

Abstract
Since the introduction of tricyclic antidepressants (TCAs) and monoamine oxidase inhibitors (MAOIs) in mid-1950’s, treatment of depression has been dominated by monoamine hypotheses. The well-established clinical efficacy of TCAs and MAOIs is due, at least in part, to the enhancement of noradrenergic or serotonergic mechanisms, or to both. Unfortunately, their very broad mechanisms of action also include many unwanted effects related to their potent activity on cholinergic, adrenergic and histaminergic receptors.
The introduction of selective serotonin reuptake inhibitors (SSRIs) over twenty years ago had been the next major step in the evolution of antidepressants to develop drugs as effective as the TCAs but of higher safety and tolerability profile. During the past two decades SSRIs (fluoxetine, fluvoxamine, paroxetine, sertraline, citalopram) gained incredible popularity and have become the most widely prescribed medication in the psychiatric practice. The evolution of antidepressants continued resulting in introduction of selective and reversible monoamine oxidase inhibitors (eg. moclobemid), selective noradrenaline (eg. reboxetine), dual noradrenaline and serotonin reuptake inhibitors (milnacipram, venlafaxin, duloxetin) and drugs with distinct neurochemical profiles such as mirtazapine, nefazadone and tianeptine. Different novel serotonin receptor ligands have also been intensively investigated. In spite of the remarkable structural diversity, most currently introduced antidepressants are ‘monoamine based’. Furthermore, these newer agents are neither more efficacious nor rapid acting than their predecessors and approximately 30% of the population do not respond to current therapies.
By the turn of the new millennium, we are all witnessing a result of innovative developmental strategies based on the better understanding of pathophysiology of depressive disorder. Several truly novel concepts have emerged suggesting that the modulation of neuropeptide (substance P, corticotrophin-releasing factor, neuropeptide Y, vasopressin V1b, melanin-concentrating hormone-1), N-methyl-D-aspartate, nicotinic acetylcholine, dopaminergic, glucocorticoid, δ-opioid, cannabinoid and cytokine receptors, gamma-amino butyric acid (GABA) and intracellular messenger systems, transcription, neuroprotective and neurogenic factors, may provide an entirely new set of potential therapeutic targets, giving hope that further major advances might be anticipated in the treatment of depressive disorder soon.
The goal of this review is to give a brief overview of the major advances from monoamine-based treatment strategies, and particularly focus on the new emerging approaches in the treatment of depression.
1. INTRODUCTION
Mood disorders such as major depressive disorders (MDDs) and bipolar disorder (manic depressive illness) are common, severe, chronic and often life-threatening illness. More than 20% of the adult population suffers from these conditions at some time during their life. Suicide is estimated to be a cause of death in up to approximately 15% of individuals with MDDs. In addition MDDs represent a major risk factor for the development of cardiovascular disease and death after myocardial infarction [1]. The World Health Organization (WHO) predicts that depression will become the second leading cause of premature death or disability worldwide by the year 2020.
The treatment of depression was dramatically changed about a half-century ago with the introduction of two classes of agents that were discovered by serendipity: monoamine oxidase inhibitors (MAOIs) and the tricyclic antidepressants (TCAs). The original tricyclic agents (e.g., imipramine) arose from antihistamine research, whereas the early monoamine oxidase inhibitors (e.g. iproniazid) were derived from work on antitubercular drug. Both classes of agents were demonstrated to dramatically improve the symptoms of depression, but had poor tolerability and risk profile. The discovery of the acute protein targets of antidepressant medications (monoamine transporters and monoamine oxidases), the recognition of the importance of the serotonergic system in the pathophysiology and treatment of depression, and the increasing need for the drugs with better tolerability and safety profile led to the development of numerous second-generation medications (e.g. selective serotonin reuptake inhibitors (SSRIs), selective and reversible monoamine oxidase inhibitors (eg. moclobemid), selective noradrenaline (e.g. reboxetine), dual noradrenaline and serotonin reuptake inhibitors (milnacipram, venlafaxin, duloxetin) and drugs with distinct neurochemical profiles such as mirtazapine, nefazadone and tianeptine (Fig. 1). Some newly introduced compounds may not offer unique spectra of action and are variations on old themes. Other new drugs are the results of innovative developmental strategies based on the better understanding of the pathophysiology of depressive disorder.
Fig. 1.
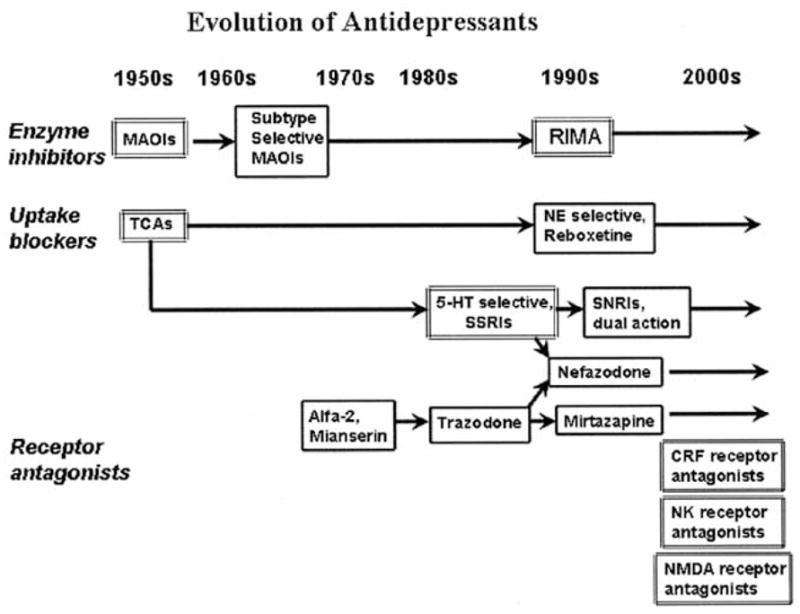
While agents modulating monoaminergic system will likely continue to dominate the treatment of major depression, a number of truly novel targets and agents [2–26] show considerable promise for refining treatment options. In this article major advances from monoamine-based treatment strategies and new promising directions in pharmacological research of depression will be reviewed.
2. MODULATION OF MONOAMINE SYSTEM
For over 4 decades, the biological approach to depression has been dominated by the monoamine hypothesis. The hypothesis proposed that depression was caused by functional deficit of monoamines (noradrenaline (NA), serotonin (5-HT), and possibly dopamine (D)) in the limbic (emotional) regions of the brain. This hypothesis was supported by the finding that treatment with reserpine (an antihypertensive drug, which depletes monoamines) caused depressive episodes in some patients and also by observations that effective antidepressant drugs exerted their primary effects by increasing intrasynaptic concentrations of these biogenic amines. Furthermore, the monoaminergic systems are extensively distributed throughout the network of limbic, striatal and prefrontal cortical neuronal circuits thought to support the behavioral and visceral manifestations of mood disorders. However, the hypothesis could not explain why up to 2–3 weeks of treatment were necessary to alleviate depressive symptoms, even though monoamine levels increased within 1–2 days. Nor could it explain several other anomalies, such as why some drugs (e.g. amphetamine and cocaine) that enhance noradrenergic or serotonergic transmission are not very effective antidepressants. Nevertheless, despite its drawbacks, monoamine hypothesis has continued to provide the predominant framework for understanding depression and has provided the rational for the development of several classes of antidepressants (see below 2.1–2.9).
The cell bodies of noradrenergic neurons are predominantly originate from the locus coeruleus (LC) with extensive connections to fear circuitry structures and corticotropin-releasing factor (CRF) system. Noradrenaline modulates 5-HT and dopamine release, particularly in the thalamocortical regions. LC noradrenergic system is vitally important in the control of concentration, attention, mood, sleep, emotions, blood pressure and pain regulation. Dysfunction of these pathways implicated in negative affect state in depression. Postsynaptic α1 and α2 recepors are likely to be implicated in the effects of NA on mood, attention and cognition in the frontal cortex.
A multitude of brain functions are influenced by serotonin, including cognition, sleep, motor activity, sensory perception, appetite, sexual behavior, temperature regulation, nociception and hormone secretion. The majority of serotonergic neuron bodies in the brain are localized in the raphe nuclei of the brainstem. Projections from these neurons to the frontal cortex may be important for regulating mood, whereas projections to basal ganglia may help control movement, obsessions and compulsions. Abnormalities in serotonergic function in anxiety may be the result of deficient or excessive innervations to key structures, and/or cellular mechanisms resulting in aberrant neurotransmission. At the cellular level, abnormalities may include abnormal regulation of 5-HT release and/or reuptake (a role of the presynaptic receptors) or abnormal responsiveness to 5-HT signal (a role of the postsynaptic receptors). The presence of 5-HT heteroreceptors on neurons of other neurochemical systems means that the end result of altering serotonin neurotransmission may be the modulation of other, nonserotonergic systems. The serotonergic neurotransmission can be pharmacologically manipulated by targeting serotonin reuptake transporter (SERT), presynaptic autoreceptors and different postsynaptic receptor subtypes [13] (see below 2.7 and Fig. 2).
Fig. 2. Targets in serotonergic synapse.
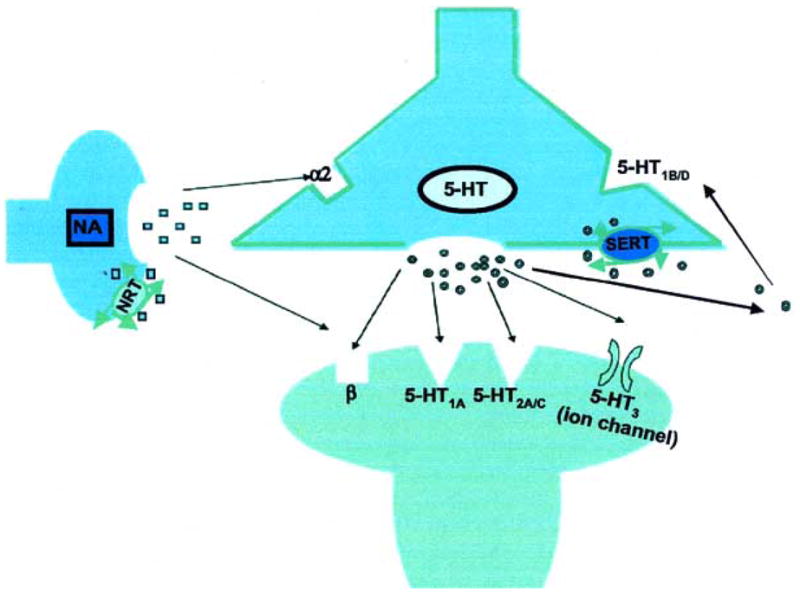
Released serotonin (5-HT) binds to target receptors and heteroreceptors.
NA- noradrenaline; SERT- serotonin reuptake transporter; NRT- noradrenaline reuptake transporter.
2.1 Tricyclic Antidepressants (TCAs)
Since the early 1960s for two decades the tricyclic antidepressants represented the major pharmacological treatment for depression. The TCAs have generally been considered a homogeneous group of drugs (Fig. 3), differing mostly in their potency to inhibit presynaptic norepinephrine or serotonin uptake and in their propensity for causing variety of unwanted effects. Their efficacy in the treatment of major depression is well established, and they also have proven to be useful in a number of other psychiatric disorders. These medications can be further divided into two main classes [12,13]. The tertiary amines (e.g. amitriptyline and imipramine) are drugs that are generally dual (serotonin (5-HT) and norepinephrine (NE)) reuptake inhibitors, are metabolized to secondary amines, and have a high burden of anticholinergic side effects. The secondary amines (e.g., nortriptyline and desipramine) are generally more selective at blocking NE reuptake with somewhat reduced anticholinergic side effects. The TCAs induce anticholinergic, antihistaminergic and cardiotoxic side effects which are related to their action on muscarinic (mainly M1), histamine (H1), adrenergic (α1) receptors and cardiac Na+ and Ca2+ channels [13,27–29]. The improved safety, tolerability, and broad therapeutic action of the newer drugs have resulted in displacement of the TCAs as the first-choice drugs for treating recurrent major depression and these compounds occupy a narrower but still important role in the psychopharmacological armamentarium.
Fig. 3.

2.2 Reversible and Selective Inhibitors of Monoamine Oxidase Type A (RIMAs)
The major goal of developing new MAO-A inhibitors is to avoid the severe life-threatening hypertensive reaction that can occur when irreversible inhibition of the A form of MAO, which is found in the gut as well as in the brain, decreases intestinal and hepatic degradation of tyramine, allowing excessive amounts of this naturally occurring pressor amine to be absorbed from the food. Minimalizing the complications of earlier generation agents inspired the development of both selective and reversible MAOAIs. The newer generation preferentially and reversibly affects the MAO-A, the enzyme system primarily responsible for deamination of those amines believed to be implicated in the ethiology of depression. Since these compounds form unstable complexes with MAO-A subtype, they can therefore be displaced from the enzyme by tyramine, making it possible for ingested tyramine to be metabolized, markedly diminishing the need for the dietary restrictions that plague the use of older irreversible MAO-A inhibitors. The new class of selective and reversible inhibitors of MAO-A includes moclobemide, brofaromine (Fig. 3), cimoxatone and befloxatone. Moclobemide is a benzamide derivative containing a morpholine ring (p-chloro-N-[2-morpho-linoethy)benzamide) (Fig. 3). The neuropharmacological profiles of cimoxatone [30] and moclobemide [31,32] have been reviewed. The development of brofaromine has been stopped because of the side effects [33]. In preclinical assays befloxatone was of higher efficacy than previous reversible (moclobemid) and irreversible (clorgyline) MAOIs, TCA (imipramine) and SSRI (fluoxetine) and is currently evaluated in clinical trial for depression [34,35]. MAOIs are generally used in patients with atypical depression or who do not respond to other antidepressants [13,36,37].
