.49; N, 4.53. Found: C, 61.96; H, 7.46; N, 4.53.
tert-Butyl 2,3,3a,4,5,6-hexahydro-3a-methyl-5,6-acetonide-2-oxoindole-1-carboxylate (56)
To a solution containing 0.1 g (0.38 mmol) of oxabicycle 24 in 2 ml of acetone was added 5 mg (0.03 mmol) of anhydrous stannousl chloride. After stirring at rt for 15 min, the solvent was removed under reduced pressure and the residue was subjected to the flash silica gel chromatography to afford 0.11 g (92%) of 56 as a white solid: mp 118–120 °C; 1H-NMR (CDCl3, 400 MHz) δ 1.18 (s, 3H), 1.40 (s, 3H), 1.48 (s, 3H), 1.56 (s, 9H), 1.51 –1.58 (m,1 H), 2.11 (dd, 1H, J = 12.4 and 6.0 Hz), 2.32 (d, 1H, J = 16.4 Hz), 2.42 (d, 1H, J = 16.4 Hz), 4.44 (dt, 1H, J = 12.0 and 6.0 Hz), 4.73 (dd, 1H, J = 6.0 and 4.0 Hz), and 6.05 (d, 1H, J =4.0 Hz); 13C-NMR (CDCl3, 100 MHz) δ 25.4, 25.6, 28.2, 28.3, 34.5, 38.8, 46.7, 71.1, 71.6, 84.6, 103.4, 109.1, 146.6, 149.3, and 172.4; HRMS Calcd. for C17H25NO5: 323.1733. Found: 323.1735.
Supplementary Material
Supporting Information Available: 1H and 13C NMR data of various key compounds lacking CHN analyses together with an ORTEP drawing for compounds 41, 51 and 53 as well as the corresponding CIFs for these compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We appreciate the financial support provided by the National Science Foundation (Grant No. CHE-0450779) and the National Institutes of Health (GM 059384). We thank our colleague, Dr. Kenneth Hardcastle, for his assistance with the X-ray crystallographic studies.
References
- 1.(a) Lautens M. Synlett. 1993:177. [Google Scholar]; (b) Chiu P, Lautens M. Top Curr Chem. 1997;190:1. [Google Scholar]; (c) Lautens M, Fagnou K, Hiebert S. Acc Chem Res. 2003;36:48. doi: 10.1021/ar010112a. [DOI] [PubMed] [Google Scholar]
- 2.(a) Vogel P, Fattori D, Gasparini F, Le Drian C. Synlett. 1990:173. [Google Scholar]; (b) Reymond J-L, Pinkerton AA, Vogel P. J Org Chem. 1991;56:2128. [Google Scholar]
- 3.(a) Renaud P, Vionnet J-P. J Org Chem. 1993;58:5895. [Google Scholar]; (b) Dean FM. Adv Heterocycl Chem. 1981;30:168. [Google Scholar]; (c) Lipshutz BH. Chem Rev. 1986;86:795. [Google Scholar]; (d) Woo S, Keay BA. Synthesis. 1996:669. [Google Scholar]
- 4.Eggelte TA, de Koning H, Huisman HO. J Chem Soc, Perkin Trans 1. 1978:980. [Google Scholar]
- 5.Ogawa S, Iwasawa Y, Nose T, Suami T, Ohba S, Ito M, Saito Y. J Chem Soc, Perkin Trans 1. 1985:903. [Google Scholar]
- 6.Just G, Kim S. Tetrahedron Lett. 1976;17:1063. [Google Scholar]
- 7.Murai A, Takahashi K, Taketsuru H, Masamune T. J Chem Soc, Chem Commun. 1981:221. [Google Scholar]
- 8.Kotsuki H, Nishizawa H. Heterocycles. 1981;16:1287. [Google Scholar]
- 9.Cox PJ, Simpkins NS. Synlett. 1991:321. [Google Scholar]
