

Abstract
Background
In allograft rejection, recipient leukocytes and alloantibodies first target donor endothelial cells. Although the leukocyte integrin Mac-1 (αMβ2, CD11b/CD18) facilitates cell-cell interactions among leukocytes and interactions between leukocytes and endothelial cells or platelets, its role in allograft survival and vasculopathy is incompletely defined.
Methods and Results
This study examined parenchymal rejection and graft arterial disease after total-allomismatched cardiac transplantation (BALB/c donor heart and B6 recipients) in wild-type (WT) and Mac-1-deficient (Mac-1−/−) recipients. Recipient Mac-1 deficiency attenuated parenchymal rejection and significantly prolonged cardiac allograft survival from 8.3 ± 1.3 days in WT recipient allografts (n=18) to 13.8 ± 2.3 days in Mac-1−/− recipient allografts (n=6), p<0.0001). Accumulation of neutrophils and macrophages significantly decreased in Mac-1−/− compared to WT recipients. Adoptive transfer of WT but not Mac-1−/− macrophages to Mac-1−/− recipients exacerbated parenchymal rejection and reduced allograft survival; in contrast, adoptive transfer of WT neutrophils did not affect graft survival. Mac-1−/− macrophages expressed significantly lower levels of co-stimulatory molecules both in vivo and in vitro, and mixed lymphocyte reaction using allo-antigen-primed Mac-1−/− macrophages resulted in significantly lower antigen-presenting function than for WT macrophages. Tumor necrosis factor-alpha (TNF-α) production also fell in cultures with Mac-1−/− macrophages. Despite attenuation of acute rejection, recipient Mac-1-deficiency did not prevent late graft arterial disease.
Conclusion
These studies demonstrate critical participation of Mac-1 in alloresponses during cellular allograft rejection. These observations establish a molecular target for modulating recipient responses to prolong graft survival.
Keywords: CD11b/CD18 (Mac-1), T-lymphocytes, macrophages, antigen presenting cells, cytokine, heart transplantation, pathogenesis
INTRODUCTION
In allografts, recipient leukocytes and alloantibodies attack donor endothelial cells early in acute rejection. Cell-cell interactions among leukocytes and interactions between leukocytes and endothelial cells or platelets critically influence immune responses. These adhesive interactions require multi-step adhesive and signaling events including selectin-mediated attachment and rolling, leukocyte activation, and integrin-mediated firm adhesion and diapedesis that result in the infiltration and accumulation of inflammatory cells into tissues.1 Firm attachment depends on members of the β2-integrin family, LFA-1 (αLβ2, CD11a/CD18), Mac-1 (αMβ2, CD11b/CD18), p150,95 (αMβ2, CD11c/CD18), and CD11d/CD18, expressed exclusively by hematopoietic cells.
Formation of the immunologic synapse requires direct interaction between T cell integrins and cognate ligands on antigen-presenting cells (APCs).2 LFA-1 binding to ICAM-1 reduces the level of antigen required to form the immune synapse, thereby lowering B-cell activation thresholds.3 Engagement of ICAM-1 on target cells leads to major histocompatibility complex class I (MHC-I) recruitment to contact areas and enhances presentation of cognate peptide/MHC-I complexes to cytotoxic T cells.4
Mac-1 resides on neutrophils, monocytes, macrophages, dendritic cells, CD8+ T cells, and natural killer (NK) cells.5–7 Endothelial ligands for Mac-1 include ICAM-1, 8–10 endothelial-associated extracellular matrix proteins (e.g., fibrinogen),11–13 CD154,14 and glycosaminoglycans.15 These adhesive interactions facilitate firm attachment and transmigration into interstitial tissue.16, 17
Numerous studies have shown that the donor endothelial cells in rejecting cardiac allografts robustly express ICAM-1, and that augmented expression of adhesion molecules precedes leukocyte accumulation within vessels.18–20 These results suggest that ICAM-1 induction during early rejection contributes to mononuclear cell recruitment and renders endothelial cells more susceptible to cell-mediated injury, including graft arterial disease.20, 21 Nevertheless, it remains unclear whether donor adhesion molecules contribute critically to allograft rejection. Treatment with monoclonal antibodies against ICAM-1 and LFA-1 resulted in indefinite cardiac allograft survival between fully histo-incompatible mice strains,22 but had no effect on allograft survival in fully incompatible rat strains.23 In one study, ICAM-1-deficient donor hearts did not exhibit prolonged graft survival or attenuated graft arterial disease;24 in another report, ICAM-1-deficient donor hearts had modest (2–2.5 days), albeit significantly prolonged, graft survival.25
A definitive role for Mac-1 in allograft rejection is uncertain. Antibody targeting of Mac-1 did not affect graft survival time, but did decrease the accumulation of graft macrophages.26 However, antibody experiments have several important limitations: 1) complete blockade is difficult, particularly for those integrins with mobilizable intracellular storage pools, such as Mac-127; 2) antibodies binding to the surface of leukocytes can stimulate changes in cellular functions and behavior (e.g., through activation of Fc receptors when IgG antibodies are used); and 3) since Mac-1 associates closely with and influences other cell surface receptors, such as the urokinase receptor,28 antibody binding may cause steric interference with the associated receptor. The present study avoided these limitations of antibody administration by use of a genetic approach, examining parenchymal rejection and graft arterial disease after cardiac transplantation in Mac-1−/− recipients.
METHODS
Animals
C57BL/6 (B6; H-2b), B6.C-H2<bm12>KhEg (bm12; H-2bm12), and BALB/c (B/c) mice were from Taconic Farm or Jackson Laboratory. Mac-1−/− C57BL/J6 mice were kindly provided by Dr. Christie M. Ballantyne (Baylor College of Medicine).29 Mice were maintained on acidified water in barrier animal facilities. Animal care and procedures were reviewed and approved by the Harvard Medical School Standing Committee on Animals and performed in accordance with the guidelines of the American Association for Accreditation of Laboratory Animal Care and the National Institutes of Health.
Vascularized heterotopic cardiac transplantation
B/c (total allo-mismatch, to assess effects on acute rejection) or bm12 (MHC class II-mismatch, to assess effects on GAD) donor hearts were transplanted heterotopically into B6 recipients without immunosuppression.30 We assessed graft function by palpation twice daily. In total allo-mismatched experiments, 2 cohorts of WT and Mac-1−/− recipients were used. The first group of animals served to determine graft survival—defined as the day of cessation of beating or when the strength of beating declined to the point that failure would occur (by prior experience) within the next 12 hours. In the second group of animals, beating cardiac allografts were harvested at the pre-specified time point of 7 d after transplantation. For single MHC class II-mismatched transplantation, grafts were harvested at 4 weeks and 12 weeks.
Parenchymal rejection and the severity and extent of GAD were quantified using a 4-point scoring system by blinded observers, as described previously.30 PR was scored on a scale of 0 to 4 (0: no rejection, 1: focal nononuclear cell infiltrates, 2: focal mononuclear infiltrates with necrosis, 3: multifocal infiltrates with necrosis, and 4: widespread infiltrates with hemorrhage and/or vasculitis) and GAD on a scale of 0 to 4 based on the extent of luminal stenoses averaged over ≥ 10 arteries (0: <10%, 1: < 10–25%, 2: 25–50%, 3: 50–75%, and 4: >75% stenosis).
Graft harvest
Harvested allografts were transversely sectioned into three parts. In sectioned hearts, the most basal part was used for routine hematoxylin and eosin morphological examination. A second mid-transverse section was frozen for immunohistochemical staining, and the apical portion was used for total RNA extraction for RNase Protection assay (RPA).30 For cellular extraction, hearts were digested at 37°C in 2 mg/mL collagenase (Sigma) and 2% bovine serum albumin in buffered saline, followed by straining, and Ficoll density gradient centrifugation (Organon Teknika).30
Immunohistochemistry
Five µm cryosections were fixed in acetone before incubation with 0.5% H2O2. Slides were incubated with 10% normal goat serum and stained with purified monoclonal anti-CD4 (RM4-5), anti-CD8 (53-6.7), anti-Mac-3 (macrophage), or anti-Gr-1 (neutrophil, RB6-8C5) antibodies. Sections were incubated with biotinylated goat anti-rat IgG antibodies (1 µg/ml, Southern Biotechnology Associates, Inc.), followed by streptavidin-peroxidase (DAKO). Antibody binding was visualized with 3-amino-9-ethyl carbazole (DAKO). Nuclei were counter-stained with Gill’s hematoxylin (Sigma).
Mixed lymphocyte reaction (MLR), T cell proliferation assay, and cytokine ELISA
One-way MLR was performed as described previously,30 using irradiated BALB/c splenocytes as stimulators and B6 splenocytes as responders. T cell or B cell proliferation assay was performed using anti-CD3 Ab-coated 96 well plates or by adding anti-CD40 mAbs (3/23, BD PharMingen).
We also performed MLR using allograft-derived macrophages as stimulators and naïve B6 T cells as responders. We isolated macrophages from the allografts of B6 WT or B6 Mac-1−/− recipient 7 days after transplantation as described previously.30 We extracted T cells from naïve B6 WT splenocytes using MACS beads (Miltenyi Biotec Inc., Auburn, CA) with negative selection according to the manufacturer’s methods. We plated irradiated allograft-derived macrophages (50,000 cells/well) and naive B6 T cells (500,000 cells/well, S:R=1:10) in the 96 well plates and measured T cell proliferation using 3H-thymidine incorporation.30 We also measured TNF-alpha levels in the supernatant of the co-culture by ELISA as described previously,30 using purified anti-TNF-alpha and biotinylated anti-TNF-α mAbs (BD PharMingen).
Total RNA extraction and RNase Protection Assay
Total RNA was isolated from allografts with TRIzol™ reagent (Invitrogen). RPA was performed (RiboQuant, PharMingen) using 10 µg of RNA from each heart according to the manufacturer’s recommendations.30
Murine peritoneal macrophage and bone marrow neutrophil isolation
Peritoneal cells were collected via peritoneal lavage using 10 ml of ice-cold RPMI 1640. Lavage cells were pelleted and resuspended in 5 ml of hypertonic RBC lysis buffer (ACK lysing buffer, Invitrogen) at 37°C for 5 min and washed with RPMI 1640 twice and resuspended in 5 ml of RPMI 1640. Cells were centrifuged in Percoll gradient solution for 30 min.31 Cells in the upper layer were resuspended in 10% FCS RPMI 1640 and plated on plastic dishes for 60 min and washed twice to remove nonadherent cells, resulting in > 95 % pure macrophages, confirmed by Mac-3 staining and flow cytometry. To isolate neutrophils from the bone marrow, we removed the femur and the tibia from both hind legs and cut off the extreme distal tip of each extremity. RPMI 1640 medium was forced through the bone with a syringe. After dispersing cell clumps and erythrolysis, the cell suspension was centrifuged (400 g, 10 min, 4°C) and resuspended in 1 ml RPMI 1640. Cells were then treated on a three-layer Percoll gradient exactly as described above for peritoneal cells. Flow cytometry using anti-Ly-6G and Gr-1 antibodies showed > 95% purity of neutrophils.