2.3 Selective Serotonin Reuptake Inhibitors (SSRIs)
Although much of the interest in the early development of antidepressant drugs was on the norepinephrine system, by the 1980s, attention focused on the importance of serotoninergic system in the pathophysiology and treatment of depression.
The search for selective serotonin reuptake inhibitors have been fruitful, and the development of such drugs has represented a revolution in the pharmacotherapy of depression, moving management from the psychiatrist’s clinic to the general practitioner [38]. This led to the introduction of SSRIs in the 1980s, which have dominated the treatment of depression for the past decade. Fluoxetine was the first truly specific serotonin uptake blocker. It containes the propylamine side chain found in most tricyclics (Fig. 3). Sertraline, the second specific serotonin uptake inhibitor to reach the market, is structurally different, being a 1-amino-tetrahydronapthalene (Fig. 3). Paroxetine is a phenylpiperidine derivative, while fluvoxamine is a 2-aminoethyl oxime arylketone (Fig. 3). Citalopram has the dimethylaminopropyl side chain (Fig. 3). These compounds selectively and powerfully block the reuptake of serotonin at central synapses [13, 39–41]. Because reuptake is the primary mechanism of serotonin inactivation, inhibition of the serotonin reuptake carrier raises the level of this neurotransmitter in the synapse. In the short term, the increased concentration of serotonin available to act on pre-synaptic autoreceptors leads to reduction in serotonin neurotransmission. However, approximately 2 weeks after the initiation of treatment, the pre-synaptic serotonin autoreceptors become desensitised, the release of serotonin from nerve terminals is increased and serotonergic neurotransmission is potentiated [13,42].
Although SSRIs differ in chemical structure [43] (Fig. 3) and pharmacokinetic characteristics, all are extensively biotransformed before excretion, initially through oxidative pathways. Fluvoxamine is metabolized by oxidation, oxidative deamination and hydrolysis to several metabolites [44]. Only few of them have been tested for their effects on monoamine uptake mechanisms, and showed no or only weak inhibition of serotonin uptake (i.e the carboxyl acid derivative) comparing to the parent drug [45]. The main metabolites of paroxetine are also weak in vitro inhibitors of serotonin uptake mechanisms. Fluoxetine, citalopram and sertraline are all first demethylated to their nor-derivatives norfluoxetine, demethylcitalopram and norsertraline, which are also SSRIs. All the parent compounds and their nor-derivatives are further biotransformed to a number of metabolites.
Paroxetine and sertraline are available only as single enantiomers. On the contrary, both fluoxetine and citalopram are available as racemic mixtures. In each case, it is the enantiomer with the greatest serotonergic activity that is marketed as an antidepressant [46]. Fluvoxamine is not chiral. The (S)- and (R)-enantiomers of fluoxetine are almost equipotent [47,48], but the (S)-enantiomer of its metabolite, norfluoxetine, is a more potent serotonin reuptake inhibitor than the (R)-enantiomer [48]. In patients, the plasma concentration of (S)-norfluoxetine usually twice exceeds that of the (R)-enantiomer, whereas the ratio of (S)-fluoxetine to (R)-fluoxetine ranges between 1 and 3.5. The (S) enantiomers of citalopram, the plasma level of which represents approximately one-third of the total plasma concentration of citalopram [49,50] is more potent serotonin reuptake inhibitors than the (R) form [51]. The (S)-enantiomer of citalopram and of fluoxetine, and its major metabolite norfluoxetine may have antimigraine effects not found with the (R)-enantiomer of fluoxetine.
Since SSRIs demonstrate minimal affinity for muscarinic cholinergic, alfa-adrenergic and histamine receptors, they are largely devoid of the adverse effects typically associated with TCAs. Numerous studies have demonstrated that the SSRIs are as effective as the TCAs in the treatment of depressive disorder but better tolerated and have superior safety profile in overdose for most patients. However, the ability of several agents in this class to inhibit the cytochrome P4502D6 enzyme increases the potential for drug-drug interactions [13,52]. Recent studies have surprisingly demonstrated that SSRIs, similarly to older TCAs may interfere with cardiovascular Ca2+, Na+ and K+ channels, which could explain cardiovascular side effects occasionally observed in patients [27,52–61]. A major problem of the SSRIs is sexual dysfunction [62]. A serotonin reuptake inhibitor that shared the good efficacy and side effect profile of available SSRIs but lacked the propensity to cause sexual dysfunction would represent a significant therapeutic advance.
2.4 Dual Serotonin and Noradrenaline Reuptake Inhibitors (SNRIs)
There is some evidence, although it is not universally accepted, that dual reuptake inhibitors have more rapid onset of action or are more efficacious than single monoamine reuptake inhibitors [63,64]. Available dual reuptake inhibitors include the tertiary TCAs, venlafaxine and milnacipran (Fig. 3). Other effective new dual reuptake blocker, duloxetine, is in clinical development in the United States [65, 66]. The venlafaxine and milnacipran (Fig. 3), have some difference in their structure from selective inhibitors of serotonin or noradrenaline uptake. From the point of the putative mechanism of action, these drugs are similar to the tertiary amine tricyclic antidepressant agents (TCA), since they affect both serotonin and noradrenaline reuptake. Venlafaxine, a phenethylamine bicyclic derivative, is a weaker inhibitor of synaptosomal noradrenaline uptake than serotonin and has also slight dopamine uptake inhibitory properties [13,67]. Venlafaxine exists as a racemic mixture of the (R)- and (S)-enantiomers. (−)-Venlafaxine inhibits reuptake of serotonin and noradrenaline, whereas the (+)-isomer primarily inhibits serotonin reuptake. Milnacipran, a 1-aryl-2-(aminomethyl)cyclopropane-carboxylic acid derivative, almost equipotently inhibits both serotonin and noradrenaline reuptake without significantly affecting dopamine transporters [68]. Milnacipran shares some structural similarities with the monoamine oxidase (MAO) inhibitor tranylcypromine [69].
2.5 The Selective Noradrenaline Reuptake Inhibitors
Reboxetine is a recently marketed compound. Chemically it is (RS)-2-[(RS)-α-(2-ethoxyphenoxy)]–benzylmorpholine methan-sulphonate (Fig. 3), shares some chemical similarities with viloxazine, and to a lesser extent fluoxetine and structurally related compounds. It exists as a racemic mixture of the (S)- and (R)-enantiomers both of which inhibit noradrenaline uptake in rats, the (S)-enantiomer being approximately 20 times as potent as the (R)-enantiomer [70,71]. As expected, it has more distinct adverse side effect profile than the SSRIs [13], with urinary and cardiovascular side effects of greater concern.
2.6 Drugs Enhancing Dopaminergic System
There is evidence for the reduction in the activity of certain dopamine (D)-containing pathways in depressed patients, which may underlie symptoms of anhedonia and anergia. In addition, drugs enhancing dopaminergic neurotransmission may have antidepressant properties. Pramipexole, a D2 receptor agonist and 5-HT2 receptor antagonist, has been reported to be as effective as fluoxetine in treating depression [72]. Nomifestine, a potent D reuptake inhibitor and an effective antidepressant, was withdrawn from the market because of adverse hematological side effects. Sertraline, an SSRI antidepressant, also possesses D reuptake antagonist properties. Tianeptine also may enhance the functional responsiveness of dopamine D2 and D3 receptors. Bupropion seems to be both a dopamine and NA reuptake inhibitor [13]. Several novel D reuptake inhibitors, including some that are also NA or 5-HT inhibitors are under development.
2.7 Drugs Targeting Serotonin Receptors
During the last two decades significant progress has been made in understanding the function of serotonergic system. Several distinct serotonin (5-HT) receptor subtypes, categorized into seven main classes (5-HT1–5HT7), are known to mediate the physiological effects of the neurotransmitter [73,74]. Given the evidence for the involvement of the 5-HT system in depression and the mechanism of action of antidepressants, much interest has focused on the role of 5-HT receptors as potential therapeutic targets in depression [75].
2.7.1 5-HT1A Receptor Ligands
Within the 5-HT1 family, at least five subtypes have been documented: 5-HT1A, 5-HT1B, 5HT1D, 5-HT1E and 5HT1F. The 5-HT1A receptor subtype is widely distributed in the central nervous system and has been the focus of considerable research effort since the early 1980s. Until recently, only nonselective 5-HT1A receptor antagonists-such as (−)-pindolol (Fig. 4) and (−)-propranolol-had been available. It has been well documented that the delayed onset of action of SSRIs may result from the time taken to desensitize 5-HT autoreceptors on cell bodies (5-HT1A) and/or presynaptic terminals (5-HT1B) during chronic SSRI administration. This raises the possibility that selective 5-HT autoreceptor antagonists, used either alone or in combination with SSRIs, may prove to be effective antidepressants with a more rapid onset of action than can be achieved with an SSRI. Pindolol, a mixed 5-HT1A and β-adrenoceptor antagonist, has recently been shown to potentiate the antidepressant effect of SSRIs and decreased the onset of action [76–78]. Further support of this hypothesis was the observation that a 5-HT1A antagonist WAY 100635 was found to potentiate the antidepressant effects of several SSRIs [79,80]. Consequently, there has been considerable interest by several groups to identify drug design strategies focusing on incorporating a 5-HT1A antagonist component into a 5-HT reuptake inhibitor to bring about a more immediate and complete antidepressant effect [81–85]. Perez et al. [81] synthesized several molecules by covalent coupling of known 5-HT1A antagonists of the aminopropanol family (pindolol, propranolol and penbutolol) with SSRIs like fluoxetine or paroxetine and with the new serotonin noradrenalin reuptake inhibitor antidepressant milnacipram (Fig. 4) [81]. Most of these hybrid molecules were as or even more potent 5-HT1A antagonists than the parent aminopropranol derivatives, however were found to interact moderately with the 5-HT uptake site [81]. Other group focused on modifying a known class of indoles which embrace the known 5-HT uptake inhibitor indalpine. This series of indoles was reported as being potent 5-HT uptake inhibitors and was used to attempt to incorporate 5-HT1A antagonistic activity [84–85]. Work is in progress to evaluate the in vivo pharmacological properties of these hybrid molecules.
Fig. 4.

The discovery that anxiolytic agent buspirone which binds with high affinity to 5-HT1A receptors has encouraged the development of 5-HT1A R ligands as potential antidepressants. Several structural classes of agents were identified and known to bind 5-HT1AR sites, such as aminotetralins, indolylalkylamines, ergolines, aporphines, aryloxyalkylamines and arylpiperazines [86]. Among these, arylpiperazine derivatives represent one of the most important classes of 5-HT1AR ligands [82,83,87,88]. For a recent comprehensive review summarizing structure-affinity relationships of agonists and antagonists, selectivity over other serotonergic, alfa1-adrenergic and dopaminergic D2 receptors and ligand-receptor interactions see Lopez-Rodriguez [89].
During last few years, several selective 5-HT1A receptor antagonists have been available just naming a few (Fig. 4) [89–96]. These include the phenylpiperazine derivative WAY-100635 [91] and its close structural analogue p-MPPI [92], the pindolol analogue LY-297996 [93], the aminomethylpiperidine SL-88.0338 [94], DU-125530 [95] and NAD-299 [96] (Fig. 4). In certain animal models of anxiety these compounds have demonstrated anxiolytic properties, however the precise mechanisms underlying this effects remain to be determined [90]. The development of such compounds as novel anxiolytics and antidepressants is being actively pursued by a number of pharmaceutical companies and data on the therapeutic potential of these compounds may be available soon.
Other investigators have evaluated 5-HT1A receptor agonists or partial agonists (e.g. buspirone, flesinoxan) (Fig. 4), which are thought to combine the ability to activate postsynaptic receptors and desensitize autoreceptors more quickly. Unfortunately, in clinical trials these compounds showed only weak antidepressant efficacy and/or significant side effects [97,98].
2.7.2 5-HT2 Receptor Antagonist
Another potential strategy might be to combine an SSRI with an antagonist of the 5-HT2 receptor(s) responsible for specific side effects such as sexual dysfunction and sleep disorder [42]. Nefazodone is a phenoxyethyl triazoline phenylpiperazine derivative (Fig. 4) is structurally related to trazodone. It has potent antagonistic effects on 5-HT2A receptors and moderate serotonin and noradrenaline reuptake inhibiting properties [99]. Nefazodone indeed does appear to have less sexual side effects than SSRIs and TCAs [99], but is considered clinically as an antidepressant of week efficacy. Mirtazapine shares this absence of sexual dysfunction as well as reduced incidence of nausea mostly due to 5-HT2A and 5-HT3 receptor antagonism [100]. Another compound YM 35992 (Fig. 4), which exhibit 5-HT2A receptor blocking properties together with more potent reuptake inhibition, may prove to be more promising [101,102].
Fig. 5.