- 10.(a) Le Drian C, Vieira E, Vogel P. Helv Chim Acta. 1989;72:338. [Google Scholar]; (b) Takahashi T, lyobe A, Arai Y, Koizumi T. Synthesis. 1989:189. [Google Scholar]; (c) Guildford AJ, Turner RW. J Chem Soc, Chem Commun. 1983:466. [Google Scholar]; (d) Yang W, Koreeda M. J Org Chem. 1992;57:3836. [Google Scholar]
- 11.(a) Suami T. Pure Appl Chem. 1987;59:1509. [Google Scholar]; (b) Harwood LM, Jackson B, Prout K, Witt FJ. Tetrahedron Lett. 1990;31:1885. [Google Scholar]; (c) Koreeda M, Jung K-Y, Hirota M. J Am Chem Soc. 1990;112:7413. [Google Scholar]; (d) Reynard E, Reymond J-L, Vogel P. Synett. 1991:469. [Google Scholar]; (e) Ogawa S, Yoshikawa M, Taki T. J Chem Soc, Chem Commun. 1992:406. [Google Scholar]; (f) Ogawa S, Tsunoda H. Liebigs Ann Chem. 1992:637. [Google Scholar]
- 12.(a) Jung ME, Street LJ. J Am Chem Soc. 1984;106:8327. [Google Scholar]; (b) Mirsadeghi S, Rickborn B. J Org Chem. 1985;50:4340. [Google Scholar]
- 13.Grieco PA, Lis R, Zelle RE, Finn J. J Am Chem Soc. 1986;108:5908. doi: 10.1021/ja00279a041. [DOI] [PubMed] [Google Scholar]
- 14.(a) Takayama H, Hayashi K, Koizumi T. Tetrahedron Lett. 1986;27:5509. [Google Scholar]; (b) Hanessian S, Beaulieu P, Dube D. Tetrahedron Lett. 1986;27:5071. [Google Scholar]
- 15.(a) Arjona O, de Dios A, Fernandez de la Pradilla R, Plumet J, Viso A. J Org Chem. 1994;59:3906. [Google Scholar]; (b) Lautens M, Dockendorff C, Fagnou K, Malicki A. Org Lett. 2002;4:1311. doi: 10.1021/ol0256062. [DOI] [PubMed] [Google Scholar]
- 16.(a) Lautens M, Fagnou K, Rovis T. J Am Chem Soc. 2000;122:5650. [Google Scholar]; (b) Lautens M, Fagnou K, Taylor M. Org Lett. 2000;2:1677. doi: 10.1021/ol005729r. [DOI] [PubMed] [Google Scholar]; (c) Lautens M, Fagnou K. Tetrahedron. 2001;57:5067. [Google Scholar]; (d) Lautens M, Chiu P, Ma S, Rovis T. J Am Chem Soc. 1995;117:532. [Google Scholar]; (e) Lautens M, Chiu P, Colucci JT. Angew Chem, Int Ed Engl. 1993;32:281. [Google Scholar]; (f) Lautens M, Di Felice C, Huboux A. Tetrahedron Lett. 1989;30:6817. [Google Scholar]
- 17.Kappé CO, Murphree SS, Padwa A. Tetrahedron. Vol. 53. 1997. p. 14179. [Google Scholar]
- 18.Sargent MV, Dean FM. In: Comprehensive Heterocyclic Chemistry. 4. Katritzky AR, Rees CW, editors. Pergamon; Oxford: 1984. p. 599. [Google Scholar]
- 19.(a) Moore JA, Partain EM., III J Org Chem. 1983;48:1105. [Google Scholar]; (b) Bailey MS, Brisdon BJ, Brown DW, Stark KM. Tetrahedron Lett. 1983;24:3037. [Google Scholar]
- 20.Moursounidis J, Wege D. Aust J Chem. 1983;36:2473. [Google Scholar]
- 21.Laszlo P. Ace Chem Res. 1986;19:121. [Google Scholar]
- 22.Dolata DP, Bergman R. Tetrahedron Lett. 1987;28:707. [Google Scholar]
- 23.Saksena AK, Girijavallabhan VM, Chen Y-T, Jao E, Pike RE, Desai JA, Rane D, Ganguly AK. Heterocycles. 1993;35:129. [Google Scholar]