Murine endothelial cell isolation and co-culture with macrophages
Endothelial cells (ECs) were isolated from the B/c murine hearts as described previously32, and incubated for 72 hours with the peritoneal macrophages extracted from naÏve B6 WT or Mac-1−/− mice. We performed flow cytometry analysis for co-stimulatory molecule expression of the B/c EC-primed macrophages as described below.
Cellular surface staining and flow cytometric analysis
Graft infiltrating cells and B/c-EC-primed macrophages were analyzed by flow cytometry after surface staining using methods described previously30; antibodies included anti-MHC II-PE (I-A/I-E, M5/114.15.2), anti-CD40-PE, CD86-PE, or ICOSL-PE antibodies, PE-conjugated rat IgG2b (negative control, PharMingen), or anti-CD80-PE and PE-conjugated hamster IgG (negative control); anti-MAC-3 antibody was used for macrophage staining.
Statistical analysis of the graft survival and cell number, proliferation, and cytokine production
Comparisons between two groups for graft survival used Kaplan-Meier and Log-rank test; frequency of graft infiltrating cells, cell proliferation, and ELISA used one-way analysis of variance. We described PR and GAD scores as mean ± SD and analyzed them by the Fisher’s exact test using the GraphPad Prism 4 software for Macintosh.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
RESULTS
Deficiency of recipient Mac-1 attenuates parenchymal rejection and prolongs graft survival in total-allomismatched allografts
In total-allomismatched murine cardiac allografts (BALB/c donor hearts and B6 recipients), survival averaged 13.8 ± 2.3 days (mean ± SD, n=6) for allografts in Mac-1−/− recipients compared to 8.3 ± 1.3 days (n=18) in WT recipients (Figure 1A). Failing grafts in WT recipients showed severe coronary arteritis with perivascular edema and confluent areas of myocardial necrosis 7 d after transplantation. In comparison, at the same time point, grafts in Mac-1−/− recipients exhibited virtually no arterial inflammation, edema, or coagulative necrosis, despite the presence of multi-focal parenchymal inflammatory infiltrates (Figure 1B). Reflecting these differences, the PR scores were significantly lower for allografts in Mac-1−/− recipients 7 d after transplantation (2.3 ± 0.4, mean ± SD, n=6) compared to WT recipients (3.0 ± 0.4, n=13, pp=0.0408). At this 7 d time point, inflammatory cells and thrombi populated vessel luminal areas in allografts of WT recipients. These lesions differ from chronic GAD lesions that consist predominantly of smooth muscle–like cells.33 Nevertheless, we scored the extent of early luminal occlusion using the GAD scoring system. Scores were significantly lower for grafts in Mac-1−/− (0.01 ± 0.02, mean ± SD, n=6) compared to WT (0.63 ± 0.44, n=13, p =0.0108) recipients (Figure 1C). Thus, deficiency of recipient Mac-1 prolongs allograft survival and is associated with diminished inflammatory cell accumulation including reduced luminal inflammatory cells. At the time of eventual graft failure in Mac-1−/− recipients (13.8 ± 2.3 days), PR scores were 3.2 ± 0.9, with luminal stenosis graded at 1.3 ± 1.1.
Figure 1. Recipient Mac-1 deficiency attenuates acute parenchymal rejection and prolongs graft survival after total-allomismatched transplantation.
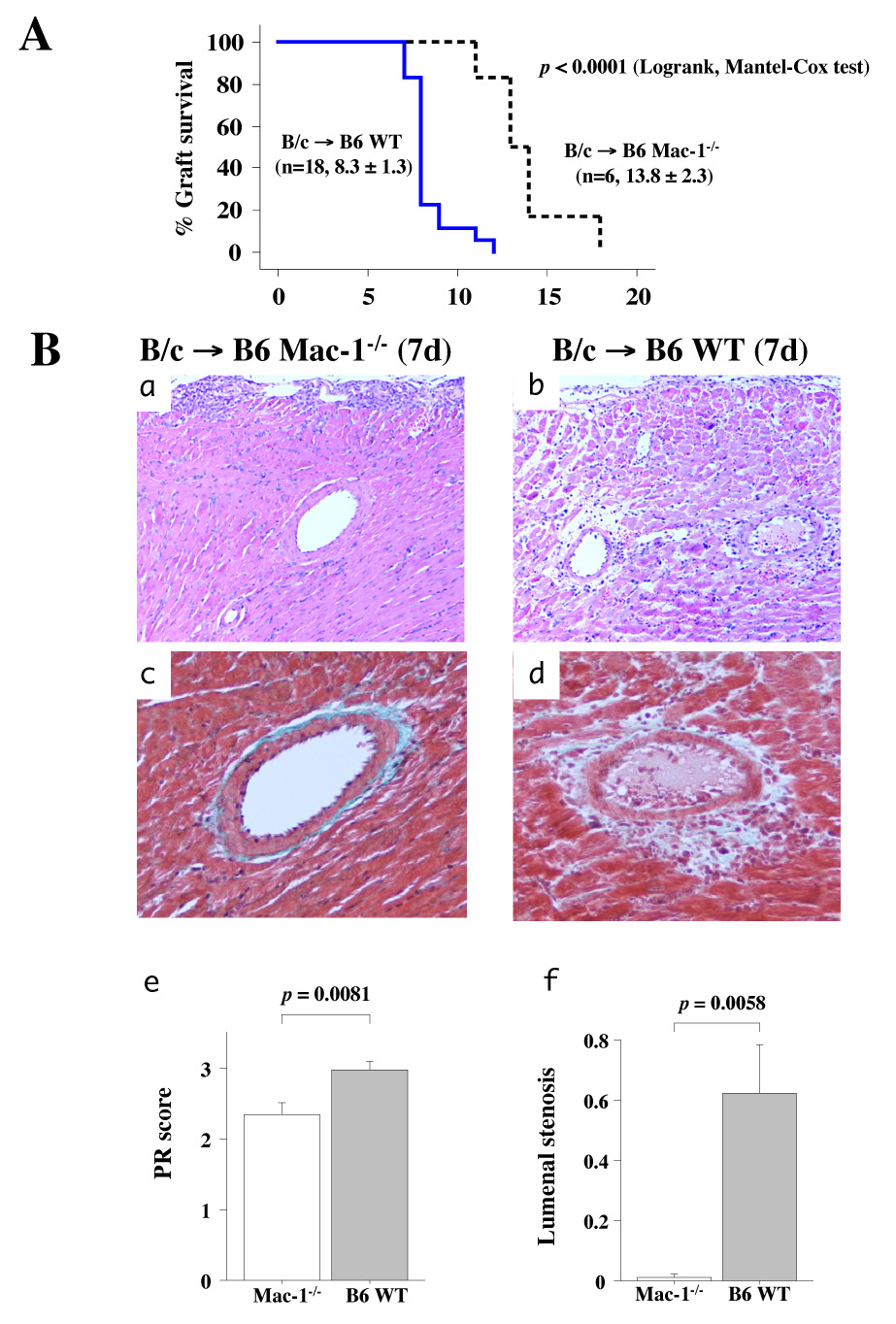
A, Kaplan-Meier analysis of graft survival after total-allomismatched heart transplantation in WT (n=18) and Mac-1−/− (n=6) recipients. B, Photomicropgraphs of B/c cardiac allografts harvested 7 d after transplantation in WT and Mac-1−/− B6 recipients: H & E staining (a, b), Masson Trichrome staining (c, d). PR (e) and luminal occlusion (f) scores (mean ± SD) were determined as described in Methods from hearts harvested from WT (solid bars, n=13) and Mac-1−/− (open bars, n=6) recipients.
Mac-1-deficiency reduces the intragraft accumulation of neutrophils, CD8+ T lymphocytes, and macrophages
Immunohistochemical analysis examined the characteristics of the immune cell infiltration in transplanted hearts in WT and Mac-1−/− recipients 7 d after allografting. Anti-Gr-1 identified neutrophils, anti-CD4 or anti-CD8 for T cells, and anti-Mac-3 for macrophages. Neutrophil (Gr-1-positive cells), CD8+ T cell, and macrophage (Mac-3-positive cells) accumulation all decreased significantly in Mac-1−/− compared to WT recipients. The transplanted hearts of both Mac-1−/− and WT recipients had comparable numbers of CD4+ T cells (Figure 2A, B). To verify that differences in leukocyte recruitment in Mac-1−/− mice were not secondary to differences in peripheral blood leukocyte counts, we performed complete blood count analysis in WT and Mac-1−/− mice (male 10–12 weeks old) using HEMAVET®850 (CBC Technologies).There were no statistical differences in the number of peripheral blood neutrophils (2.12 ± 0.67 vs. 2.10 ± 0.53 × 103/µL, mean ± SD, p=.96), monocytes (0.11 ± 0.06 vs. 0.09 ± 0.04 × 103/µL, p=0.53), and lymphocytes (6.85 ± 0.67 vs. 6.52 ± 0.28, p=0.40) between WT (n=4) and Mac-1−/− (n=4), respectively.
Figure 2. Accumulation of neutrophils, T lymphocytes, and macrophages 7 d after transplantation.