The use of several selective 5-HT2C receptor antagonists has been investigated in depression, including SB-200646, SB 221284, SB 206553, SB-242084 and SB-243213 [103]. Unfortunately, the 5-HT2C antagonists have tended to stimulate the mesolimbic dopamine system and interfere with cytochrome P450 isoenzymes. Deramciclane is a 5-HT2A and 5-HT2C antagonist currently in phase III trials for anxiety [104]. A combined 5-HT2C antagonist and melatonin agonist, agomelatine (S 20098), is also under development [105].
In contrast, the 5-HT2C agonist Ro 60-0175 and Ro 60-0332 have demonstrated activity in various animal models of depression and panic disorders. Such results are consistent with hypothesis that 5-HT has complex, dual action on the neural mechanism of anxiety by either facilitating or inhibiting different kinds of anxiety in different brain regions [104].
2.7.3 Other Serotonin Receptor Targets
There is also interest in several other 5-HT receptor subtypes (e.g. 5HT6, 5HT7 ) as potential target for antidepressant treatment [43,89,106]. 5-HT7 receptor is positively coupled to the cyclic adenosine monophosphate (cAMP) signal transduction cascade [75], which has contributed to the interest in agonists for this receptor subtype. 5-HT7 receptor is a proposed target for the treatment of depression and possibly jet lag. Studies of the selective 5-HT7 receptor antagonist SB-269970 have demonstrated antidepressant effects in animal models. Similar agents, including DR 4004, DR4365, LY 215840 and SB-258719 may have some potential in depression [107].
2.8 Tianeptine: a Serotonin Reuptake Enhancer
Unlike antidepressants which facilitate postsynaptic effects of central neurotransmitters by blocking their reuptake or by inhibiting their catabolism (i.e. MAOIs), tianeptine increases presynaptic serotonin reuptake, thereby reducing synaptic availability of the neurotransmitter [108–110]. Tianeptine also reduces stress-induced atrophy of neuronal dendrites and basal and stress-evoked activity of HPA axis. The fact that enhancers as well as inhibitors of 5-HT uptake act as antidepressants, challenges any simple explanation of their mechanism of action [111,112]. Chemically it is a modified tricyclic (i.e. the sodium salt of 3-[(3-cloro-6-methyl-5,5-dioxo–6,11-dihydrodibenzo(c,f(1,2-thiazepin))11-yl)amino]-heptaonic acid (Fig. 4). Tianeptine is a racemate and its antidepressant-like properties associated with its effect on serotonin disposition appear to depend on the (−)isomer [113].
2.9 Mirtazapine: an α2-Adrenergic Auto- and Hetero-Receptor Blocker
This is the only antidepressant that increases noradrenergic and serotonergic (5-HT1-mediated-) neurotransmission through a blockade of central α2-adrenergic auto- and hetero-receptors. [13,114,115] Chemically, mirtazapine is 1,2,3,4,10,14b-hexahydro-2-methyl-pyrazino[2,1-a]-pyrido-[2,3-c]benzazepin(Z)-2 butenedioate (Fig. 4). Mirtazapine is an antidepressant with faster onset of action than SSRIs and with similar tolerability and safety profile. The introduction of mirtazapine has led to renewed interest in adrenergic receptors as a target for antidepressants [116]; however, it remains unclear whether this property is responsible for mirtazapine’s antidepressant effects. Mirtazapine also inhibits several postsynaptic serotonin receptor types (including 5-HT2A, 5-HT2C and 5-HT3 receptors) and can produce gradual downregulation of 5-HT2A receptors [117]. Mirtazapine is also a potent histamine H1 -receptor antagonist, and correspondingly relatively sedative [13].
3. MODULATION OF NEUROPEPTIDE RECEPTORS
During the latter half of this century, many fascinating discoveries about neuropeptides have been made, but the clinical rewards have been less; thus, skepticism remains as to the medical benefits of neuropeptide research. Inspite of the difficulties and first disappointing clinical data, the development of selective and stable neuropeptide receptor antagonists with better bioavailability has not lost its position as one of the most promising pharmacological research directions. Antagonists of substance P, corticotrophin-releasing factor (CRF), neuropeptide-Y, vasopressin, cholecystokinin-B and melanin-concentrating hormone receptors are all being investigated. Clinical development, however, will depend upon finding non-peptide analogues that have good oral bioavailability and can penetrate the blood-brain barrier.
3.1 Neurokinin (Substance-P) Receptor Antagonists
Substance P (SP), discovered in 1931 by von Euler and Gaddum, is one of the best-known neuropeptides and the most abundant so-called neurokinin (NK) in the mammalian brain and peripheral (notably sensory) neurons. SP was also localized in brain regions that coordinate stress responses and receive convergent monoaminergic innervation (e.g. the amygdala and locus coeruleus). Recent behavioral study using a SP receptor (SPR) knockout mouse has also suggested that SP plays an important role in the adaptive response to stress and emotional behavior [118]. The substance P receptor, called NK1, has been pursued as a therapeutic target for diverse conditions including pain, inflammation, emesis, migraine, schizophrenia and depression (for recent reviews see [7,8,119–120]. Substance P and other neurokinins are, hypothetically, mediators of emotional distress in depression, anxiety, or schizophrenia. Since the first report on synthetic selective substance P receptor antagonist (SPA) in 1991 [121], several antagonists have been synthesized, but none has been proven as an effective one in reducing pain. However, one of the latest antagonists, MK-869 (Fig. 5), has been reported to have antidepressant properties [122]. In a six-week randomized, double blind placebo-controlled study, MK-869 was as effective as SSRI, paroxetine in patients with moderate to severe depressive disorder, and differed in adverse effects. Nausea and altered sexual function had been caused by paroxetine, whereas the substance-P antagonist produced less of these effects but more irritability [122,123]. Preclinical studies showed that MK-0869 and its metabolites had no significant affinity for monoamine reuptake sites or transporters, or for MAO, or monoamine receptors; conversely, established antidepressants showed no significant affinity for the human NK1 receptor. Interestingly, chronic treatment with conventional antidepressant drugs reduced SP synthesis [122–124] suggesting that SP may be a final common pathway for the action of various classes of antidepressants [125]. There is also preliminary evidence that substance P and NK1 receptor density may be altered in depressive disorder, suggesting a possible link between substance P and pathophysiology of depression [reviewed in 120]. Interestingly, results of some animal studies have suggested that stimulation of hippocampal neurogenesis by blockade of NK1 receptors in the amygdale and interaction with monoamines is involved in the psychotherapeutic effects of SPAs [119,120,126].
Currently several new SPAs antagonists are synthesized and being tested (Fig. 5) in both preclinical and clinical models of depression and schizophrenia [7,8,119,120,126–129]. It must be emphasized, however, that ongoing trials of various neurokinin antagonists are too early in testing to be conclusive but neurokinin antagonists as a novel psychotropics are still a very provocative possibility.
3.2 Dual Neurokinin-1 Receptor Antagonist-Serotonin Reuptake Inhibitors
The mode of action of NK1 antagonists is believed to involve an indirect modulation of 5-HT function via noradrenergic pathways [130,131] and NK1 receptor knockout mice and wild-type mice treated with NK1 antagonist have an attenuated presynaptic 5HT1A receptor function [131,132]. These observations led to an interesting idea that the combination of serotonin reuptake inhibition with NK1 antagonism (modulating 5HT1A function) may result in a new class of antidepressants with an improved onset of action and better efficacy [133,134]. Ryckmans and his co-workers designed and synthesized several of these dual NK1 antagonist-serotonin reuptake inhibitor compounds of the family of benzyloxyphenethyl piperazines and some of them were demonstrated to be orally active and validated in animal models of depression sensitive to both mechanisms [133,134].
3.3 Corticotropin-Releasing Factor (CRF) Receptor Antagonists
The CRF family of neuropeptides has undergone considerable expansion during the past couple of years. Recently, in mammalian brain, this family of neuropeptides consists of CRF, urocortin (Ucn), Ucn II (also known as stress-copin-related peptide) and Ucn III (also called stresscopin) [135–141].
Two major CRF G-protein-coupled receptor types (CRFR1 and CRFR1) have been identified: CRFR1 is highly expressed in the brain, including the anterior pituitary, locus coeruleus, amygdala, neocortex, hippocampus and cerebellum. CRFR2 is more abundant in peripheral tissues. Whereas CRF is relatively selective for CRF1 over CRF2 receptors, Ucn binds to both CRF1 and CRF2 with high affinity. Ucn II and Ucn III are selective ligands for CRF2 receptors. Neither Ucn II nor Ucn III binds to non-receptor CRF-binding protein (CRFBP), whereas CRF and Ucn do [139–141].
After its isolation and characterization in the 80s, the corticotropin-releasing factor, a 41 amino acid-containing Neurokinin1 (Substance P) Receptor Antagonists peptide, has been found to mediate not only the endocrine, but also the autonomic, immunological and behavioral responses of mammalian organisms to stress [135]. There is evidence that CRF is hypersecreted from hypothalamic as well as from extrahypothalamic neurons in depression. CRF acts both as a neurohormon resulting in hyperactivity of the hypothalamic-pituitary-adrenal axis and a neuromodulator in other brain regions [10,11,135,142–144]. Recent research results have indicated that CRF mediates the responses to stress and anxiety via the CRF1 receptor [138,139].
Within the last few years, a number of pharmaceutical companies have developed selective, small molecule CRFR1 antagonists, including R121919, SC 241, NBI-103, NBI-104, NBI 37582, NBI-27914, NBI 34041, NBI-29356, NBI-31199, NBI-31200, NBI 30545, CRA 1000, CRA 1001, CRR 0165, PD 171729 and SSR125543A, which are currently being intensively investigated [10,11,26,145,146] (see some on Fig. 5). These compounds block the effects of CRF both in vitro and in vivo. There is also evidence that these agents possess anxiolytic and antidepressant activity in animal behavioral models [10,11,144–148]. Structure of several promising compounds is shown in (Fig. 5). Some of these CRFR1 antagonists are currently undergoing clinical trials.
3.4 Vasopressine Receptor Antagonists
Arginin vasopressin (AVP), a cyclic nonapeptide that is synthesized centrally in the hypothalamus, was identified in 1954. Two major subtypes of AVP receptors have been identified [149–151]. V1a receptors are found in blood vessels and in CNS [152], while V2 receptors are predominantly located in the cells of the renal collecting system, although there is some evidence for central V2 receptors also. AVP participates in the hypothalamic-pituitary-adrenal axis, regulating pituitary ACTH (corticotrophin) secretion by potentiating the stimulatory effects of CRF. The ACTH releasing properties occur via V1b receptor subtype. Extra-hypothalamic AVP-containing neurons have also been identified and characterized in rat medial amygdala that innervates lateral septum and ventral hippocampus [153]. In these structures, AVP was suggested to act as a neurotransmitter, exerting its action by binding to specific G-protein-coupled AVP receptors, i.e., V1a and V1b [154–156]. It has been established that AVP is involved in various types of behavioral processes [157,158]. In animal model of anxiety the intra-septal application of a mixed V1a/b receptor antagonist d(CH2)5Tyr(Et)VAVP was found to produce anxiolytic effects, in contrast infusion of an antisense oligodeoxynucleotide to the V1a subtype mRNA reduced anxiety [159,160]. In addition, chronic immobilization stress has been shown to increase V1b receptor mRNA level in the rat brain [161]. Griebel and his colleagues have recently demonstrated potent anxiolytic- and antidepressant effects using the first orally active non-peptide V1b receptor antagonist, SSR149415, in a variety of rodent models of anxiety and depression, providing the framework for a novel perspective on potential antidepressant effects of V1b receptor antagonists [162].
3.5 Neuropeptide Y (NPY) Receptor Ligands
NPY is a 36-amino acid containing peptide is widely distributed in the central nervous system. In addition to its multiple effects NPY is thought to play a role in the pathophysiology of certain mood disorders and in the mechanism of action of antidepressant drugs [163–165]. At present five G protein-coupled NPY receptor subtypes have been cloned (Y1–Y5). Pre-clinical studies have demonstrated multi-level changes in NPY immunoreactivity, NPY receptor mRNA expression and NPY receptor functioning both in animal models of depression and following antidepressant treatment [165]. Available data suggest that the NPY Y1 receptor subtype seems to be the major player. As NPY has been shown to display behavioral activity similar to that of SSRIs in an animal model of depression [166], studies investigating the possible contribution of the serotonergic system would be of interest. There is also clinical evidence suggesting a role for NPY in depression [165].
3.6 Melanin-Concentrating Hormone (MCH) Receptor Antagonists
Melanin-concentrating hormone (MCH), a cyclic 19-aminoacid polypeptide, is produced predominantly by neurons in the lateral hypothalamus and zona incerta, which project broadly throughout the brain [167]. Several lines of evidence implicate MCH as an important mediator in the regulation of energy balance and body weight. The effects of MCH are mediated through G protein-coupled receptors. At present two main MCH receptor subtypes were identified (MCH1 and MCH2 receptors)[25,168–170]. In a recent study Borowsky et al. [25] have demonstrated potent anxiolytic-and antidepressant effects using a novel selective, high-affinity MCH1 receptor antagonist SNAP-7941 (Fig. 6.), in a variety of rodent models of anxiety and depression. The compound also exerted potent anorectic effects.
Fig. 6.