- 24.Matsumoto K, Sera A. Synthesis. 1985:999. [Google Scholar]
- 25.Oppolzer W. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, editors. Vol. 5. Pergamon; Oxford: 1991. p. 315. [Google Scholar]
- 26.Keay BA, Hunt IR. Advances in Cycloaddition. 1999;6:173. [Google Scholar]
- 27.(a) Roush WR. Advances in Cycloaddition. 1990;2:91. [Google Scholar]; (b) Fallis AG. Can J Chem. 1984;62:183. [Google Scholar]; (c) Craig D. Chem Soc Rev. 1987;16:187. [Google Scholar]
- 28.(a) Padwa A, Brodney MA, Dimitroff M. J Org Chem. 1998;63:5304. doi: 10.1021/jo010020z. [DOI] [PubMed] [Google Scholar]; (b) Padwa A, Brodney MA, Satake K, Straub CS. J Org Chem. 1999;64:4617. doi: 10.1021/jo982061+. [DOI] [PubMed] [Google Scholar]
- 29.(a) Sternbach DD, Rossana DM, Onan KD. J Org Chem. 1984;49:3427. [Google Scholar]; (b) Klein LL. J Org Chem. 1985;50:1770. [Google Scholar]; (c) Jung ME, Gervay J. J Am Chem Soc. 1989;111:5469. [Google Scholar]
- 30.(a) Padwa A, Brodney MA, Lynch SM. J Org Chem. 2001;66:1716. doi: 10.1021/jo0014109. [DOI] [PubMed] [Google Scholar]; (b) Padwa A, Brodney MA, Dimitroff M, Liu B, Wu T. J Org Chem. 2001;66:3119. doi: 10.1021/jo010020z. [DOI] [PubMed] [Google Scholar]
- 31.Padwa A, Ginn JD, Bur SK, Eidell CK, Lynch SM. J Org Chem. 2002;67:3412. doi: 10.1021/jo0111816. [DOI] [PubMed] [Google Scholar]; (b) Wang Q, Padwa A. Org Lett. 2004;6:2189. doi: 10.1021/ol049348f. [DOI] [PubMed] [Google Scholar]
- 32.(a) Bur SK, Lynch SM, Padwa A. Org Lett. 2002;4:473. doi: 10.1021/ol016804g. [DOI] [PubMed] [Google Scholar]; (b) Ginn JD, Padwa A. Org Lett. 2002;4:1515. doi: 10.1021/ol025746b. [DOI] [PubMed] [Google Scholar]
- 33.For examples: see Oppolzer W, Fröstl W. Helv Chim Acta. 1975;58:590. doi: 10.1002/hlca.19750580232.Oppolzer W, Fröstl W, Weber HP. Helv Chim Acta. 1975;58:593. doi: 10.1002/hlca.19750580232.White JD, Demnitz FWJ, Oda H, Hassler C, Snyder JP. Org Lett. 2000;2:3313. doi: 10.1021/ol000200f.Tantillo DJ, Houk KN, Jung ME. J Org Chem. 2001;66:1938. doi: 10.1021/jo001172h.Weingarten MD, Prein M, Price AT, Snyder JP, Padwa A. J Org Chem. 1997;62:2001. doi: 10.1021/jo962184z.
- 34.Jung and Gervay, however, reported data for intramolecular Diels-Alder reactions which suggest that substitution on the tether results in an increase in the population of reactive rotamers. The rate enhancement was suggested to originate from a restricted rotation, producing a lowest energy ground state conformer that is more energetically similar to the reactive conformer, see: Jung ME, Gervay J. J Am Chem Soc. 1991;113:224.See also: Sternbach DD, Rossana DM, Onan KD. Tetrahedron Lett. 1985;26:591.Giessner-Prettre C, Hükel S, Maddaluno J, Jung ME. J Org Chem. 1997;62:1439.