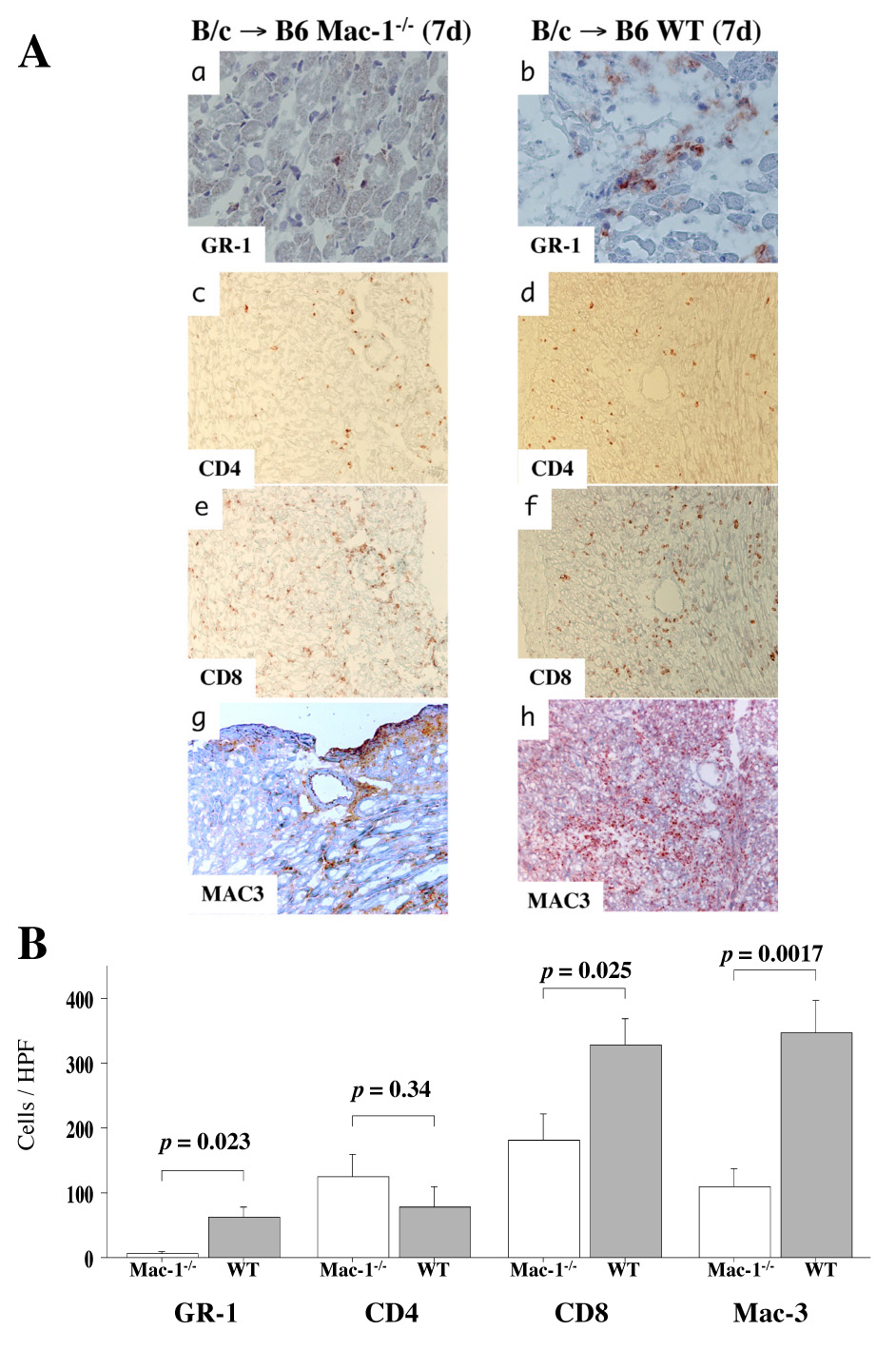
A, Immunohistochemistry was performed to examine immune cell infiltration into transplanted hearts in WT and Mac-1−/− recipients 7 d after transplantation. Anti-Gr-1 staining was used to identify neutrophils (a, b), anti-CD4 (c, d) or anti-CD8 e, f) for T cells, and anti-Mac-3 (g, h) for macrophages. B, Immune cell accumulation was quantified (mean ± SD) by determining the average number of cells per high power field (x 100) in WT (n=13, closed bars) and Mac-1−/− (n=6, open bars) recipients.
WT macrophage but not neutrophil adoptive transfer reduces allograft survival in Mac-1−/− recipients
Neutrophils and monocytes/macrophages can both participate in acute graft failure. Deficiency of Mac-1 reduces the recruitment of both neutrophils and monocytes/macrophages at sites of vascular injury.34 To determine the cell type involved in acute graft dysfunction, we performed adoptive transfer experiment using neutrophils and macrophages from WT donors. Mac-1−/− recipients of B/c heart allografts were injected intravenously with either neutrophils (5 × 106) or macrophages (5 × 106) extracted from WT or Mac-1−/− mice 1 d after transplantation. Neither WT nor Mac-1−/− neutrophil adoptive transfer affected graft survival in Mac-1−/− recipients (Figure 3A). However, adoptive transfer of WT but not Mac-1−/− macrophages significantly reduced graft survival of Mac-1−/− recipients (Figure 3B). PR scores of allografts also increased significantly in Mac-1−/− recipients receiving WT (2.3 ± 0.4, mean ± SD, n=6) versus Mac-1−/− (1.5 ± 0.5, n=5, p =0.024) macrophages (Figure 3C). These observations indicate that macrophages expressing Mac-1 participate in graft inflammatory cell accumulation and influence graft survival in total allo-mismatched allografts.
Figure 3. Neutrophil and macrophage adoptive transfer.
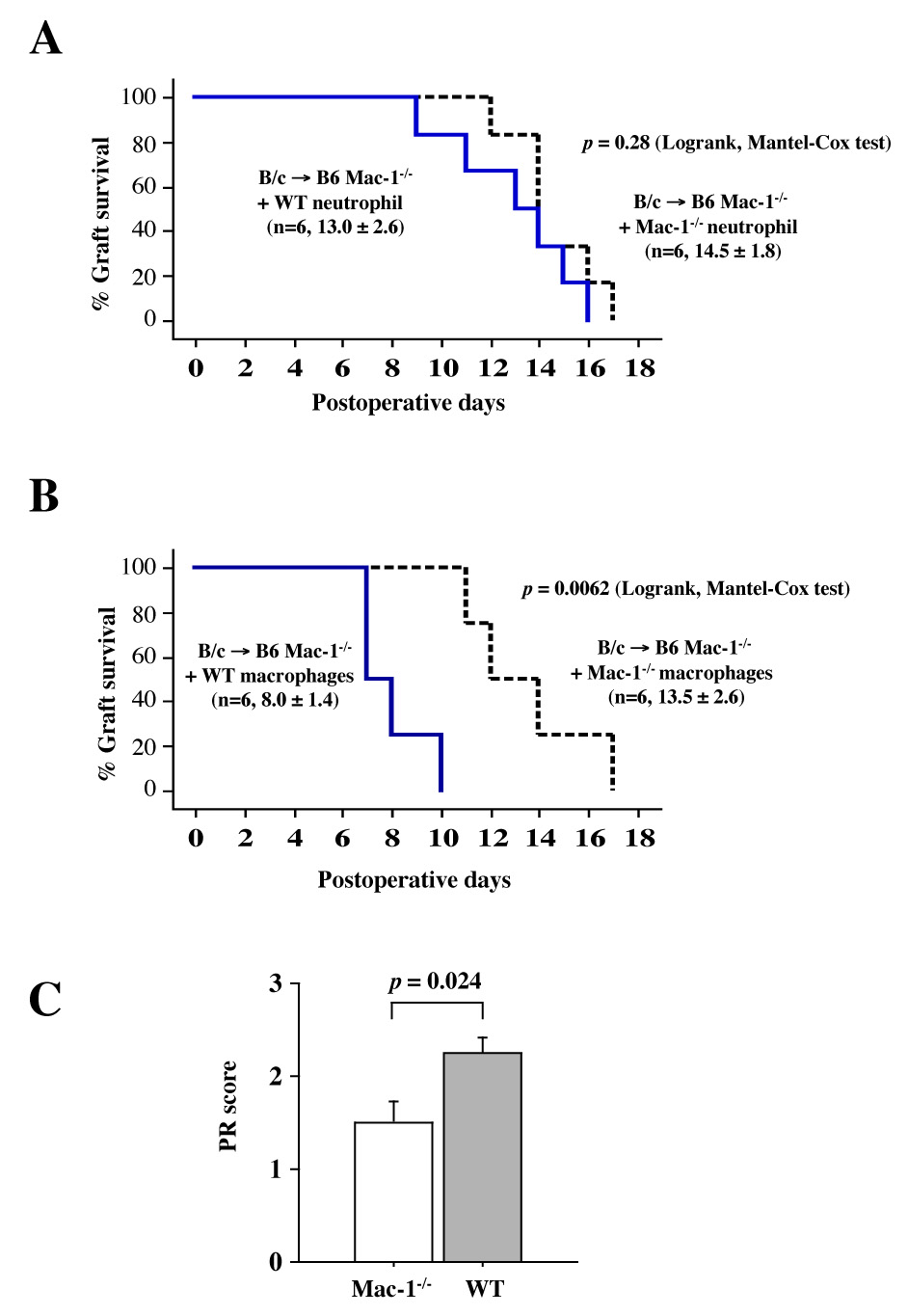
A, Mac-1−/− and WT B6 neutrophil adoptive transfer did not affect graft survival of Mac-1−/− recipient cardiac allografts: solid line, graft survival of WT neutrophil recipients; dashed line, graft survival of Mac-1−/− recipients.B and C, WT B6 macrophage adoptive transfer significantly reduced graft survival (B) and increased PR score (C, mean ± SD) of cardiac allografts in Mac-1−/− recipients receiving Mac-1−/− (n=5, open bars) or WT (n=6, closed bars) macrophages.
Mac-1 lack reduced early parenchymal rejection and graft arterial disease, but not chronic graft arterial disease in MHC class II-mismatched cardiac transplants
We examined parenchymal rejection (PR) and graft arterial disease (GAD) after MHC class II-mismatched murine heart transplantation using bm12 donor hearts and WT or Mac-1−/− B6 recipients without immunosuppression. Single MHC class mismatch permits graft survival for the assessment of GAD. Grafts were harvested at 4 and 12 weeks after transplantation. The 12-week time point is commonly utilized to evaluate arterial lesions at a more chronic stage when GAD lesions typically are well-developed.32, 35 Four weeks after transplantation, both PR (Mac-1−/−: 1.9 ± 0.5, n=6 vs. WT: 2.8 ± 0.8, n=8; p=0.026) and GAD (Mac-1−/−: 0.0 ± 0.0, n=6 vs. 1.2 ± 1.1, n=8; p=0.031) scores were reduced in Mac-1−/− compared to WT recipients. However, at 12 weeks, GAD lesions were comparable in Mac-1−/− (1.7 ± 1.1, n=8) and WT (2.2 ± 1.5, n=11; p=0.65) recipients (Figure 4). Parenchymal rejection at 12 weeks was reduced compared to 4 weeks, but was similar in Mac-1−/− (1.5 ± 1.1) and WT (2.0 ± 1.0, p=0.63) recipients.