4. MODULATION OF NICOTINIC ACETYLCHOLINE RECEPTORS (NACHR)
There is accumulating evidence suggesting that the hypercholinergic neurotransmission associated with depressed mood state, may be mediated through excessive neuronal nicotinic receptor activation and conventionally used antidepressants including imipramine, nortriptyline, amitriptyline, desipramine, fluoxetine, sertraline, paroxetine, nefazodone, nisoxetine, citalopram, nomifensine and bupropion are potent nAChR antagonists [171]. Furthermore, preliminary evidence suggests that the potent centrally acting nAChR antagonists, mecamylamine, which is devoid of monoamine reuptake inhibition, may reduce symptoms of depression and mood instability in patients with comorbid depression and bipolar disorder [172, 173]. With this in mind, future preclinical and clinical research is warranted to investigate the therapeutic potential of selective nicotinic receptor antagonists for the treatment of mood disorders.
5. MODULATION OF GLUTAMATE RECEPTORS
L-glutamic acid (glutamate), primarily derived from intermediary glucose metabolism, is the major excitatory amino acid in the central nervous system. Several glutamate transporters regulate synaptic transmission and synaptic cleft levels of glutamate and several subtypes of glutamate receptors with different function and distribution are identified. Ionotropic glutamate receptors, comprise of N-methyl-D-aspartate (NMDA), alpha-amino-3-hydroxy-5-methylis-oxazole propionate (AMPA), and kainite, are generally postsynaptic and mediate fast excitations and synaptic plasticity associated with the opening of sodium and calcium-permeable ligand-gated ion channels. Metabotropic glutamate receptors are present in various sites (presynaptic, postsynaptic, glial, and heterosynaptic) and modulate postsynaptic excitability to glutamate by providing positive or negative feedback to dectrease release of presynaptic glutamate.
There is accumulating preclinical and clinical evidence showing structural and functional abnormalities of glutamatergic transmission in depressive disorder [reviewed in 15,21], suggesting that the modulation of this system may represent a suitable strategy for the development of novel antidepressants [reviewed in 23,174,175].
The NMDA receptor is a ligand gated ion channel, which mediates excitatory synaptic transmission in the CNS. Channel opening is triggered by synaptically released glutamate; however, the NMDA receptor is unique in that it also has an absolute requirement for the binding of glycine, a co-agonist, for receptor activation [176,177]. NMDA receptor is composed of multiple protein subunits each encoded by a unique gene [178,179]. The physiological receptor is a heteromer containing both NR1 and NR2 subunits. The NR1 subunits contain the glicin binding sites whereas the NR2 subunit contains glutamate binding sites [180,181]. There are regional differences in expression of NMDA receptor subunits throughout the CNS [182]. The subunit composition significantly affects the sensitivity to a group of allosteric modulators, which includes protons, polyamines, Zn2+, and oxidizing/reducing agents. The ion channel associated with the NMDA receptor is permeable to Na+ and Ca2+. Ion flux through the NMDA channel is also regulated by the polarization state of the postsynaptic membrane. The channel pore has a binding site for Mg2+, which occludes the pore when occupied. The degree of Mg2+ binding is dependent on polarization state: upon depolarization, Mg2+ binding is reduced, decreasing the block and allowing ion flux. Thus, both presynaptic glutamate release and postsynaptic depolarization is required for NMDA receptor activation. Glutamate plays an essential role as a neurotransmitter in many physiological functions; however, under various conditions neurons can become so sensitive to glutamate that it actually kills them through receptor-mediated depolarization and/or calcium influx. Increased susceptibility can be governed by a number of factors, such as energy deficits, neuronal depolarization, an increase in glutamate release, malfunctioning of neuronal and glial uptake, or changes in glutamate receptor properties or expression pattern.
The hypothesis that aberrant activation of NMDA receptors underlies a number of neurodegenerative (e.g. stroke, Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, ALS) and psychiatric disorders (depression and anxiety) has generated considerable interest in the NMDA receptor as a target for new pharmacotherapies [183–185].
Interestingly, several studies showed that chronic antidepressant treatment with conventional drugs can modulate NMDA receptor subunit expression and in turn NMDAR function [186–190]. Preclinical studies demonstrated the antidepressant-like actions of different NMDAR antagonists such as 2-amino-7-phosphoheptanoic acid (AP-7), 1-aminocyclopropancarboxylic acid (ACPC), memantine, eliprodil and fenprodil [175]. AP-7 is a competitive NMDAR antagonist, memantine is a low affinity uncompetitive channel blocker, ACPC is a partial glycin agonist, eliprodil and ifenprodil act at the polyamine binding sites associated with NMDARs [175]. This is consistent with a case report demonstrating that amantadine, an antiviral agent with NMDA receptor antagonistic properties, has clinical efficacy in depression [191]. A placebo-contolled pilot study has suggested that the administration of single dose of ketamine, an NMDA receptor antagonist, produced a rapid antidepressant effects in depressed patients [192]. NMDA-receptor antagonists are also reported to block the atrophy of hippocampal pyramidal neurons and the down-regulation of neurogenesis that occurs in hippocampus in response to stress [17,18]. The chemical structure of several NMDA receptor antagonists is shown in (Fig. 6).
Interestingly, an effective dual serotonin and noradrenalin reuptake blocker antidepressant, milnacipran was found to be a non-competitive NMDA-receptor antagonist [193]. By increasing its specifical affinity for the NMDA receptor and decreasing its inhibitory effect on the 5-HT/NE reuptake, Shuto and his co-workers developed a new class of efficient NMDA-receptor antagonists [193].
AMPA receptor potentiators such LY392098, LY451646 also showed antidepressant-like effects in several animal models [23].
New pharmacological agents that modulate glutamatergic activity are expected in the near future, with both metabotropic and ionotropic receptors as targets. Several NMDAR antagonists with potential antidepressant properties are already in the phase of development or preclinical evaluation.
6. MODULATION OF GABA (GAMMA-AMINOBUTYRIC ACID) RECEPTORS
Gamma-aminobutyric acid, formed by the decarboxylation of glutamate, is the predominant inhibitory neurotransmitter in the central nervous system. There are two different superfamilies of GABA receptors: GABAA and GABAB. The protein of GABAA receptor contains binding sites for benzodiazepines, barbiturates, neurosteroids, picrotoxin and other anesthetics and is a chloride channel regulated by GABA. This receptor subtype is targeted by anxiolytic and sedative medications. GABAB is a G-protein coupled receptor that has been implicated in presynaptic inhibition of GABA release as well as postsynaptic inhibition on some cell bodies or dendrites.
There has been a renewed interest in GABA receptors as a potential therapeutic target in depression with the discovery that major depressive disorder is associated with reduction in GABAergic transmission in the brain. Furthermore, electroconvulsive therapy and antidepressant treatment raise GABA levels in depressed patients, suggesting that normalizing GABA function may contribute or reflect effective antidepressant treatment [194]. Thus, correction of GABAergic dysfunction by modulating multiple subtypes of ionotropic GABAA receptors and G protein-coupled GABAB receptors could be an important therapeutic target for development of new antidepressants.
7. MODULATION OF GLUCOCORTICOID RECEPTORS (GRS)
It is well-known that Cushing's syndrome (hypersecretion of cortisol) is associated with a high incidence of depression and impairment in memory. Similarly, pharmacological usage of glucocorticoids may also lead to mood changes and impaired memory. Furthermore, in depressive patients hypercortisolemia is associated with cognitive dysfunction and reduced number and/or function of GRs. In each of these conditions, reduction of glucocorticoid level, either through discontinuation of steroid treatment or through usage of agents that block glucocorticoid synthesis, ameliorates the adverse behavioural effects. Although clinical usage of the currently available antiglucocorticoid drugs is limited by significant adverse effects, development of drugs specifically targeting the glucocorticoid receptor may lead to innovative strategies in the treatment of mood disorders [reviewed in 195,196]. A recent study has reported favorable results with short term use of GR antagonist, mifepristone (17(-hydroxy-11(-(4-dimethylaminiophenyl)17((1-propynyl)estra-4,9-dien-3-one; RU486; C-1073), in major psychotic depression [197].
8. MODULATION OF CYTOKINE RECEPTORS
Cytokines are low molecular-weight proteins or glycoproteins produced by different immune cells in response to a wide range of physiological and pathophysiological stimuli. Initially cytokines were considered to be mediators of communication between immune cells, but recently it is well accepted that they play important role in the central nervous system [198]. During the last decade cytokines have been implicated in mood disorders and a psychoneuroendocrinoimmunological model of depression was suggested. This model was built on: 1) depressive effects of various cytokines in experimental animals; 2) experimental work investigating the depressive effects of immunological therapies (interferon-alpha and interleukine-2 therapy) in patients suffering from infectious viral disease (chronic hepatitis B or C, AIDS, etc.) or cancer; 3) studies assessing circulatory levels or cytokine production in stimulated lymphocytes of depressed patients; 4) investigations showing interaction between proinflammatory cytokines such as IL-1 and the hypothalamo-pituitary-adreno-corticotrope axis in depressed patients or in experimental animals; 5) studies showing modulatory effects of different antidepressants on production of proinflammatory cytokines; 6) accumulated experimental evidence showing that many of the functions that altered in depressed patients, including eating, sleeping, cognition, and endocrine activity, are influenced by cytokines [for recent reviews see 199–201].
Depression was associated with increased production of several proinflammatory cytokines such as IL-1, IL-6, IFN, TNF both in experimental animals and in humans. One of the most interesting cytokine with respect to stress and depression is interleukin-1-beta (IL-1β). IL-1 is a potent activator of the HPA axis via the stimulation of the CRF and may also modulate noradrenergic and serotonergic mechanisms in the brain by various mechanisms [reviewed in 199–201]. Should further work further establish the relationship between cytokines and depression, medications targeting cytokine receptors might represent novel antidepressants.
9. MODULATION OF OPIOID RECEPTORS
Increasing evidence have implicated the involvement of opioid receptor system in the pathophysiology of depressive disorder and in the mechanism of action of known antidepressants. Early clinical experiments demonstrated that opioid peptides had antidepressant activity in human patients. Also, enkephalinase inhibitors, which prevent the degradation of endogenous enkephalins, produced antidepressant-like effects mediated through the δ-opioid receptor in animal models of depression. Recent studies have demonstrated antidepressant effects of non-peptidic δ-opioid receptor agonists SNC80, BW373U86, (+)BW373U86 and BU48 (Fig. 6) in animal models [reviewed in 202].
10. MODULATION OF CANNABINOID RECEPTORS
Recently, there has been renewed interest in cannabis and its active constituents, cannabinoids, as therapeutic agents [203,204]. The most important natural cannabinoid is the psychoactive Δ9-Tetrahydrocannabinol (THC); others include cannabidiol and cannabigerol. Physiological actions of endocannabinoids in the CNS are mediated by the activation of a specific cannabinoid receptor, the CB1 receptor [205]. This receptor is abundant in the limbic system and in the brain areas related to stress responses [206]. Anandamide is the endogenous ligand [207]. Among other multiple roles, the endogenous cannabinoid system is involved in the control of anxiety, mood, aggressive behaviour, memory and cognition, perception, movement, appetite and sleep regulation [208]. The evidence for cannabis as an antidepressant is conflicting. In 5 cases, cannabis appeared to have antidepressant effects [209], while another study has suggested that it may actually provoke anxiety attacks [210]. Two trials, one of which was randomized and controlled, suggest that THC may have antidepressant properties in cancer patients [211,212]. Controlled studies with nabilone, a synthetic THC analogue, have also shown anxiolytic and hypnotic effects [213,214].
11. MODULATION OF THE MECHANISMS BEYOND THE RECEPTORS
11.1 Modulation of Intracellular Signal Transduction Proteins
Several lines of evidence have suggested that protein phosphorylation, a prominent regulatory process used by most signaling pathways, is involved in the long term action of conventional antidepressants. These findings stimulated the interest in the investigation of the role of various signal transduction cascades in the pathophysiology of depressive disorder and in the identification of potential novel therapeutic targets [17–19,215].
11.2 cAMP Signal Transduction Cascade
Chronic treatment with most currently used antidepressants increasing synaptic concentrations of norepinephrine and/or serotonin, stimulating adenyl-cyclase via G-protein coupled receptors and up-regulating the cAMP cascade at several levels, including increased levels of cAMP-dependent protein kinase (PKA) and up-regulation of the function and expression of the cAMP-response-element-binding protein (CREB) [Reviewed in 14,17–19,195]. Among the multiple target genes that could be regulated by CREB and that could be involved in pathophysiology of depression and antidepressant actions is a brain derived neurotrophic factor (BDNF), a major neurotrophin in brain, suggesting that agents that activate this pathway might be beneficial in the treatment of depression.
cAMP-specific phosphodiesterase (PDE4) is responsible for the breakdown of cAMP. Clinical trials with rolipram (Fig. 6), a selective inhibitor of phosphodiesterase IV have shown that the compound exhibit antidepressant effects, however, administration and development may be limited by reports of nausea as an adverse effect [216–220]. Recent molecular cloning studies have demonstrated 4 separate genes of PDE4, three of which are expressed in brain (PDE4A, PDE4B and PDE4D). Current evidence suggests that PDE4A and PDE4B may be suitable targets for novel antidepressant medications.
11.3 Mitogen-Activated Protein Kinase (MAP) Cascade
Recent studies have suggested the involvement of the MAP kinase cascade in response to antidepressant medications [reviewed in 17–19]. One of the multiple functional effects of MAP kinase activation is the phosphorylation of CREB, which is involved in the regulation of BDNF expression. BDNF acts via activation of tyrosine kinase receptors, MAP kinase and other intracellular cascades. Stress reduces BDNF expression in hippocampus, whereas chronic antidepressant treatment up-regulates BDNF expression suggesting that one important mechanism of antidepressants may include the up-regulation of BDNF-MAP kinase pathway.