- 35.(a) Woo S, Keay B. Tetrahedron:Asymmetry. 1994;5:1411. [Google Scholar]; (b) Rogers C, Keay BA. Tetrahedron Lett. 1991;32:6477. [Google Scholar]; (c) Rogers C, Keay BA. Can J Chem. 1992;70:2929. [Google Scholar]; (d) De Clercq PJ, Van Royen LA. Synth Commun. 1979;9:771. [Google Scholar]; (e) Van Royen LA, Mijngvheer R, De Clercq PJ. Bull Soc Chim Belg. 1984;93:1019. [Google Scholar]; (f) Fischer K, Hunig S. J Org Chem. 1987;52:564. [Google Scholar]
- 36.(a) Vogel P, Cossy J, Plumet J, Arjona O. Tetrahedron. 1999;55:13521. [Google Scholar]; (b) Vogel P. Bull Soc Chim Belg. 1990;99:395. [Google Scholar]
- 37.Baker RW, Baker TM, Birkbeck AA, Giles RGF, Sargent MV, Skelton BW, White AH. J Chem Soc, Perkin Trans 1. 1991:1589. and references cited therein. [Google Scholar]
- 38.(a) Arjona O, Borrallo C, Iradier F, Medel R, Plumet J. Tetrahedron Lett. 1998;39:1977. [Google Scholar]; (b) Arjona O, Fernandez de la Pradilla R, Garcia E, Martin-Domenech A, Plumet J. Tetrahedron Lett. 1989;30:6437. [Google Scholar]; (c) Caple R, Chen GM-S, Nelson JD. J Org Chem. 1971;36:2874. [Google Scholar]; (d) Jeffrey AM, Yeh HJC, Jerina DM, DeMarinis RM, Foster CH, Piccolo DE, Berchtold GA. J Am Chem Soc. 1974;96:6929. [Google Scholar]
- 39.(a) Lautens M, Belter RK. Tetrahedron Lett. 1992;33:2617. [Google Scholar]; (b) Lautens M, Chiu P. Tetrahedron Lett. 1991;32:4827. [Google Scholar]; (c) Lautens M, Kumanovic S. J Am Chem Soc. 1995;117:1954. [Google Scholar]
- 40.With the oxabenzonorbornene system studied by the Lautens’ group, 16 the rhodium metal undergoes initial exo-coordination with the Π-bond and this is followed by C-O insertion and then a subsequent displacement of the allyl rhodium species via endo attack of the nucleophile.
- 41.(a) Hayashi T. Synlett. 2001:879. [Google Scholar]; (b) Sakai M, Hayashi H, Miyaura N. Organometallics. 1997;16:4229. [Google Scholar]; (c) Senda T, Ogasawara M, Hayashi T. J Org Chem. 2001;66:6852. doi: 10.1021/jo0103930. [DOI] [PubMed] [Google Scholar]
- 42.Takaya Y, Ogasawara M, Hayashi T, Sakai M, Miyaura N. J Am Chem Soc. 1998;120:5579. [Google Scholar]
- 43.Hayashi T, Senda T, Takaya Y, Ogasawara M. J Am Chem Soc. 1999;727:11591. [Google Scholar]
- 44.Hayashi T, Senda T, Ogasawara M. J Am Chem Soc. 2000;122:10716. [Google Scholar]
- 45.Murakami M, Igawa H. Chem Commun. 2002:390. doi: 10.1039/b108808d. [DOI] [PubMed] [Google Scholar]
- 46.Bertounesque E, Florent J-C, Monneret C. Synthesis. 1991:270. [Google Scholar]
- 47.Vyvyan JR, Meyer JA, Meyer KD. J Org Chem. 2003;68:9144. doi: 10.1021/jo035112y. [DOI] [PubMed] [Google Scholar]
- 48.Smith AB, III, Toder BH, Branca SJ, Dieter RK. J Am Chem Soc. 1981;103:1996. [Google Scholar]
- 49.Itoh K, Harada T, Nagashima H. Bull Chem Soc Jpn. 1991;64:3746. [Google Scholar]
- 50.Miyashi T, Nishizawa Y, Fujii Y, Yamakawa K, Kamata M, Akao S, Mukai T. J Am Chem Soc. 1986;108:1617. [Google Scholar]
- 51.Burness DM. Org Syn Coll. 1963;4:649. [Google Scholar]
- 52.Harayama T, Cho H, Inubushi Y. Chem Pharm Bull. 1978;26:1201. [Google Scholar]
- 53.Padwa A, Brodney MA, Liu B, Satake K, Wu T. J Org Chem. 1999;64:3595. doi: 10.1021/jo982453g. [DOI] [PubMed] [Google Scholar]
- 54.Friedrich LE, Cormier RA. J Org Chem. 1971;36:3011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: 1H and 13C NMR data of various key compounds lacking CHN analyses together with an ORTEP drawing for compounds 41, 51 and 53 as well as the corresponding CIFs for these compounds. This material is available free of charge via the Internet at http://pubs.acs.org.