Figure 4. Recipient Mac-1 deficiency, parenchymal rejection, and GAD in MHC class II mismatched allografts (bm12 allografts in WT and B6 Mac-1−/− recipients).
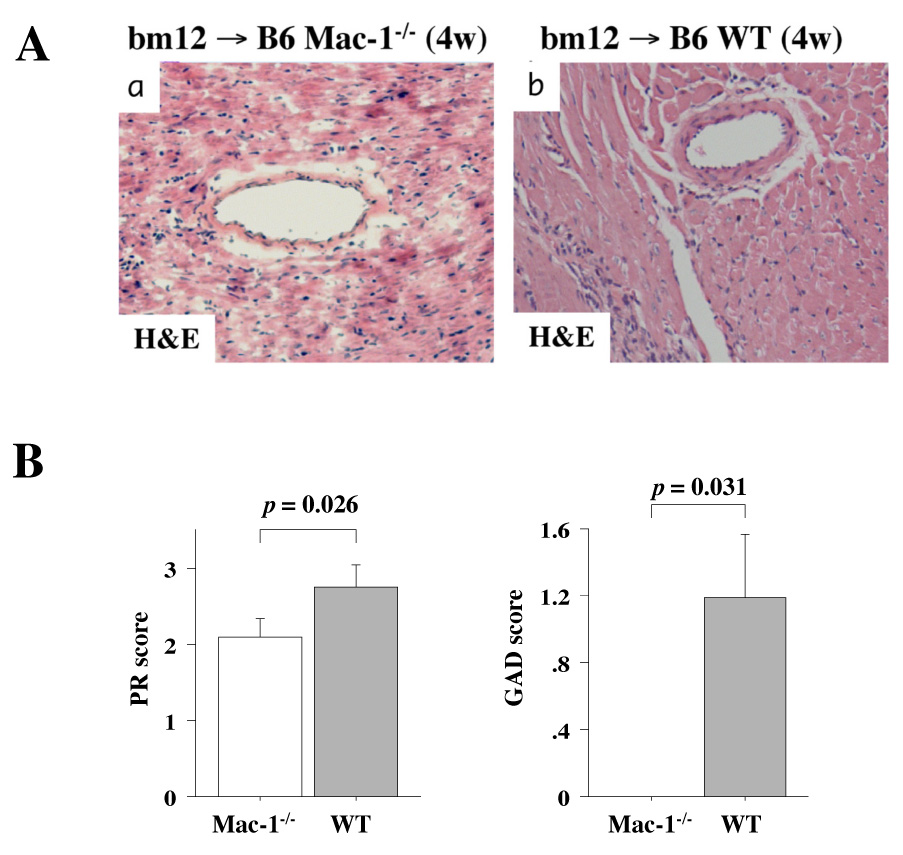
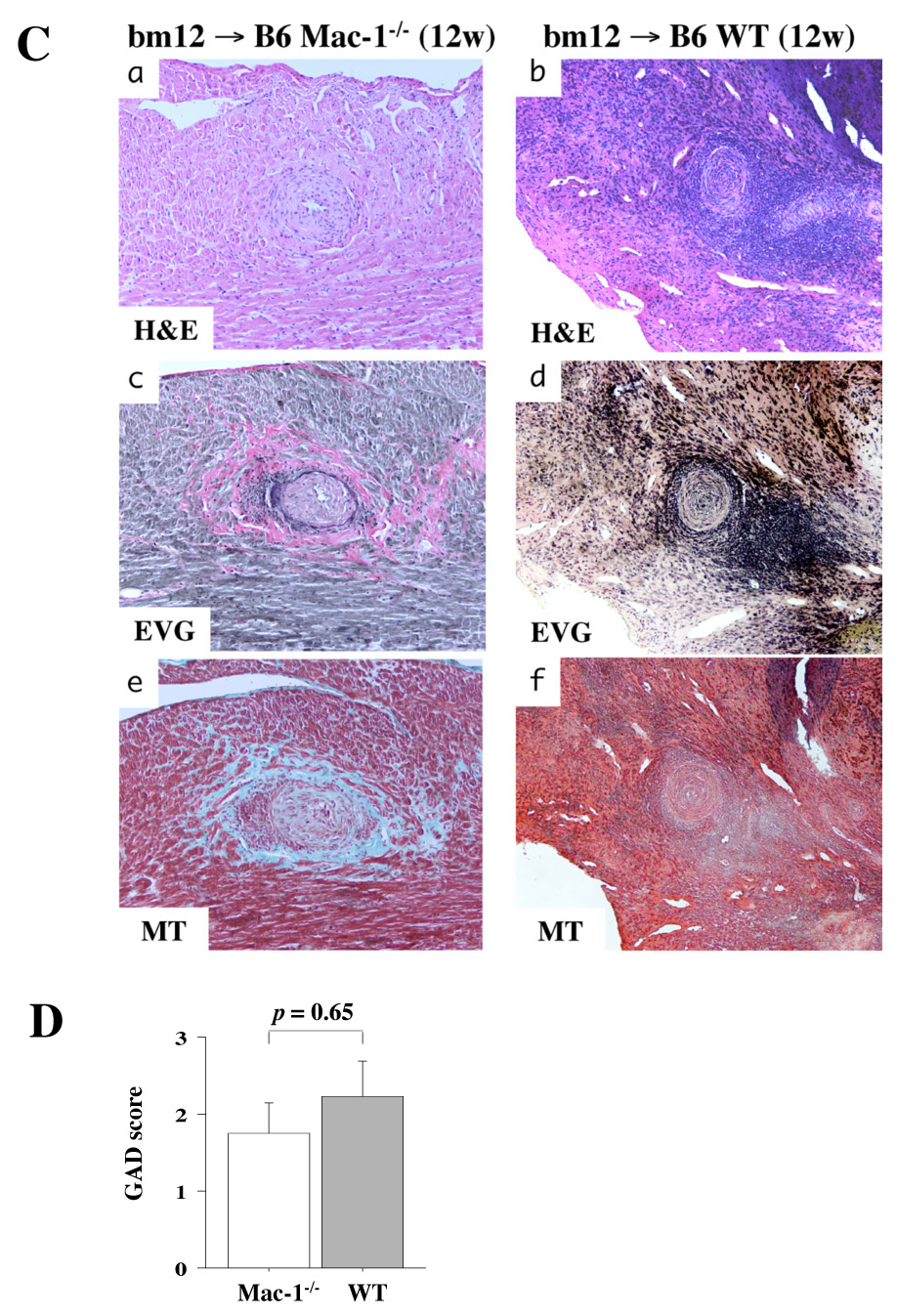
Photomicrographs after H and E staining of 4-week bm12 allografts in Mac-1−/− (A-a) or in WT (A-b) recipients, and photomicrographs after H and E (C-a, b), elastica Van Gieson (C-c, d), and Masson Trichrome (C-e, f) staining of 12-week bm12 allografts in Mac-1−/− (C-a, c, and e) or in WT (C-b, d, and f) recipients. B, GAD and PR was scored in 4 week transplanted hearts (bm12 allografts into B6 recipients) harvested from WT and Mac-1−/− recipients. D, GAD score of 12-week transplantation.
Chemokine mRNA expression is comparable in allografts from WT and Mac-1−/− recipients
The reduction in graft immune cell infiltration in Mac-1−/− recipients could result from diminished endothelial cell adhesion via leukocyte Mac-1 or from changes in local chemokine and cytokine expression. To test the latter possibility, we performed RPA to measure chemokine and cytokine mRNA expression from allografts harvested 7 d after transplantation. Allograft expression of RANTES, MCP-1, MIP-1α, MIP-1β, MIP-2, and IP-10 mRNA was similar in WT and Mac-1−/− recipients. Among cytokine mRNA expression profiles, only tumor necrosis factor-alpha (TNF-α) decreased significantly in Mac-1−/− compared to WT recipients (Figure 5).
Figure 5. Chemokine and cytokine expression in transplanted hearts.
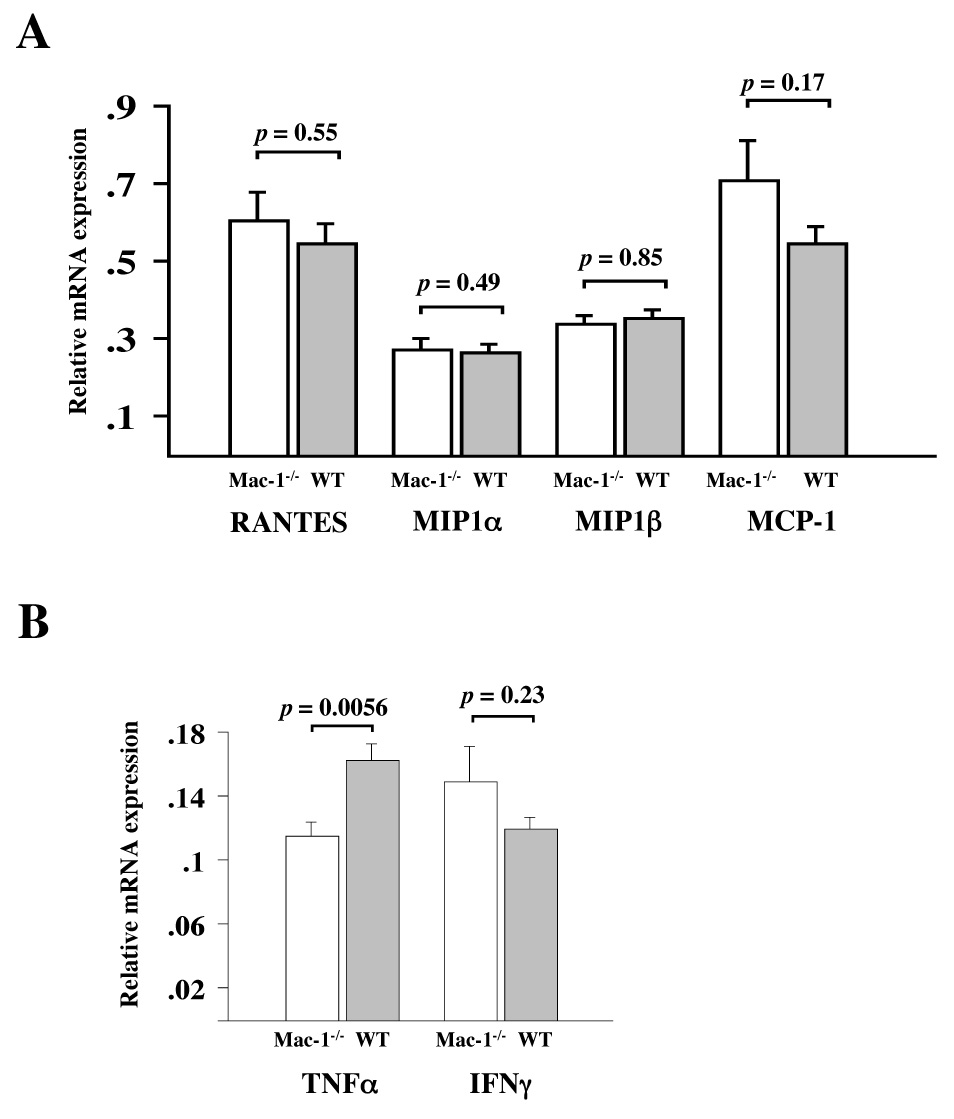
Chemokine (RANTES, MIP1α, MIP1β, and MCP-1, panel A) cytokine (TNFα and IFNγ, panel B) were examined by RPA in day 7 cardiac allografts harvested from WT (solid bars) and Mac-1−/− (open bars) recipients. Data represent mean ± SEM, n=6 per group.