11.4 Modulation of Cell Survival Pathways
There is increasing evidence suggesting that chronic-stress and depression is associated with reduction in number or size of neurons and glia of specific brain regions, which could be reversed by chronic antidepressant therapies [reviewed in 17–19]. These studies have implicated processes of neurogenesis and cell survival in the pathophysiology and treatment of depressive disorders.
11.4.1 Neurogenesis
In contrast to earlier dogma, it has recently been demonstrated that neurogenesis occurs in the adult brain. Moreover, there is evidence that stress and depression may decrease neurogenesis, while chronic treatment with conventional antidepressants and electroconvulsive therapy increases this process. The exact molecular and cellular mechanisms that control adult neurogenesis have not yet been identified, however, there are reports that certain neurotrophic factors may be involved, such as BDNF, insulin-like growth factor-1 (IGF-1) and fibroblast growth factor-2, and their downstream signaling pathways.
Recent findings suggest that conventional antidepressant medications might work by increasing the production of the braińs own neurotrophic factors and depression might also be associated with the compromised production of these factors [221–223]. BDNF is an endogenous protein, a member of structurally related family of trophic factors, which includes nerve growth factor, neurotrophin-3 and neurotrophin-4. BDNF was shown to promote the function and growth of 5-HT-containing neurons. Notable, 5-HT receptors (including the 5-HT2A subtype), phosphodiesterase inhibition, and β-adrenoreceptors appear to be positively coupled to the production of BDNF mRNA in some brain areas [223–225]. Interestingly, exposure of neurons to the BDNF resulted in reduction in NMDA receptor subunit mRNA and protein and marked decrease in NMDA-evoked Ca2 increase [226].
Small molecule agonists that selectively target BDNF mRNA expression could represent a new generation of antidepressants with greater specificity than currently used 5-HT or NA reuptake blocking compounds.
11.4.2 Apoptosis
Apoptosis, or programmed cell death, which is highly regulated by neurotrophic factors and the MAP kinase pathway, is a major determinant of neural survival during development and in the adult brain. Work could focus on the development of new strategies aimed to decrease the expression of pro-apoptotic proteins (e.g. Bax, Bid) and increase the expression of anti-apoptotic ones (e.g. Bcl-2, Bcl-x).
12. CONCLUSIONS
The extended knowledge of mood disorders and the increased interest in the development of better antidepressants resulted in the introduction of great number of new compounds. Although, most currently available antidepressants in the market are still monoamin based and modulating monoamine activity as a therapeutic strategy continues to dominate antidepressant research, several truly novel concepts have emerged by the turn of the new millennium suggesting that the modulation of neuropeptide (substance P, corticotrophin-releasing factor, neuropeptide Y, vasopressin V1b, melanin-concentrating hormone-1), N- methyl-D-aspartate (NMDA), nicotinic acetylcholine (nACh), dopaminergic, glucocorticoid, δ-opioid, cannabinoid and cytokine receptors, gamma-amino butyric acid (GABA), intracellular messenger systems, transcription, neuroprotective and neurogenic factors, may provide an entirely new set of potential therapeutic targets, giving hope that further major advances might be anticipated in the treatment of depressive disorder soon. However, the heterogenous nature of anxiety and depression makes it unlikely that the hope for the ideal antidepressant will ever be realized. Instead, each new addition to the armamentarium of antidepressants eventually is found to have its own application and limitations.
Acknowledgments
Authors are indebted to Szilvia Bercsenyi-Pacher for her efforts and assistance on the preparation of this manuscript.
ABBREVIATIONS
- MDD(s)
Major depressive disorder(s)
- TCA
Tricyclic antidepressants
- MAOIs
Monoamine oxidase inhibitors
- RIMAs
Reversible monoamine oxidase inhibitors
- SSRIs
Selective serotonin reuptake inhibitors
- NA
Noradrenaline, norepinephrine
- 5-HT
5-Hydroxytryptamine, serotonin
- 5-HTR
5-Hydroxytryptamine receptor
- LC
Locus coeruleus
- SNRIs
Serotonin and noradrenaline reuptake inhibitors
- SP
Substance
- P
NK-neurokinin
- SPR(s)
Substance P receptor(s)
- SPA(s)
Substance P receptor antagonist(s)
- CRF
Corticotropin-releasing factor
- CRFR(s)
Corticotropin-releasing factor receptor(s)
- NMDA
N-Methyl-D-aspartate
- NMDAR(s)
N-Methyl-D-aspartate receptor(s)
- CREB
cAMP-response-element-binding protein
- BDNF
Brain derived neurotrophic factor
- GABA
Gamma-amino butyric acid
- GR(s)
Glucocorticoid receptor(s)
References
- 1.Musselman DL, Evans DL, Nemeroff CB. Arch Gen Psychiatry. 1998;55:580–592. doi: 10.1001/archpsyc.55.7.580. [DOI] [PubMed] [Google Scholar]
- 2.Pacher P, Kohegyi E, Kecskemeti V, Furst S. Curr Med Chem. 2001;8(2):89–100. doi: 10.2174/0929867013373796. [DOI] [PubMed] [Google Scholar]
- 3.Reid IC, Stewart CA. Br J Psychiatry. 2001;178:299–303. doi: 10.1192/bjp.178.4.299. [DOI] [PubMed] [Google Scholar]
- 4.Bourin M, Hascoet M. Curr Opin Investig Drugs. 2001;2(2):259–265. [PubMed] [Google Scholar]
- 5.Manji HK, Drevets WC, Charney DS. Nat Med. 2001;7(5):541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- 6.Popoli M, Brunello N, Perez J, Racagni G. J Neurochem. 2000;74(1):21–33. doi: 10.1046/j.1471-4159.2000.0740021.x. [DOI] [PubMed] [Google Scholar]
- 7.Stout SC, Owens MJ, Nemeroff CB. Annu Rev Pharmacol Toxicol. 2001;41:877–906. doi: 10.1146/annurev.pharmtox.41.1.877. [DOI] [PubMed] [Google Scholar]
- 8.DeVane CL. Pharmacotherapy. 2001;21(9):1061–1069. doi: 10.1592/phco.21.13.1061.34612. [DOI] [PubMed] [Google Scholar]
- 9.O'Brien D, Skelton KH, Owens MJ, Nemeroff CB. Hum Psychopharmacol. 2001;16(1):81–87. doi: 10.1002/hup.187. [DOI] [PubMed] [Google Scholar]
- 10.Reul JM, Holsboer F. Curr Opin Pharmacol. 2002;2(1):23–33. doi: 10.1016/s1471-4892(01)00117-5. [DOI] [PubMed] [Google Scholar]
- 11.Nemeroff CB. Psychopharmacol Bull. 2002;36(S2):6–23. [PubMed] [Google Scholar]
- 12.Artigas F, Nutt DJ, Shelton R. Psychopharmacol Bull. 2002;36(S2):123–132. [PubMed] [Google Scholar]
- 13.Baldessarini RJ. The pharmacological basis of the therapeutics. McGrawe-Hills; New York: 2001. Goodman & Gilmańs. [Google Scholar]
- 14.Nestler EJ, Gould E, Manji H, Buncan M, Duman RS, Greshenfeld HK, Hen R, Koester S, Lederhendler I, Meaney M, Robbins T, Winsky L, Zalcman S. Biol Psychiatry. 2002;52(6):503–528. doi: 10.1016/s0006-3223(02)01405-1. [DOI] [PubMed] [Google Scholar]
- 15.Kent JM, Mathew SJ, Gorman JM. Biol Psychiatry. 2002;52(10):1008–1030. doi: 10.1016/s0006-3223(02)01672-4. [DOI] [PubMed] [Google Scholar]
- 16.Manji HK, Duman RS. Psychopharmacol Bull. 2001;35(2):5–49. [PubMed] [Google Scholar]
- 17.Duman RS. Mol Psychiatry. 2002;7(S1):29–34. [Google Scholar]
- 18.D'Sa C, Duman RS. Bipolar Disord. 2002;4(3):183–194. doi: 10.1034/j.1399-5618.2002.01203.x. [DOI] [PubMed] [Google Scholar]
- 19.Bezchlibnyk Y, Young LT. Can J Psychiatry. 2002;47(2):135–148. doi: 10.1177/070674370204700203. [DOI] [PubMed] [Google Scholar]
- 20.Gainetdinov RR, Sotnikova TD, Caron MG. Trends Pharmacol Sci. 2002;23(8):367–373. doi: 10.1016/s0165-6147(02)02044-8. [DOI] [PubMed] [Google Scholar]
- 21.Krystal JH, Sanacora G, Blumberg H, Anand A, Charney DS, Marek G, Epperson CN, Goddard A, Mason GF. Mol Psychiatry. 2002;7(S1):71–80. doi: 10.1038/sj.mp.4001021. [DOI] [PubMed] [Google Scholar]
- 22.Scott LV, Dinan TG. J Affect Disord. 2002;72(2):113–124. doi: 10.1016/s0165-0327(02)00026-5. [DOI] [PubMed] [Google Scholar]
- 23.Skolnick P. Amino Acids. 2002;23(1–3):153–159. doi: 10.1007/s00726-001-0121-7. [DOI] [PubMed] [Google Scholar]
- 24.Shytle RD, Silver AA, Lukas RJ, Newman MB, Sheehan DV, Sanberg PR. Mol Psychiatry. 2002;7(6):525–535. doi: 10.1038/sj.mp.4001035. [DOI] [PubMed] [Google Scholar]
- 25.Borowsky B, Durkin MM, Ogozalek K, Marzabadi MR, DeLeon J, Lagu B, Heurich R, Lichtblau H, Shaposhnik Z, Daniewska I, Blackburn TP, Branchek TA, Gerald C, Vaysse PJ, Forray C. Nat Med. 2002;8:825–830. doi: 10.1038/nm741. [DOI] [PubMed] [Google Scholar]
- 26.Farvolden P, Kennedy SH, Lam RW. Expert Opin Investig Drugs. 2003;12:65–86. doi: 10.1517/13543784.12.1.65. [DOI] [PubMed] [Google Scholar]
- 27.Pacher P, Ungvari Z, Nanasi PP, Furst S, Kecskemeti V. Curr Med Chem. 1999;6:469–80. [PubMed] [Google Scholar]
- 28.Glassman AH. Annu Rev Med. 1984;35:503–511. doi: 10.1146/annurev.me.35.020184.002443. [DOI] [PubMed] [Google Scholar]
- 29.Leonard BE, Healy D. Differential Effects of Antidepressants. Martin Dunitz; London: 1999. [Google Scholar]
- 30.Poirier MF, Olie JP, Loo H, Deniker P, Strolin Benedetti M, Rovei V, Lesage A. Encephale. 1983;9(4):331–343. [PubMed] [Google Scholar]
- 31.Fitton A, Faulds D, Goa KL. Drugs. 1992;43(4):561–96. doi: 10.2165/00003495-199243040-00009. [DOI] [PubMed] [Google Scholar]
- 32.Fulton B, Benfield P. Drugs. 1996;52(3):450–74. doi: 10.2165/00003495-199652030-00013. [DOI] [PubMed] [Google Scholar]
- 33.Volz HP, Muller H, Moller HJ. Neuropsychobiology. 1995;32(1):23–30. doi: 10.1159/000119208. [DOI] [PubMed] [Google Scholar]
- 34.Caille D, Bergis OE, Fankhauser C, Gardes A, Adam R, Charieras T, Grosset A, Rovei V, Jarreau FX. J Pharmacol Exp Ther. 1996;277(1):265–277. [PubMed] [Google Scholar]
- 35.Rosenzweig P, Patat A, Curet O, Durrieu G, Dubruc C, Zieleniuk I, Legangneux EJ. Affect Disord. 1998;51(3):305–312. doi: 10.1016/s0165-0327(98)00226-2. [DOI] [PubMed] [Google Scholar]
- 36.McGrath PJ, Stewart JW, Nunes EV, Ocepek-Welikson K, Rabkin JG, Quitkin FM, Klein DF. Am J Psychiatry. 1993;150(1):118–123. doi: 10.1176/ajp.150.1.118. [DOI] [PubMed] [Google Scholar]