Mac-1−/− splenocytes have attenuated mixed lymphocyte reactions (MLR) but normal responses to anti-CD3 and anti-CD40 antibodies
Interaction of antigen-presenting cells and T cells, and subsequent strength of the alloresponse, may affect cellular recruitment.36 Mac-1 can also directly limit T cell and/or dendritic cell function and may alter the local cytokine milieu.5, 6, 37 Therefore, we examined whether deficiency of Mac-1 modulates mixed lymphocyte reaction (MLR) using WT or Mac-1−/− splenocytes as responders and irradiated BALB/c splenocytes as stimulator cells. Mac-1−/− splenocytes showed significantly less proliferation compared to WT splenocytes after 3–5 d of culture (Figure 6A). To dissect the mechanisms by which Mac-1−/− splenocytes show defective proliferative responses, we stimulated splenocytes with anti-CD3 mAbs (i.e., direct T cell stimulation) or anti-CD40 (3/23) mAbs (direct B cell stimulation). Interestingly, both WT and Mac-1−/− splenocytes proliferated comparably under these conditions (Figure 6B, C), suggesting that Mac-1-deficient T and B cells have normal intrinsic proliferative capability. The results indicate that Mac-1−/− splenocytes have attenuated antigen-presenting functions.
Figure 6. Mac-1 splenocytes have significantly reduced proliferation in MLR, but comparable responses to anti-CD3 and anti-CD40 antibodies.
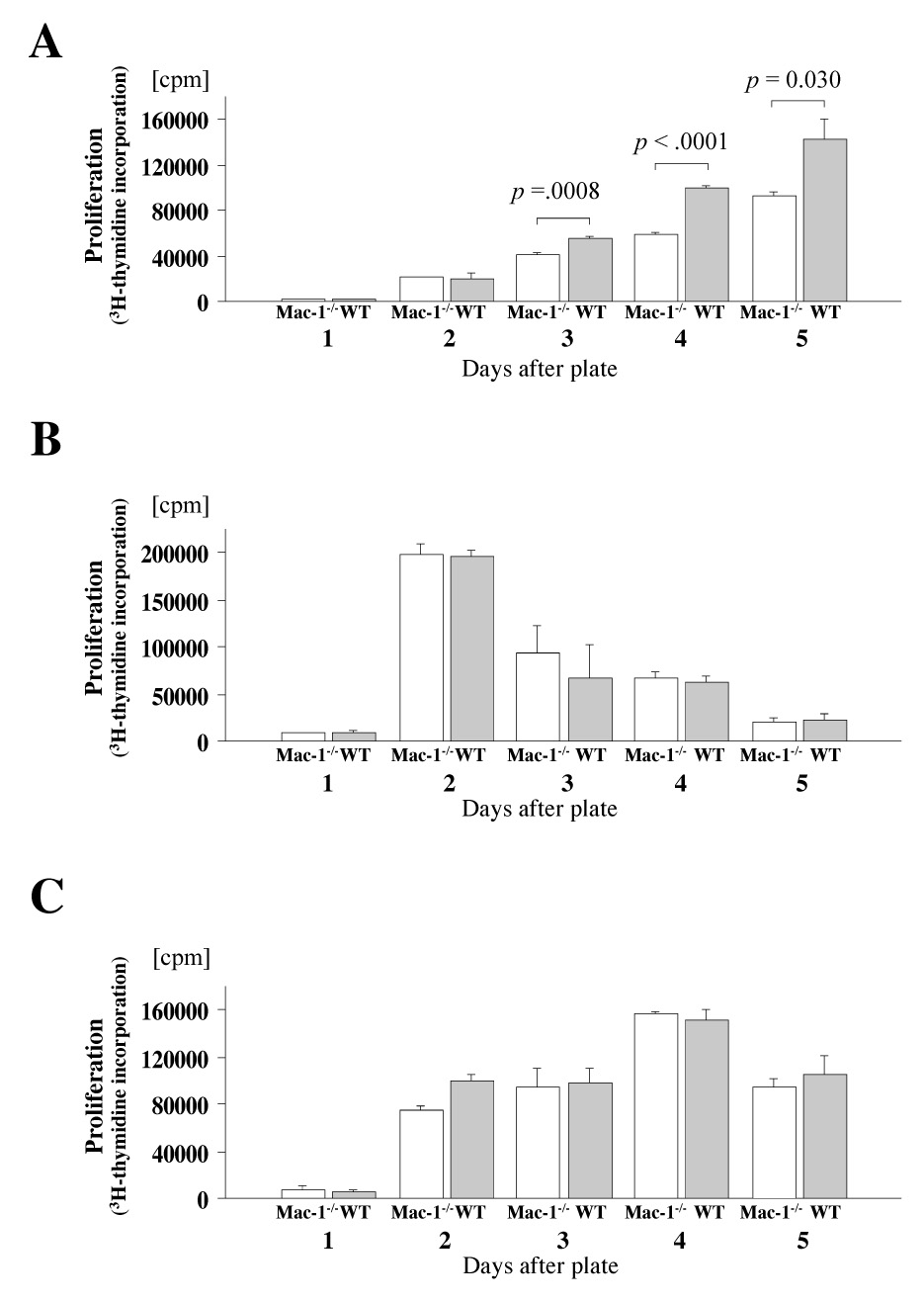
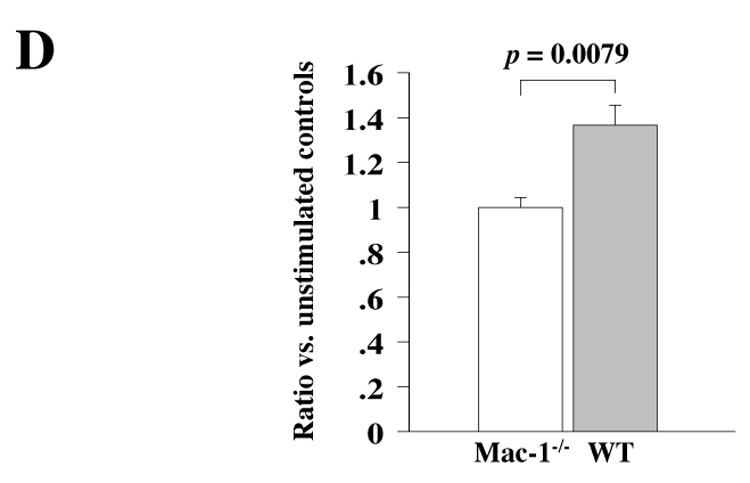
A, MLR using WT or Mac-1−/− B6 splenocyte responders and irradiated B/c splenocytes stimulators. B, T cell responses of WT and Mac-1−/− B6 splenocytes against immobilized anti-CD3 TCR stimulating antibodies. C, B cell responses of WT and Mac-1−/− B6 splenocytes against anti-CD40 stimulating antibodies. In vivo B/c-primed Mac-1−/− splenocytes show reduced responses to B/c stimulation. D, primed-MLR using Mac-1−/− or WT B6 splenocyte responders extracted from recipients of B/c donor heart 6 days after transplantation. The ratio of 3H-thymidine incorporation (cpm) of MLR stimulated by irradiated B/c splenocytes to T cell proliferation without stimulation was calculated. Data represent peak response 4 d after plating and average values (mean ± SEM) of three independent experiments from WT (solid bars) and Mac-1−/− (open bars) splenocytes.
To examine whether Mac-1-deficient splenocytes ineffectively prime T cells, we performed B/c-primed MLR. B/c-primed splenocytes from WT or Mac-1−/− recipients of B/c cardiac allografts were recovered 6 d after transplantation, and were stimulated with or without (control) irradiated naïve B/c splenocytes. Interestingly, splenocytes harvested from Mac-1−/− transplant recipients showed significantly less proliferation compared to splenocytes recovered from WT recipients (Figure 6D). The results suggest that Mac-1−/− splenocytes lead to ineffective T cell priming in vivo.
Mac-1−/− macrophages express reduced levels of co-stimulatory molecules and have diminished allo-antigen-presenting function
Macrophages comprise the majority of inflammatory cells at the later phases of acute rejection.30, 38 Macrophages can exhibit direct cytotoxicity against certain cells,39 and can also function in allograft rejection as antigen-presenting cells.40 We tested the hypothesis that intragraft Mac-1−/− macrophages have impaired activation and exhibit reduced antigen-presenting function compared to WT macrophages. To do so, immune cells were extracted from cardiac allografts harvested from WT and Mac-1−/− recipients 6 d after transplantation; flow cytometry was performed to assess macrophage MHC class II and co-stimulatory molecule expression. Graft macrophages in Mac-1−/− recipients showed significantly lower expression of CD40, CD80, CD86, ICOSL, and MHC II compared to those in WT recipients (Figure 7A, B).
Figure 7. Mac-1−/− macrophages have diminished APC function.
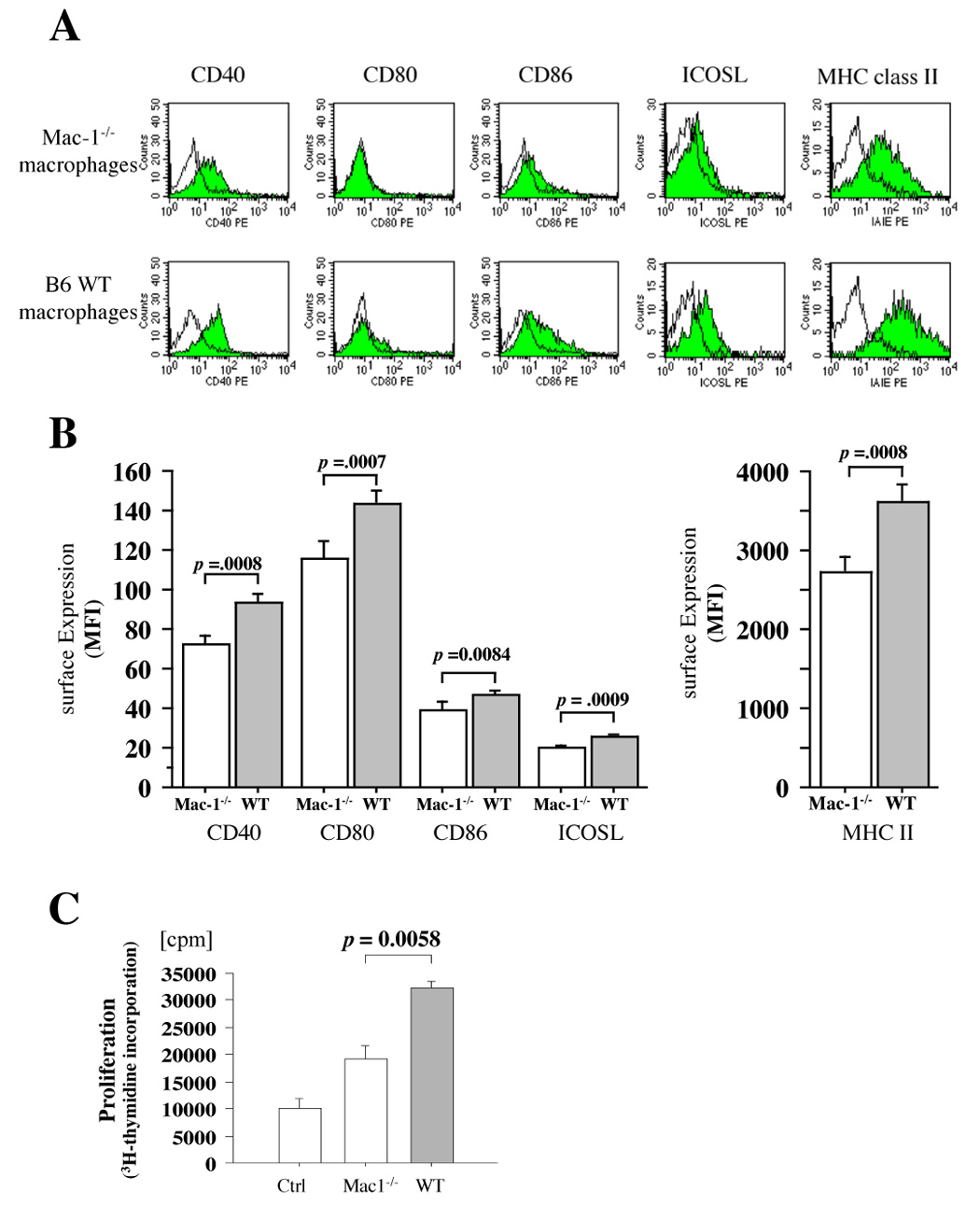
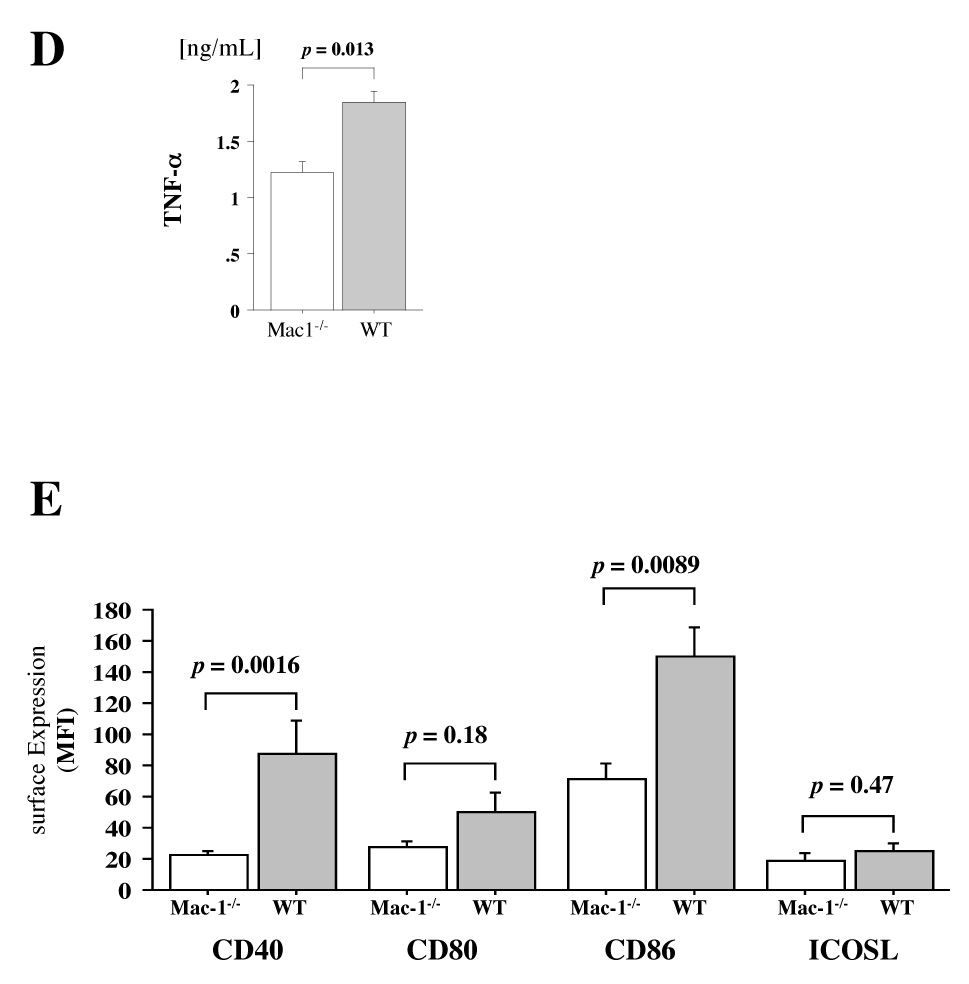
A, Representative figure shows expression of co-stimulatory molecules and MHC class II (solid graph) on macrophages extracted from day 7 transplanted cardiac allografts in Mac-1−/− (upper panel) and WT (lower panel) recipients assessed by flow cytometry and double staining with PE-conjugated anti-CD40, anti-CD80, anti-CD86, anti-ICOSL, or anti-MHC II (IA/IE), and FITC-conjugated anti-Mac-3. Clear graph shows negative control using PE-conjugated rat IgG2a (negative control for CD40, CD86, ICOSL, and MHC II) or hamster IgG (negative control for CD80). B, The bar graph shows values representing mean fluorescent intensity ± SEM of the Mac-3+ macrophages extracted from WT (solid bars) and Mac-1−/− (open bars) recipient allografts (n=4 per group). C and D, Proliferation of naïve wild-type B6 T cells stimulated by the macrophages extracted from B/c allografts of B6 WT (solid bars) and Mac-1−/− (open bars) recipients (C) and production of TNF-α from those T cells (D) were assessed as described in Methods. Macrophage antigen presentation after interaction with allogeneic EC. E, Expression of co-stimulaory molecules on B/c EC-primed WT (solid) and Mac-1−/− (open) macrophages was assessed by flow cytometry, as described above. Data represent average values (mean ± SEM) of three independent experiments (n=4 per group).
We also used the macrophages extracted from B/c allografts of B6 WT or Mac-1−/− recipients as a stimulator of B6 naïve T cells and performed primed MLR (primed macrophages as stimulator and naïve T cells as responder). The results demonstrated that naïve T cells stimulated by the primed macrophages extracted from the allografts of Mac-1−/− recipients had a lower proliferative capacity (Figure 7C) and secreted less TNF-a (Figure 7D) compared to those stimulated by the primed macrophages extracted from the allografts of WT recipients. These results suggested that Mac-1−/− macrophages have lower antigen-presenting capacity.
Because recipient leukocytes first encounter donor ECs after transplantation, the ability of donor ECs to facilitate antigen presentation is likely important. Thus, to assess whether macrophages (B6) can process and present a foreign antigen of the endothelial cells (B/c) in vitro, we performed B/c-EC and B6-macrophage co-culture and examined antigen-presenting capacity of the macrophages to B6 T cells. Naïve B6 peritoneal macrophages were also extracted from WT and Mac-1−/− B6 animals, and co-cultured with B/c ECs. After 3 days, macrophages were isolated by gradient centrifugation and used for flow cytometry analysis. Mac-1−/− macrophages expressed significantly lower levels of CD40 (mean fluorescent intensity, 22.7 ± 4.5) and CD86 (71.7 ± 9.5) compared to WT macrophages (CD40, 87.7 ± 21.3, p =0.0016, n=4; CD86, 150 ± 18.2, p =0.0089, n=4) (Figure 7E). Of note, no significant differences occurred in the expression (mean fluorescence intensity ± SD) of CD40 (Mac-1−/−: 19.8 ± 5.9 vs. WT: 20.0 ± 7.0, p=0.96), CD80 (20.8 ± 10.4 vs. 22.8 ± 7.5, p=0.77), CD86 (48.3 ± 14.9 vs. 50.5 ± 24.0, p=0.77), and ICOSL (15.0 ± 6.5 vs. 13.5 ± 5.1, p=0.73) on naïve peritoneal macrophages isolated from Mac-1−/−, compared to WT mice. These findings indicate that Mac-1−/− macrophages co-cultured with B/c ECs have weaker antigen-presenting capacity than WT macrophages.