- 37.Stewart JW, Tricamo E, McGrath PJ, Quitkin FM. Am J Psychiatry. 1997;154(1):31–36. doi: 10.1176/ajp.154.1.31. [DOI] [PubMed] [Google Scholar]
- 38.Nutt DJ. Int Clin Psychopharmacol. 2002;17(S1):1–12. doi: 10.1097/00004850-200206001-00002. [DOI] [PubMed] [Google Scholar]
- 39.Wong DT, Bymaster FP, Reid LR, Threlkeld PG. Biochem Pharmacol. 1983;32(7):1287–1293. doi: 10.1016/0006-2952(83)90284-8. [DOI] [PubMed] [Google Scholar]
- 40.Wong DT, Bymaster FP, Engleman EA. Life Sci. 1995;57(5):411–441. doi: 10.1016/0024-3205(95)00209-o. [DOI] [PubMed] [Google Scholar]
- 41.Wong DT, Bymaster FP. Adv Exp Med Biol. 1995;363:77–95. [PubMed] [Google Scholar]
- 42.Goodnick PJ, Goldstein BJ. J Psychopharmacol. 1998;12(3SB):5–20. doi: 10.1177/0269881198012003041. [DOI] [PubMed] [Google Scholar]
- 43.Spinks D, Spinks G. Curr Med Chem. 2002;9(8):799–810. doi: 10.2174/0929867024606795. [DOI] [PubMed] [Google Scholar]
- 44.Caccia S. Clin Pharmacokinet. 1998;34(4):281–302. doi: 10.2165/00003088-199834040-00002. [DOI] [PubMed] [Google Scholar]
- 45.Benfield P, Ward A. Drugs. 1986;32(4):313–334. doi: 10.2165/00003495-198632040-00002. [DOI] [PubMed] [Google Scholar]
- 46.Baumann P. Int Clin Psychopharmacol. 1992;(S5):13–20. [PubMed] [Google Scholar]
- 47.Wong DT, Fuller RW, Robertson DW. Acta Pharm Nord. 1990;2(3):171–180. [PubMed] [Google Scholar]
- 48.Fuller RW, Snoddy HD, Krushinski JH, Robertson DW. Neuropharmacology. 1992;31(10):997–1000. doi: 10.1016/0028-3908(92)90100-4. [DOI] [PubMed] [Google Scholar]
- 49.Rochat B, Amey M, Baumann P. Ther Drug Monit. 1995;17(3):273–279. doi: 10.1097/00007691-199506000-00011. [DOI] [PubMed] [Google Scholar]
- 50.Rochat B, Amey M, Van Gelderen H, Testa B, Baumann P. Chirality. 1995;7(6):389–395. doi: 10.1002/chir.530070602. [DOI] [PubMed] [Google Scholar]
- 51.Hyttel J, Bogeso KP, Perregaard J, Sanchez CJ. Neural Transm Gen Sect. 1992;88(2):157–160. doi: 10.1007/BF01244820. [DOI] [PubMed] [Google Scholar]
- 52.Pacher P, Ungvari Z, Kecskemeti V, Furst S. Curr Med Chem. 1998;5(5):381–390. [PubMed] [Google Scholar]
- 53.Pacher P, Magyar J, Szigligeti P, Banyasz T, Pankucsi C, Korom Z, Ungvari Z, Kecskemeti V, Nanasi PP. Naunyn Schmiedebergs Arch Pharmacol. 2000;361(1):67–73. doi: 10.1007/s002109900154. [DOI] [PubMed] [Google Scholar]
- 54.Pacher P, Bagi Z, Lako-Futo Z, Ungvari Z, Nanasi PP, Kecskemeti V. Gen Pharmacol. 2000;34(1):17–23. doi: 10.1016/s0306-3623(99)00048-8. [DOI] [PubMed] [Google Scholar]
- 55.Pacher P, Ungvari Z, Kecskemeti V, Koller A. Br J Pharmacol. 1999;127(3):740–746. doi: 10.1038/sj.bjp.0702571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ungvari Z, Pacher P, Koller A. J Cardiovasc Pharmacol. 2000;35(6):849–854. doi: 10.1097/00005344-200006000-00004. [DOI] [PubMed] [Google Scholar]
- 57.Magyar J, Rusznak Z, Harasztosi C, Kortvely A, Pacher P, Banyasz T, Pankucsi C, Kovacs L, Szucs G, Nanasi PP, Kecskemeti V. Int J Mol Med. 2003;11(4):535–542. [PubMed] [Google Scholar]
- 58.Kecskemeti V, Pacher P. Neuropsychopharmacologica Hungarica. 2002;IV4:197–206. [Google Scholar]
- 59.Pacher P, Ungvari Z. Med Hypotheses. 2001;57(4):469–471. doi: 10.1054/mehy.2001.1366. [DOI] [PubMed] [Google Scholar]
- 60.Pacher P, Ungvari Z. Circulation. 2002;105(14):e84. doi: 10.1161/01.cir.0000012607.30379.f5. [DOI] [PubMed] [Google Scholar]
- 61.Rodriguez de la Torre B, Dreher J, Malevany I, Bagli M, Kolbinger M, Omran H, Luderitz B, Rao ML. Ther Drug Monit. 2001;23(4):435–440. doi: 10.1097/00007691-200108000-00019. [DOI] [PubMed] [Google Scholar]
- 62.Rosen RC, Lane RM, Menza M. J Clin Psychopharmacol. 1999;19(1):67–85. doi: 10.1097/00004714-199902000-00013. [DOI] [PubMed] [Google Scholar]
- 63.Thompson C. Hum Psychopharmacol. 2002;(S1):27–32. [Google Scholar]
- 64.Thase ME, Nierenberg AA, Keller MB, Panagides J The Relapse Prevention Study Group. J Clin Psychiatry. 2001;62(10):782–788. doi: 10.4088/jcp.v62n1006. [DOI] [PubMed] [Google Scholar]
- 65.Pitsikas N. Curr Opin Investig Drugs. 2000;1(1):116–121. [PubMed] [Google Scholar]
- 66.Detke MJ, Lu Y, Goldstein DJ, Hayes JR, Demitrack MA. J Clin Psychiatry. 2002;63(4):308–315. doi: 10.4088/jcp.v63n0407. [DOI] [PubMed] [Google Scholar]
- 67.Muth EA, Haskins JT, Moyer JA, Husbands GE, Nielsen ST, Sigg EB. Biochem Pharmacol. 1986;35(24):4493–4497. doi: 10.1016/0006-2952(86)90769-0. [DOI] [PubMed] [Google Scholar]
- 68.Briley M, Prost JF, Moret C. Int Clin Psychopharmacol. 1996;11(S4):9–14. doi: 10.1097/00004850-199609004-00002. [DOI] [PubMed] [Google Scholar]
- 69.Bonnaud B, Cousse H, Mouzin G, Briley M, Stenger A, Fauran F, Couzinier JP. J Med Chem. 1987;30(2):318–325. doi: 10.1021/jm00385a013. [DOI] [PubMed] [Google Scholar]
- 70.Dostert P, Benedetti MS, Poggesi I. Eur Neuropsychopharmacol. 1997;7(S1):23–35. doi: 10.1016/s0924-977x(97)00417-3. [DOI] [PubMed] [Google Scholar]
- 71.Cocchiara G, Battaglia R, Pevarello P, Strolin Benedetti M. Eur J Drug Metab Pharmacokinet. 1991;16(3):231–239. doi: 10.1007/BF03189965. [DOI] [PubMed] [Google Scholar]
- 72.Corrigan MH, Denahan AQ, Wright CE, Ragual RJ, Evans DL. Depress Anxiety. 2000;11:58–65. doi: 10.1002/(sici)1520-6394(2000)11:2<58::aid-da2>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 73.Hoyer D, Clarke DE, Fozard JR, Hartig PR, Martin GR, Mylecharane EJ, Saxena PR, Humphrey PP. Pharmacol Rev. 1994;46(2):157–203. [PubMed] [Google Scholar]
- 74.Barnes NM, Sharp T. Neuropharmacology. 1999;38(8):1083–1152. doi: 10.1016/s0028-3908(99)00010-6. [DOI] [PubMed] [Google Scholar]
- 75.Wood MD, Thomas DR, Watson JM. Expert Opin Investig Drugs. 2002;11(4):457–467. doi: 10.1517/13543784.11.4.457. [DOI] [PubMed] [Google Scholar]
- 76.Perez V, Gilaberte I, Faries D, Alvarez E, Artigas F. Lancet. 1997;349(9065):1594–1597. doi: 10.1016/S0140-6736(96)08007-5. [DOI] [PubMed] [Google Scholar]
- 77.Artigas F, Perez V, Alvarez E. Arch Gen Psychiatry. 1994;51(3):248–251. doi: 10.1001/archpsyc.1994.03950030084009. [DOI] [PubMed] [Google Scholar]
- 78.Artigas F, Romero L, de Montigny C, Blier P. Trends Neurosci. 1996;19(9):378–383. doi: 10.1016/S0166-2236(96)10037-0. [DOI] [PubMed] [Google Scholar]
- 79.Romero L, Hervas I, Artigas F. Neurosci Lett. 1996;219(2):123–126. doi: 10.1016/s0304-3940(96)13199-2. [DOI] [PubMed] [Google Scholar]
- 80.Romero L, Artigas F. J Neurochem. 1997;68(6):2593–2603. doi: 10.1046/j.1471-4159.1997.68062593.x. [DOI] [PubMed] [Google Scholar]
- 81.Perez M, Pauwels PJ, Pallard-Sigogneau I, Fourrier C, Chopin P, Palmier C, Colovray V, Halazy S. Bioorg Med Chem Lett. 1998;8(23):3423–3428. doi: 10.1016/s0960-894x(98)00619-2. [DOI] [PubMed] [Google Scholar]
- 82.Oficialdegui AM, Martinez J, Perez S, Heras B, Irurzun M, Palop JA, Tordera R, Lasheras B, del Rio J, Monge A. Farmaco. 2000;55(5):345–353. doi: 10.1016/s0014-827x(00)00050-1. [DOI] [PubMed] [Google Scholar]
- 83.Martinez-Esparza J, Oficialdegui AM, Perez-Silanes S, Heras B, Orus L, Palop JA, Lasheras B, Roca J, Mourelle M, Bosch A, Del Castillo JC, Tordera R, Del Rio J, Monge A. J Med Chem. 2001;44(3):418–428. doi: 10.1021/jm001059j. [DOI] [PubMed] [Google Scholar]
- 84.Meagher KL, Mewshaw RE, Evrard DA, Zhou P, Smith DL, Scerni R, Spangler T, Abulhawa S, Shi X, Schechter LE, Andree TH. Bioorg Med Chem Lett. 2001;11(14):1885–1888. doi: 10.1016/s0960-894x(01)00334-1. [DOI] [PubMed] [Google Scholar]
- 85.Mewshaw RE, Meagher KL, Zhou P, Zhou D, Shi X, Scerni R, Smith D, Schechter LE, Andree TH. Bioorg Med Chem Lett. 2002;12(3):307–310. doi: 10.1016/s0960-894x(01)00746-6. [DOI] [PubMed] [Google Scholar]
- 86.Olivier B, Soudijn W, van Wijngaarden I. The 5-HT1A receptor and its ligands: structure and function. Prog Drug Res. 1999;52:103–165. doi: 10.1007/978-3-0348-8730-4_3. [DOI] [PubMed] [Google Scholar]
- 87.Martinez J, Perez S, Oficialdegui AM, Heras B, Orus L, Villanueva H, Palop JA, Roca J, Mourelle M, Bosch A, Del Castillo JC, Lasheras B, Tordera R, del Rio J, Monge A. Eur J Med Chem. 2001;36(1):55–61. doi: 10.1016/s0223-5234(00)01198-3. [DOI] [PubMed] [Google Scholar]
- 88.Orus L, Perez-Silanes S, Oficialdegui AM, Martinez-Esparza J, Del Castillo JC, Mourelle M, Langer T, Guccione S, Donzella G, Krovat EM, Poptodorov K, Lasheras B, Ballaz S, Hervias I, Tordera R, Del Rio J, Monge A. J Med Chem. 2002;45(19):4128–4139. doi: 10.1021/jm0111200. [DOI] [PubMed] [Google Scholar]
- 89.Lopez-Rodriguez ML, Ayala D, Benhamu B, Morcillo MJ, Viso A. Curr Med Chem. 2002;9(4):443–469. doi: 10.2174/0929867023371030. [DOI] [PubMed] [Google Scholar]
- 90.Griebel G. Drug News Perspect. 1999;12(8):484–490. [Google Scholar]
- 91.Forster EA, Cliffe IA, Bill DJ, Dover GM, Jones D, Reilly Y, Fletcher A. Eur J Pharmacol. 1995;281(1):81–88. doi: 10.1016/0014-2999(95)00234-c. [DOI] [PubMed] [Google Scholar]
- 92.Kung MP, Zhuang ZP, Frederick D, Kung HF. Synapse. 1994;18(4):359–366. doi: 10.1002/syn.890180412. [DOI] [PubMed] [Google Scholar]
- 93.Wong DT, Mayle DN, Delapp NW, Calligaro DO, Robertson DW. Soc Neurosci Abstr. 1994;20:1542. [Google Scholar]
- 94.Cohen C, Perrault G, Claustre Y, Curet O, Griebel G, Deporteere R, Lourdelet J, et al. Soc Neurosci Abstr. 1998;24:1364. [Google Scholar]
- 95.Mos J, Van Hest A, Van Drimmelen M, Herremans AH, Olivier B. Eur J Pharmacol. 1997;325(2–3):145–153. doi: 10.1016/s0014-2999(97)00131-3. [DOI] [PubMed] [Google Scholar]
- 96.Johansson L, Sohn D, Thorberg SO, Jackson DM, Kelder D, Larsson LG, Renyi L, Ross SB, Wallsten C, Eriksson H, Hu PS, Jerning E, Mohell N, Westlind-Danielsson A. J Pharmacol Exp Ther. 1997;283(1):216–225. [PubMed] [Google Scholar]
- 97.Dimitriou EC, Dimitriou CE. J Clin Psychopharmacol. 1998;18(6):465–469. doi: 10.1097/00004714-199812000-00009. [DOI] [PubMed] [Google Scholar]
- 98.Landen M, Bjorling G, Agren H, Fahlen T. J Clin Psychiatry. 1998;59(12):664–668. [PubMed] [Google Scholar]