DISCUSSION
This study provides in vivo evidence that Mac-1 promotes acute allograft rejection. Recipient Mac-1-deficiency reduced allograft accumulation of immune cells and prolonged allograft survival, but did not prevent graft arterial disease. The results demonstrated an important role for Mac-1 in macrophage antigen presentation by showing that Mac-1−/− macrophages expressed significantly lower levels of the co-stimulatory molecules both in vivo and in vitro, and that mixed lymphocyte reaction using allo-primed Mac-1−/− macrophages resulted in significantly lower antigen-presenting function than WT macrophages. Furthermore, WT but not Mac-1−/− macrophage adoptive transfer to Mac-1−/− recipients exacerbated parenchymal rejection and reduced allograft survival. In comparison, adoptive transfer of WT neutrophils did not affect graft survival. We have also observed that blockade of neutrophil recruitment using anti-Gr1 antibody (RB6-8C5) treatment does not prevent acute rejection (unpublished data).
Possible mechanism of Mac-1 action in acute rejection
Mac-1 localizes on neutrophils, monocytes, macrophages, NK cells, and CD8+ T cells.5, 7 The functional relevance of β2-integrins such as Mac-1 (CD11b/CD18) on APCs remains unclear. Varga and coworkers reported recently that Mac-1 on APCs, including DCs and macrophages, directly inhibited T cell activation.41 In that study, CD18−/− macrophages stimulate allogeneic T cells more potently than their WT counterparts. However, Mac-1−/− (CD11b−/−) APCs were not examined, thereby precluding definitive conclusions regarding the precise role of Mac-1 given possible contributions of other β2-integrins, including LFA-1 (CD11a/CD18), p150,95 (CD11c/CD18), and CD11d/CD18, to immune synapse function.
In contrast, the current work shows unambiguously that Mac-1−/− macrophages have defective APC function. Direct TCR stimulation by anti-CD3 Abs and direct B cell stimulation with anti-CD40 Abs induced comparable proliferation from WT and Mac-1−/− splenocytes, indicating that Mac-1−/− T and B cells have normal TCR and BCR responses. At the same time, graft-infiltrating macrophages in Mac-1−/− recipients and Mac-1−/− macrophages co-cultured with allo-ECs express significantly lower levels of co-stimulatory molecules and MHC II compared to WT macrophages. Mac-1−/− macrophages also stimulated T cell proliferation more poorly than WT macrophages. Over all, our data suggest that the diminished APC function of Mac-1−/− macrophages may prolong allograft survival by reducing T cell priming, but may also do so by reducing TNF-α expression.
Mac-1 and graft arterial disease
By virtue of binding diverse ligands such as fibrinogen,42, 43 ICAM-1,44 factor X,45 C3bi,42 platelet GP Ibα, 46, 47 CD154,14 and JAM-3,48 Mac-1 potentially regulates important leukocyte functions relevant to vascular injury, including adhesion, migration, coagulation, proteolysis, phagocytosis, oxidative burst, and signaling.29, 31, 49–52
Our prior report identified Mac-1 as a molecular determinant of neointimal thickening after endothelial-denuding injury; selective absence of Mac-1 impaired transplatelet leukocyte migration into the vessel wall, diminishing medial leukocyte accumulation and neointimal thickening after experimental angioplasty.34 In the present study, deficiency of Mac-1 attenuated allograft leukocyte accumulation and rejection, but had no effect on neointimal thickening in graft arterial disease at 12 weeks. These discrepant effects on neointimal thickening likely reflect a distinct cell recruitment mechanism provoked by mechanical arterial injury (i.e., platelet-dependent neutrophil recruitment mediated in part by integrin interactions between neutrophil Mac-1 and platelet GP Ibα) compared to the pathways downstream of immunologic injury involving intact, activated endothelium (i.e., endothelium-dependent mononuclear cell recruitment mediated by mononuclear β1- and β2-integrins and endothelial ICAM-1 and VCAM-1). Similar to graft arterial disease, deficiency of Mac-1 has no effect on high-fat diet–induced atherosclerotic lesion formation in LDL receptor-deficient mice.53
Clinical Implications
Allograft rejection, both cellular and antibody-mediated, remains a clinically important limitation to human cardiac transplantation. More than 40% of patients will experience an episode of rejection in the first year after transplant that requires therapy; the average patient will experience close to 2 episodes of rejection in the first year.54 Severe rejection episodes can cause heart failure, malignant arrhythmias, and sudden cardiac death. Higher grades of rejection (ISHT grade 3 or 4) portend poor outcomes.54 Current treatment strategies often resolve the acute episode effectively and typically include the use of augmented doses of glucocorticoids or calcineurin inhibitors, cytolytic agents such as OKT3, and plasmapheresis. However, they are limited by the inherent toxicities of immunosuppression, including fatal infections, aggressive malignancies, and renal failure. Furthermore, antibody-mediated rejection, in particular, has no consensus treatment to date.55 Novel therapies are greatly needed to not only decrease the morbidity of conventional immunosuppression, but to improve the outcomes of patients with specific forms of allograft rejection, especially acute antibody-mediated rejection. Although deficiency of Mac-1 did not prevent chronic rejection in our study, targeting Mac-1 is an attractive strategy to modulate acute rejection following cardiac transplantation. Indeed, previous clinical trials demonstrated improved renal allograft survival in high-risk patients (either highly immunized or re-transplant recipients) who received a monoclonal antibody to one of the Mac-1 cognate ligands, ICAM-1.56
Limitations
We examined the effect of Mac-1 deficiency on PR and GAD after MHC class II-mismatched murine heart transplantation without background immunosuppression. Co-treatment with Mac-1 inhibitor and a classical immunosuppressant such as cyclosporine or tacrolimus may synergistically attenuate GAD. Clinical application will require future studies evaluating the effect of Mac-1 deficiency and immunosuppressive therapy, alone or in combination, on rejection scores and GAD.
Mac-1-deficiency may affect the development and function of immune cells. The present experiments using adoptive transfer of normal cells provide compelling evidence for the importance of macrophage Mac-1 in acute rejection. Nonetheless, we cannot rule out an additional role of Mac-1 in the development and function of immune cells.
Conclusion
In summary, this study demonstrates that recipient Mac-1 deficiency reduces accumulation of graft infiltrating cells, macrophage APC function, T cell proliferation, and TNF-α production, and attenuates acute allograft rejection.
Acknowledgements
We thank D. Cameron, E. Shvartz, J. N. Vaisviliene, M. Rodrigue, G. Suliman, and E. Simon-Morrissey for their technical expertise, and J. Perry for editorial assistance.
Sources of Funding
This work was supported by an American Heart Association Scientist Development grant (KS), a grant award from the Roche Organ Transplantation Research Foundation (KS), and a Harvard Medical School BWH Fellowship Award (KS), and also by grants from the Donald W. Reynolds Foundation (PL) and the National Institutes of Health (HL-43364 to PL, RNM; GM-67049 to RNM, KS, PL; HL-67249 to KS, RNM; HL-67283 to KS; HL-34636 to PL; HL57506 and HL60942 to DIS).
Abbreviations
- B6
C57BL/6
- B/c
BALB/c
- WT
wild-type
- TNF-α
Tumor necrosis factor-alpha
- PR
parenchymal rejection
- GAD
graft arterial disease
References
- 1.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Sims TN, Dustin ML. The immunological synapse: integrins take the stage. Immunol Rev. 2002;186:100–117. doi: 10.1034/j.1600-065x.2002.18610.x. [DOI] [PubMed] [Google Scholar]
- 3.Carrasco YR, Fleire SJ, Cameron T, et al. LFA-1/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity. 2004;20:589–599. doi: 10.1016/s1074-7613(04)00105-0. [DOI] [PubMed] [Google Scholar]
- 4.Lebedeva T, Dustin ML, Sykulev Y. ICAM-1 co-stimulates target cells to facilitate antigen presentation. Curr Opin Immunol. 2005;17:251–258. doi: 10.1016/j.coi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Wagner C, Hansch GM, Stegmaier S, et al. The complement receptor 3, CR3 (CD11b/CD18), on T lymphocytes: activation-dependent up-regulation and regulatory function. Eur J Immunol. 2001;31:1173–1180. doi: 10.1002/1521-4141(200104)31:4<1173::aid-immu1173>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 6.Muto S, Vetvicka V, Ross GD. CR3 (CD11b/CD18) expressed by cytotoxic T cells and natural killer cells is upregulated in a manner similar to neutrophil CR3 following stimulation with various activating agents. J Clin Immunol. 1993;13:175–184. doi: 10.1007/BF00919970. [DOI] [PubMed] [Google Scholar]
- 7.Ross GD, Vetvicka V. CR3 (CD11b, CD18): a phagocyte and NK cell membrane receptor with multiple ligand specificities and functions. Clin Exp Immunol. 1993;92:181–184. doi: 10.1111/j.1365-2249.1993.tb03377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith CW, Marlin SD, Rothlein R, et al. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J Clin Invest. 1989;83:2008–2017. doi: 10.1172/JCI114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diamond MS, Staunton DE, de Fougerolles AR, et al. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18) J Cell Biol. 1990;111:3129–3139. doi: 10.1083/jcb.111.6.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunne JL, Collins RG, Beaudet AL, et al. Mac-1, but not LFA-1, uses intercellular adhesion molecule-1 to mediate slow leukocyte rolling in TNF-alpha-induced inflammation. J Immunol. 2003;171:6105–6111. doi: 10.4049/jimmunol.171.11.6105. [DOI] [PubMed] [Google Scholar]