- 99.Davis R, Whittington R, Bryson HM. Drugs. 1997;53(4):608–636. doi: 10.2165/00003495-199753040-00006. [DOI] [PubMed] [Google Scholar]
- 100.Gelenberg AJ, McGahuey C, Laukes C, Okayli G, Moreno F, Zentner L, Delgado P. J Clin Psychiatry. 2000;61(5):356–360. doi: 10.4088/jcp.v61n0506. [DOI] [PubMed] [Google Scholar]
- 101.Takeuchi H, Yatsugi S, Hatanaka K, Nakato K, Hattori H, Sonoda R, Koshiya K, Fujii M, Yamaguchi T. Eur J Pharmacol. 1997;329(1):27–35. doi: 10.1016/s0014-2999(97)10108-x. [DOI] [PubMed] [Google Scholar]
- 102.Hatanaka K, Nomura T, Hidaka K, Takeuchi H, Yatsugi S, Fujii M, Yamaguchi T. Neuropharmacology. 1996;35(11):1621–1626. doi: 10.1016/s0028-3908(96)00079-2. [DOI] [PubMed] [Google Scholar]
- 103.Wood MD, Reavill C, Trail B, Wilson A, Stean T, Kennett GA, Lightowler S, Blackburn TP, Thomas D, Gager TL, Riley G, Holland V, Bromidge SM, Forbes IT, Middlemiss DN. Neuropharmacology. 2001;41:186–199. doi: 10.1016/s0028-3908(01)00054-5. [DOI] [PubMed] [Google Scholar]
- 104.Jenck F, Moreau JL, Wichmann J, Stadler H, Martin, Bos M. In: Anxiolytics: Milestones in Drug Therapy. Briley M, Nutt D, editors. Birkhauser Verlag AG; Switzerland: 2000. [Google Scholar]
- 105.Papp M, Gruca P, Boyer PA, Mocaer E. Neuropsychopharmacology. 2003;28:694–703. doi: 10.1038/sj.npp.1300091. [DOI] [PubMed] [Google Scholar]
- 106.Pauwels PJ. Biochem Pharmacol. 2000;60(12):1743–1750. doi: 10.1016/s0006-2952(00)00476-7. [DOI] [PubMed] [Google Scholar]
- 107.BASF to merge Boots Pharma under Knoll name. Scrip. 1995;2014:10. [Google Scholar]
- 108.Mocaer E, Rettori MC, Kamoun A. Clin Neuropharmacol. 1988;11(S2):32–42. [PubMed] [Google Scholar]
- 109.Mennini T, Garattini S. Presse Med. 1991;20(37):1823–1827. [PubMed] [Google Scholar]
- 110.Wilde MI, Benfield P. Drugs. 1995;49(3):411–439. doi: 10.2165/00003495-199549030-00007. [DOI] [PubMed] [Google Scholar]
- 111.Wagstaff AJ, Ormrod D, Spencer CM. CNS Drugs. 2001;15:231–259. doi: 10.2165/00023210-200115030-00006. [DOI] [PubMed] [Google Scholar]
- 112.Dziedzicka-Wasylewska M, Dlaboga D, Pierzchala-Koziec K, Rogoz Z. J Physiol Pharmacol. 2002;53:117–125. [PubMed] [Google Scholar]
- 113.Oluyomi AO, Datla KP, Curzon G. Neuropharmacology. 1997;36(3):383–387. doi: 10.1016/s0028-3908(97)00016-6. [DOI] [PubMed] [Google Scholar]
- 114.Nutt D. Acta Psychiatr Scand Suppl. 1997;391:31–37. doi: 10.1111/j.1600-0447.1997.tb05956.x. [DOI] [PubMed] [Google Scholar]
- 115.Sitsen JMA, Zivkov MZ. CNS Drugs. 1995;4(S1):39–48. [Google Scholar]
- 116.Blier P. J Clin Psychiatry. 2001;62(S4):7–11. 37–40. [PubMed] [Google Scholar]
- 117.Kent JM. Lancet. 2000;355(9207):911–918. doi: 10.1016/S0140-6736(99)11381-3. [DOI] [PubMed] [Google Scholar]
- 118.De Felipe C, Herrero JF, O'Brien JA, Palmer JA, Doyle CA, Smith AJ, Laird JM, Belmonte C, Cervero F, Hunt SP. Nature. 1998;392(6674):394–397. doi: 10.1038/32904. [DOI] [PubMed] [Google Scholar]
- 119.Rupniak NM. Curr Opin Investig Drugs. 2002;3(2):257–261. [PubMed] [Google Scholar]
- 120.Rupniak NM. Can J Physiol Pharmacol. 2002;80(5):489–494. doi: 10.1139/y02-048. [DOI] [PubMed] [Google Scholar]
- 121.Snider RM, Constantine JW, Lowe JA, Longo KP, LebelWSWoody HA, Drozda SE, Desai MC, Vinick FJ, Spencer RW, et al. Science. 1991;251(4992):435–437. doi: 10.1126/science.1703323. [DOI] [PubMed] [Google Scholar]
- 122.Kramer MS, Cutler N, Feighner J, Shrivastava R, Carman J, Sramek JJ, et al. Science. 1998;281(5383):1640–1645. doi: 10.1126/science.281.5383.1640. [DOI] [PubMed] [Google Scholar]
- 123.Rupniak NMJ, Kramer SM. Trends Pharmacol Sci. 1999;(244 20):471–509. doi: 10.1016/s0165-6147(99)01396-6. [DOI] [PubMed] [Google Scholar]
- 124.Shirayama Y, Mitsushio H, Takashima M, Ichikawa H, Takahashi K. Brain Res. 1996;739(1–2):70–78. doi: 10.1016/s0006-8993(96)00812-8. [DOI] [PubMed] [Google Scholar]
- 125.Nutt D. Lancet. 1998;352(9141):1644–1646. doi: 10.1016/S0140-6736(05)61444-4. [DOI] [PubMed] [Google Scholar]
- 126.Maubach KA, Martin K, Smith DW, Hewson L, Frankshun RA, Harrison T, Seabrook GR. Neuropharmacology. 2001;40(6):806–817. doi: 10.1016/s0028-3908(00)00209-4. [DOI] [PubMed] [Google Scholar]
- 127.Millet R, Domarkas J, Rigo B, Goossens L, Goossens JF, Houssin R, Henichart JP. Bioorg Med Chem. 2002;10(9):2905–2912. doi: 10.1016/s0968-0896(02)00144-x. [DOI] [PubMed] [Google Scholar]
- 128.Lieb K, Treffurth Y, Berger M, Fiebich BL. Neuropsychobiology. 2002;45(S1):2–6. doi: 10.1159/000049254. [DOI] [PubMed] [Google Scholar]
- 129.Steinberg R, Alonso R, Griebel G, Bert L, Jung M, Oury-Donat F, Poncelet M, Gueudet C, Desvignes C, Le Fur G, Soubrie P. J Pharmacol Exp Ther. 2001;299(2):449–458. [PubMed] [Google Scholar]
- 130.Haddjeri N, Blier P. Neuroreport. 2000;11(6):1323–1327. doi: 10.1097/00001756-200004270-00035. [DOI] [PubMed] [Google Scholar]
- 131.Santarelli L, Gobbi G, Debs PC, Sibille ET, Blier P, Hen R, Heath MJ. Proc Natl Acad Sci USA. 2001;98(4):1912–1917. doi: 10.1073/pnas.041596398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Froger N, Gardier AM, Moratalla R, Alberti I, Lena I, Boni C, De Felipe C, Rupniak NM, Hunt SP, Jacquot C, Hamon M, Lanfumey L. J Neurosci. 2001;21(20):8188–8197. doi: 10.1523/JNEUROSCI.21-20-08188.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ryckmans T, Balancon L, Berton O, Genicot C, Lamberty Y, Lallemand B, Pasau P, Pirlot N, Quere L, Talaga P. Bioorg Med Chem Lett. 2002;12(2):261–264. doi: 10.1016/s0960-894x(01)00727-2. [DOI] [PubMed] [Google Scholar]
- 134.Ryckmans T, Berton O, Grimee R, Kogej T, Lamberty Y, Pasau P, Talaga P, Genicot C. Bioorg Med Chem Lett. 2002;12(21):3195–3198. doi: 10.1016/s0960-894x(02)00563-2. [DOI] [PubMed] [Google Scholar]
- 135.Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB. J Endocrinol. 1999;160(1):1–12. doi: 10.1677/joe.0.1600001. [DOI] [PubMed] [Google Scholar]
- 136.Vaughan J, Donaldson C, Bittencourt J, Perrin MH, Lewis K, Sutton S, Chan R, Turnbull AV, Lovejoy D, Rivier C, et al. Nature. 1995;378(6554):287–292. doi: 10.1038/378287a0. [DOI] [PubMed] [Google Scholar]
- 137.Chen R, Lewis KA, Perrin MH, Vale WW. Proc Natl Acad Sci USA. 1993;90(19):8967–8971. doi: 10.1073/pnas.90.19.8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lovenberg TW, Liaw CW, Grigoriadis DE, Clevenger W, Chalmers DT, De Souza EB, Oltersdorf T. Proc Natl Acad Sci USA. 1995;92(3):836–840. doi: 10.1073/pnas.92.3.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chalmers DT, Lovenberg TW, De Souza EB. J Neurosci. 1995;15(10):6340–6350. doi: 10.1523/JNEUROSCI.15-10-06340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Reyes TM, Lewis K, Perrin MH, Kunitake KS, Vaughan J, Arias CA, Hogenesch JB, Gulyas J, Rivier J, Vale WW, Sawchenko PE. Proc Natl Acad Sci USA. 2001;98(5):2843–2848. doi: 10.1073/pnas.051626398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hsu SY, Hsueh AJ. Nat Med. 2001;7(5):605–611. doi: 10.1038/87936. [DOI] [PubMed] [Google Scholar]
- 142.Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, et al. Nat Genet. 1998;19(2):162–166. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- 143.Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, et al. Neuron. 1998;20(6):1093–1102. doi: 10.1016/s0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- 144.Holsboer F. Biol Psychol. 2001;57(1–3):47–65. doi: 10.1016/s0301-0511(01)00089-8. [DOI] [PubMed] [Google Scholar]
- 145.Owens MJ, Nemeroff CB. Expert Opin Investig Drugs. 1999;8(11):1849–1858. doi: 10.1517/13543784.8.11.1849. [DOI] [PubMed] [Google Scholar]
- 146.Mansbach RS, Brooks EN, Chen YL. Eur J Pharmacol. 1997;323(1):21–26. doi: 10.1016/s0014-2999(97)00025-3. [DOI] [PubMed] [Google Scholar]
- 147.Dautzenberg FM, Hauger RL. Trends Pharmacol Sci. 2002;23(2):71–77. doi: 10.1016/s0165-6147(02)01946-6. [DOI] [PubMed] [Google Scholar]
- 148.Preti A. Curr Opin Investig Drugs. 2001;2(2):274–279. [PubMed] [Google Scholar]
- 149.Jard S, Gaillard RC, Guillon G, Marie J, Schoenenberg P, Muller AF, Manning M, Sawyer WH. Mol Pharmacol. 1986;30(2):171–177. [PubMed] [Google Scholar]
- 150.Barberis C, Audigier S, Durroux T, Elands J, Schmidt A, Jard S. Ann NY Acad Sci. 1992;652:39–45. doi: 10.1111/j.1749-6632.1992.tb34344.x. [DOI] [PubMed] [Google Scholar]
- 151.Birnbaumer M. Trends Endocrinol Metab. 2000;11(10):406–410. doi: 10.1016/s1043-2760(00)00304-0. [DOI] [PubMed] [Google Scholar]
- 152.Tribollet E, Barberis C, Dubois-Dauphin M, Dreifuss JJ. Brain Res. 1992;589(1):15–23. doi: 10.1016/0006-8993(92)91156-9. [DOI] [PubMed] [Google Scholar]
- 153.de Vries GJ, Miller MA. Prog Brain Res. 1998;119:3–20. doi: 10.1016/s0079-6123(08)61558-7. [DOI] [PubMed] [Google Scholar]
- 154.Lolait SJ, O'Carroll AM, Mahan LC, Felder CC, Button DC, Young WS, 3rd, Mezey E, Brownstein MJ. Proc Natl Acad Sci USA. 1995;92(15):6783–6787. doi: 10.1073/pnas.92.15.6783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Young LJ, Toloczko D, Insel TR. J Neuroendocrinol. 1999;11(4):291–297. doi: 10.1046/j.1365-2826.1999.00332.x. [DOI] [PubMed] [Google Scholar]
- 156.Vaccari C, Lolait SJ, Ostrowski NL. Endocrinology. 1998;139(12):5015–5033. doi: 10.1210/endo.139.12.6382. [DOI] [PubMed] [Google Scholar]
- 157.Engelmann M, Wotjak CT, Neumann I, Ludwig M, Landgraf R. Neurosci Biobehav Rev. 1996;20(3):341–358. doi: 10.1016/0149-7634(95)00059-3. [DOI] [PubMed] [Google Scholar]
- 158.Scott LV, Dinan TG. J Affect Disord. 2002;72(2):113–124. doi: 10.1016/s0165-0327(02)00026-5. [DOI] [PubMed] [Google Scholar]
- 159.Liebsch G, Wotjak CT, Landgraf R, Engelmann M. Neurosci Lett. 1996;217(2–3):101–104. [PubMed] [Google Scholar]
- 160.Landgraf R, Gerstberger R, Montkowski A, Probst JC, Wotjak CT, Holsboer F, Engelmann M. J Neurosci. 1995;15(6):4250–4258. doi: 10.1523/JNEUROSCI.15-06-04250.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Aguilera G, Rabadan-Diehl C. Regul Pept. 2000;96(1–2):23–29. doi: 10.1016/s0167-0115(00)00196-8. [DOI] [PubMed] [Google Scholar]