- 11.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 12.Scharffetter-Kochanek K, Lu H, Norman K, et al. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J Exp Med. 1998;188:119–131. doi: 10.1084/jem.188.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Languino LR, Duperray A, Joganic KJ, et al. Regulation of leukocyte-endothelium interaction and leukocyte transendothelial migration by intercellular adhesion molecule 1-fibrinogen recognition. Proc Natl Acad Sci U S A. 1995;92:1505–1509. doi: 10.1073/pnas.92.5.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zirlik A, Maier C, Gerdes N, et al. CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation. 2007;115:1571–1580. doi: 10.1161/CIRCULATIONAHA.106.683201. [DOI] [PubMed] [Google Scholar]
- 15.Diamond MS, Alon R, Parkos CA, et al. Heparin is an adhesive ligand for the leukocyte integrin Mac-1 (CD11b/CD1) J Cell Biol. 1995;130:1473–1482. doi: 10.1083/jcb.130.6.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 17.Dunne JL, Ballantyne CM, Beaudet AL, et al. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood. 2002;99:336–341. doi: 10.1182/blood.v99.1.336. [DOI] [PubMed] [Google Scholar]
- 18.Taylor PM, Rose ML, Yacoub MH, et al. Induction of vascular adhesion molecules during rejection of human cardiac allografts. Transplantation. 1992;54:451–457. doi: 10.1097/00007890-199209000-00013. [DOI] [PubMed] [Google Scholar]
- 19.Steinhoff G, Behrend M, Haverich A. Signs of endothelial inflammation in human heart allografts. Eur Heart J. 1991;12 Suppl D:141–143. doi: 10.1093/eurheartj/12.suppl_d.141. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka H, Sukhova GK, Swanson SJ, et al. Endothelial and smooth muscle cells express leukocyte adhesion molecules heterogeneously during acute rejection of rabbit cardiac allografts. Am J Pathol. 1994;144:938–951. [PMC free article] [PubMed] [Google Scholar]
- 21.Gibbs P, Berkley LM, Bolton EM, et al. Adhesion molecule expression (ICAM-1, VCAM-1, E-selectin and PECAM) in human kidney allografts. Transpl Immunol. 1993;1:109–113. doi: 10.1016/0966-3274(93)90003-q. [DOI] [PubMed] [Google Scholar]
- 22.Isobe M, Yagita H, Okumura K, et al. Specific acceptance of cardiac allograft after treatment with antibodies to ICAM-1 and LFA-1. Science. 1992;255:1125–1127. doi: 10.1126/science.1347662. [DOI] [PubMed] [Google Scholar]
- 23.Brandt M, Steinmann J, Steinhoff G, et al. Treatment with monoclonal antibodies to ICAM-1 and LFA-1 in rat heart allograft rejection. Transpl Int. 1997;10:141–144. doi: 10.1007/s001470050028. [DOI] [PubMed] [Google Scholar]
- 24.Raisky O, Morrison KJ, Obadia JF, et al. Acute rejection and cardiac graft vasculopathy in the absence of donor-derived ICAM-1 or P-selectin. J Heart Lung Transplant. 2001;20:340–349. doi: 10.1016/s1053-2498(00)00192-3. [DOI] [PubMed] [Google Scholar]
- 25.Lacha J, Bushell A, Smetana K, et al. Intercellular cell adhesion molecule-1 and selectin ligands in acute cardiac allograft rejection: a study on gene-deficient mouse models. J Leukoc Biol. 2002;71:311–318. [PubMed] [Google Scholar]
- 26.Paul LC, Davidoff A, Benediktsson H, et al. Anti-integrin (LFA-1, VLA-4, and Mac-1) antibody treatment and acute cardiac graft rejection in the rat. Transpl Int. 1996;9:420–425. doi: 10.1007/BF00335706. [DOI] [PubMed] [Google Scholar]
- 27.Miller LJ, Bainton DF, Borregaard N, et al. Stimulated mobilization of monocyte Mac-1 and p150,95 adhesion proteins from an intracellular vesicular compartment to the cell surface. J Clin Invest. 1987;80:535–544. doi: 10.1172/JCI113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon DI, Rao NK, Xu H, et al. Mac-1 (CD11b/CD18) and the urokinase receptor (CD87) form a functional unit on monocytic cells. Blood. 1996;88:3185–3194. [PubMed] [Google Scholar]
- 29.Lu H, Smith CW, Perrard J, et al. LFA-1 is sufficient in mediating neutrophil emigration in Mac-1-deficient mice. J Clin Invest. 1997;99:1340–1350. doi: 10.1172/JCI119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimizu K, Schonbeck U, Mach F, et al. Host CD40 ligand deficiency induces long-term allograft survival and donor-specific tolerance in mouse cardiac transplantation but does not prevent graft arteriosclerosis. J Immunol. 2000;165:3506–3518. doi: 10.4049/jimmunol.165.6.3506. [DOI] [PubMed] [Google Scholar]
- 31.Coxon A, Rieu P, Barkalow FJ, et al. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 32.Shimizu K, Aikawa M, Takayama K, et al. Direct anti-inflammatory mechanisms contribute to attenuation of experimental allograft arteriosclerosis by statins. Circulation. 2003;108:2113–2120. doi: 10.1161/01.CIR.0000092949.67153.74. [DOI] [PubMed] [Google Scholar]
- 33.Shimizu K, Sugiyama S, Aikawa M, et al. Host bone-marrow cells are a source of donor intimal smooth- muscle-like cells in murine aortic transplant arteriopathy. Nature Med. 2001;7:738–741. doi: 10.1038/89121. [DOI] [PubMed] [Google Scholar]
- 34.Simon DI, Dhen Z, Seifert P, et al. Decreased neointimal formation in Mac-1(−/−) mice reveals a role for inflammation in vascular repair after angioplasty. J Clin Invest. 2000;105:293–300. doi: 10.1172/JCI7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feinberg MW, Shimizu K, Lebedeva M, et al. Essential role for Smad3 in regulating MCP-1 expression and vascular inflammation. Circ Res. 2004;94:601–608. doi: 10.1161/01.RES.0000119170.70818.4F. [DOI] [PubMed] [Google Scholar]
- 36.Leyva-Cobian F, Carrasco-Marin E. Participation of intracellular oxidative pathways in antigen processing by dendritic cells, B cells and macrophages. Immunol Lett. 1994;43:29–37. doi: 10.1016/0165-2478(94)00146-4. [DOI] [PubMed] [Google Scholar]
- 37.Wu H, Rodgers JR, Perrard XY, et al. Deficiency of CD11b or CD11d results in reduced staphylococcal enterotoxin-induced T cell response and T cell phenotypic changes. J Immunol. 2004;173:297–306. doi: 10.4049/jimmunol.173.1.297. [DOI] [PubMed] [Google Scholar]
- 38.MacPherson GG, Christmas SE. The role of the macrophage in cardiac allograft rejection in the rat. Immunol Rev. 1984;77:143–166. doi: 10.1111/j.1600-065x.1984.tb00720.x. [DOI] [PubMed] [Google Scholar]
- 39.Ratliff NB, McMahon JT. Activation of intravascular macrophages within myocardial small vessels is a feature of acute vascular rejection in human heart transplants. J Heart Lung Transplant. 1995;14:338–345. [PubMed] [Google Scholar]
- 40.Unanue ER. Antigen-presenting function of the macrophage. Annu Rev Immunol. 1984;2:395–428. doi: 10.1146/annurev.iy.02.040184.002143. [DOI] [PubMed] [Google Scholar]
- 41.Varga G, Balkow S, Wild MK, et al. Active MAC-1 (CD11b/CD18) on DCs inhibits full T-cell activation. Blood. 2007;109:661–669. doi: 10.1182/blood-2005-12-023044. [DOI] [PubMed] [Google Scholar]
- 42.Wright SD, Weitz JI, Huang AJ, et al. Complement receptor type three (CD11b/CD18) of human polymorphonuclear leukocytes recognizes fibrinogen. Proc Natl Acad Sci U S A. 1988;85:7734–7738. doi: 10.1073/pnas.85.20.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altieri DC, Bader R, Mannucci PM, et al. Oligospecificity of the cellular adhesion receptor Mac-1 encompasses an inducible recognition specificity for fibrinogen. J Cell Biol. 1988;107:1893–1900. doi: 10.1083/jcb.107.5.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diamond MS, Staunton DE, Marlin SD, et al. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65:961–971. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- 45.Altieri DC, Morrissey JH, Edgington TS. Adhesive receptor Mac-1 coordinates the activation of factor X on stimulated cells of monocytic and myeloid differentiation: an alternative initiation of the coagulation protease cascade. Proc Natl Acad Sci U S A. 1988;85:7462–7466. doi: 10.1073/pnas.85.20.7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simon DI, Chen Z, Xu H, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18) J Exp Med. 2000;192:193–204. doi: 10.1084/jem.192.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ehlers R, Ustinov V, Chen Z, et al. Targeting platelet-leukocyte interactions: identification of the integrin Mac-1 binding site for the platelet counter receptor glycoprotein Ibalpha. J Exp Med. 2003;198:1077–1088. doi: 10.1084/jem.20022181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santoso S, Sachs UJ, Kroll H, et al. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med. 2002;196:679–691. doi: 10.1084/jem.20020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arnaout MA. Leukocyte adhesion molecules deficiency: its structural basis, pathophysiology and implications for modulating the inflammatory response. Immunol Rev. 1990;114:145–180. doi: 10.1111/j.1600-065x.1990.tb00564.x. [DOI] [PubMed] [Google Scholar]
- 50.Plow EF, Zhang L. A MAC-1 attack: integrin functions directly challenged in knockout mice. J Clin Invest. 1997;99:1145–1146. doi: 10.1172/JCI119267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi C, Zhang X, Chen Z, et al. Leukocyte integrin Mac-1 recruits toll/interleukin-1 receptor superfamily signaling intermediates to modulate NF-kappaB activity. Circ Res. 2001;89:859–865. doi: 10.1161/hh2201.099166. [DOI] [PubMed] [Google Scholar]
- 52.Shi C, Zhang X, Chen Z, et al. Integrin engagement regulates monocyte differentiation through the forkhead transcription factor Foxp1. J Clin Invest. 2004;114:408–418. doi: 10.1172/JCI21100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kubo N, Boisvert WA, Ballantyne CM, et al. Leukocyte CD11b expression is not essential for the development of atherosclerosis in mice. J Lipid Res. 2000;41:1060–1066. [PubMed] [Google Scholar]
- 54.Taylor DO, Edwards LB, Boucek MM, et al. Registry of the International Society for Heart and Lung Transplantation: twenty-third official adult heart transplantation report-- 2006. J Heart Lung Transplant. 2006;25:869–879. doi: 10.1016/j.healun.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 55.Fishbein MC, Kobashigawa J. Biopsy-negative cardiac transplant rejection: etiology, diagnosis, and therapy. Curr Opin Cardiol. 2004;19:166–169. doi: 10.1097/00001573-200403000-00018. [DOI] [PubMed] [Google Scholar]
- 56.Haug CE, Colvin RB, Delmonico FL, et al. A phase I trial of immunosuppression with anti-ICAM-1 (CD54) mAb in renal allograft recipients. Transplantation. 1993;55:766–772. doi: 10.1097/00007890-199304000-00016. [DOI] [PubMed] [Google Scholar]