- 162.Griebel G, Simiand J, Serradeil-Le Gal C, Wagnon J, Pascal M, Scatton B, Maffrand JP, Soubrie P. Proc Natl Acad Sci USA. 2002;99(9):6370–6375. doi: 10.1073/pnas.092012099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Grundemar L, Hakanson R. Trends Pharmacol Sci. 1994;15(5):153–159. doi: 10.1016/0165-6147(94)90076-0. [DOI] [PubMed] [Google Scholar]
- 164.Heilig M, Thorsell A. Rev Neurosci. 2002;13(1):85–94. doi: 10.1515/revneuro.2002.13.1.85. [DOI] [PubMed] [Google Scholar]
- 165.Redrobe JP, Dumont Y, Quirion R. Life Sci. 2002;71(25):2921–2937. doi: 10.1016/s0024-3205(02)02159-8. [DOI] [PubMed] [Google Scholar]
- 166.Stogner KA, Holmes PV. Eur J Pharmacol. 2000;387(2):R9–10. doi: 10.1016/s0014-2999(99)00800-6. [DOI] [PubMed] [Google Scholar]
- 167.Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, Vale W, Sawchenko PE. J Comp Neurol. 1992;319(2):218–245. doi: 10.1002/cne.903190204. [DOI] [PubMed] [Google Scholar]
- 168.Saito Y, Nothacker HP, Wang Z, Lin SH, Leslie F, Civelli O. Nature. 1999;400(6741):265–269. doi: 10.1038/22321. [DOI] [PubMed] [Google Scholar]
- 169.Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, Dytko GM, Foley JJ, Martin J, Liu WS, Park J, Ellis C, Ganguly S, Konchar S, Cluderay J, Leslie R, Wilson S, Sarau HM. Nature. 1999;400(6741):261–265. doi: 10.1038/22313. [DOI] [PubMed] [Google Scholar]
- 170.Sailer AW, Sano H, Zeng Z, McDonald TP, Pan J, Pong SS, Feighner SD, Tan CP, Fukami T, Iwaasa H, Hreniuk DL, Morin NR, Sadowski SJ, Ito M, Ito M, Bansal A, Ky B, Figueroa DJ, Jiang Q, Austin CP, MacNeil DJ, Ishihara A, Ihara M, Kanatani A, Van der Ploeg LH, Howard AD, Liu Q. Proc Natl Acad Sci USA. 2001;98(13):7564–7569. doi: 10.1073/pnas.121170598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Shytle RD, Silver AA, Lukas RJ, Newman MB, Sheehan DV, Sanberg PR. Mol Psychiatry. 2002;7(6):525–535. doi: 10.1038/sj.mp.4001035. [DOI] [PubMed] [Google Scholar]
- 172.Shytle RD, Silver AA, Sanberg PR. Biol Psychiatry. 2000;48(10):1028–1031. doi: 10.1016/s0006-3223(00)00945-8. [DOI] [PubMed] [Google Scholar]
- 173.Shytle RD, Silver AA, Sheehan KH, Sheehan DV, Sanberg PR. Depress Anxiety. 2002;16(3):89–92. doi: 10.1002/da.10035. [DOI] [PubMed] [Google Scholar]
- 174.Skolnick P, Legutko B, Li X, Bymaster FP. Pharmacol Res. 2001;43(5):411–423. doi: 10.1006/phrs.2000.0806. [DOI] [PubMed] [Google Scholar]
- 175.Skolnick P. Eur J Pharmacol. 1999;375(1–3):31–40. doi: 10.1016/s0014-2999(99)00330-1. [DOI] [PubMed] [Google Scholar]
- 176.Johnson JW, Ascher P. Nature. 1987;325(6104):529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- 177.Kleckner NW, Dingledine R. Science. 1988;241(4867):835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- 178.Seeburg PH. Trends Pharmacol Sci. 1993;14(8):297–303. doi: 10.1016/0165-6147(93)90047-N. [DOI] [PubMed] [Google Scholar]
- 179.Seeburg PH. Trends Neurosci. 1993;16(9):359–365. doi: 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- 180.Kuryatov A, Laube B, Betz H, Kuhse J. Neuron. 1994;12(6):1291–1300. doi: 10.1016/0896-6273(94)90445-6. [DOI] [PubMed] [Google Scholar]
- 181.Laube B, Hirai H, Sturgess M, Betz H, Kuhse J. Neuron. 1997;18(3):493–503. doi: 10.1016/s0896-6273(00)81249-0. [DOI] [PubMed] [Google Scholar]
- 182.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Neuron. 1994;12(3):529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 183.Parsons CG, Danysz W, Bartmann A, Spielmanns P, Frankiewicz T, Hesselink M, Eilbacher B, Quack G. Neuropharmacology. 1999;38(1):85–108. doi: 10.1016/s0028-3908(98)00161-0. [DOI] [PubMed] [Google Scholar]
- 184.Parsons CG, Danysz W, Hesselink M, Hartmann S, Lorenz B, Wollenburg C, Quack G. Amino Acids. 1998;14(1–3):207–216. doi: 10.1007/BF01345264. [DOI] [PubMed] [Google Scholar]
- 185.Parsons CG, Danysz W, Quack G. Drug News Perspect. 1998;11(9):523–569. doi: 10.1358/dnp.1998.11.9.863689. [DOI] [PubMed] [Google Scholar]
- 186.Nowak G, Trullas R, Layer RT, Skolnick P, Paul IA. J Pharmacol Exp Ther. 1993;265(3):1380–1386. [PubMed] [Google Scholar]
- 187.Nowak G, Li Y, Paul IA. Eur J Pharmacol. 1996;295(1):75–85. doi: 10.1016/0014-2999(95)00585-4. [DOI] [PubMed] [Google Scholar]
- 188.Paul IA, Layer RT, Skolnick P, Nowak G. Eur J Pharmacol. 1993;247(3):305–311. doi: 10.1016/0922-4106(93)90199-j. [DOI] [PubMed] [Google Scholar]
- 189.Boyer PA, Skolnick P, Fossom LH. J Mol Neurosci. 1998;10(3):219–233. doi: 10.1007/BF02761776. [DOI] [PubMed] [Google Scholar]
- 190.Pallotta M, Segieth J, Whitton PS. Neurosci Lett. 1999;262(3):187–190. doi: 10.1016/s0304-3940(99)00058-0. [DOI] [PubMed] [Google Scholar]
- 191.Huber TJ, Dietrich DE, Emrich HM. Pharmacopsychiatry. 1999;32(2):47–55. doi: 10.1055/s-2007-979191. [DOI] [PubMed] [Google Scholar]
- 192.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Biol Psychiatry. 2000;47(4):351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 193.Shuto S, Yoshii K, Matsuda A. Jpn J Pharmacol. 2001;85(3):207–213. doi: 10.1254/jjp.85.207. [DOI] [PubMed] [Google Scholar]
- 194.Krystal JH, Sanacora G, Blumberg H, Anand A, Charney DS, Marek G, Epperson CN, Goddard A, Mason GF. Mol Psychiatry. 2002;7(S1):71–80. doi: 10.1038/sj.mp.4001021. [DOI] [PubMed] [Google Scholar]
- 195.Reus VI, Wolkowitz OM. Expert Opin Investig Drugs. 2001;10(10):1789–1796. doi: 10.1517/13543784.10.10.1789. [DOI] [PubMed] [Google Scholar]
- 196.Pariante CM, Miller AH. Biol Psychiatry. 2001;49(5):391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- 197.Belanoff JK, Rothschild AJ, Cassidy F, DeBattista C, Baulieu EE, Schold C, Schatzberg AF. Biol Psychiatry. 2002;52(5):386–392. doi: 10.1016/s0006-3223(02)01432-4. [DOI] [PubMed] [Google Scholar]
- 198.Hasko G, Szabo C. Biochem Pharmacol. 1998;56(9):1079–1087. doi: 10.1016/s0006-2952(98)00153-1. [DOI] [PubMed] [Google Scholar]
- 199.Corcos M, Guilbaud O, Hjalmarsson L, Chambry J, Jeammet P. Biomed Pharmacother. 2002;56(2):105–110. doi: 10.1016/s0753-3322(01)00156-1. [DOI] [PubMed] [Google Scholar]
- 200.Castanon N, Leonard BE, Neveu PJ, Yirmiya R. Brain Behav Immun. 2002;16(5):569–574. doi: 10.1016/s0889-1591(02)00008-9. [DOI] [PubMed] [Google Scholar]
- 201.Konsman JP, Parnet P, Dantzer R. Trends Neurosci. 2002;25(3):154–159. doi: 10.1016/s0166-2236(00)02088-9. [DOI] [PubMed] [Google Scholar]
- 202.Broom DC, Jutkiewicz EM, Rice KC, Traynor JR, Woods JH. Jpn J Pharmacol. 2002;90(1):1–6. doi: 10.1254/jjp.90.1. [DOI] [PubMed] [Google Scholar]
- 203.Williamson EM, Evans FJ. Drugs. 2000;60:1303–1314. doi: 10.2165/00003495-200060060-00005. [DOI] [PubMed] [Google Scholar]
- 204.Robson P. Br J Psychiatry. 2001;178:107–115. doi: 10.1192/bjp.178.2.107. [DOI] [PubMed] [Google Scholar]
- 205.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 206.Herkenham M. NIDA Res Monogr. 1991;112:129–145. [PubMed] [Google Scholar]
- 207.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 208.Martin M, Ledent C, Parmentier M, Maldonado R, Valverde O. Psychopharmacology (Berl) 2002;159:379–387. doi: 10.1007/s00213-001-0946-5. [DOI] [PubMed] [Google Scholar]
- 209.Gruber AJ, Pope HG, Jr, Brown ME. Depression. 1996;4:77–80. doi: 10.1002/(SICI)1522-7162(1996)4:2<77::AID-DEPR7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 210.Fishman SM, Rosenbaum JF, Yabusaki DI. Res Comm Subst Abuse. 1988;9:219–226. [Google Scholar]
- 211.Grinspoon L, Bakalar JB. Marihuana, The Forbidden Medicine. Yale University Press; New Haven and London: 1993. [Google Scholar]
- 212.Braude MC, Szara S, editors. The pharmacology of Marihuana. Raven Press; New York: 1976. pp. 763–775. [Google Scholar]
- 213.Fabrel LF, McLendon D. J Clin Pharmacol. 1981;21:377S–382S. doi: 10.1002/j.1552-4604.1981.tb02617.x. [DOI] [PubMed] [Google Scholar]
- 214.Ilaria RL, Thornby JI, Fann WE. Curr Ther Res. 1981;29:943–949. [Google Scholar]
- 215.Popoli M, Brunello N, Perez J, Racagni G. J Neurochem. 2000;74(1):21–33. doi: 10.1046/j.1471-4159.2000.0740021.x. [DOI] [PubMed] [Google Scholar]
- 216.Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neuron. 2002;34(1):13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 217.Zeller E, Stief HJ, Pflug B, Sastre-y-Hernandez M. Pharmacopsychiatry. 1984;17(6):188–190. doi: 10.1055/s-2007-1017435. [DOI] [PubMed] [Google Scholar]
- 218.Scott AI, Perini AF, Shering PA, Whalley LJ. Eur J Clin Pharmacol. 1991;40(2):127–129. doi: 10.1007/BF00280065. [DOI] [PubMed] [Google Scholar]
- 219.Hebenstreit GF, Fellerer K, Fichte K, Fischer G, Geyer N, Meya U, Sastre-y-Hernandez M, Schony W, Schratzer M, Soukop W, et al. Pharmacopsychiatry. 1989;22(4):156–160. doi: 10.1055/s-2007-1014599. [DOI] [PubMed] [Google Scholar]
- 220.Fleischhacker WW, Hinterhuber H, Bauer H, Pflug B, Berner P, Simhandl C, Wolf R, Gerlach W, Jaklitsch H, Sastre-y-Hernandez M, et al. Neuropsychobiology. 1992;26(1–2):59–64. doi: 10.1159/000118897. [DOI] [PubMed] [Google Scholar]
- 221.Altar CA. Trends Pharmacol Sci. 1999;20(2):59–61. doi: 10.1016/s0165-6147(99)01309-7. [DOI] [PubMed] [Google Scholar]
- 222.Nibuya M, Morinobu S, Duman RS. J Neurosci. 1995;15(11):7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Karege F, Perret G, Bondolfi G, Schwald M, Bertschy G, Aubry JM. Psychiatry Res. 2002;109(2):143–148. doi: 10.1016/s0165-1781(02)00005-7. [DOI] [PubMed] [Google Scholar]
- 224.Duman RS, Heninger GR, Nestler EJ. Arch Gen Psychiatry. 1997;54(7):597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 225.Vaidya VA, Marek GJ, Aghajanian GK, Duman RS. J Neurosci. 1997;17(8):2785–2795. doi: 10.1523/JNEUROSCI.17-08-02785.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Brandoli C, Sanna A, De Bernardi MA, Follesa P, Brooker G, Mocchetti I. J Neurosci. 1998;18(19):7953–7961. doi: 10.1523/JNEUROSCI.18-19-07953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]