

Abstract
IL-17 is the hallmark cytokine for the newly identified subset of T helper cells, Th17. Th17 cells are important instigators of inflammation in several models of autoimmune disease; in particular, collagen induced arthritis (CIA) and experimental autoimmune encephalomyelitis (EAE), which were previously characterized as Th1-mediated diseases. Although high levels of IFN-γ are secreted in CIA and EAE, disease is exacerbated in IFN-γ or IFN-γ receptor deficient mice due to the ability of IFN-γ to suppress IL-17 secretion. However, in proteoglycan-induced arthritis (PGIA), severe arthritis is dependent on the production of IFN-γ. We were therefore interested in determining the role of IL-17 in PGIA. We assessed the progression of arthritis in IL-17-deficient (IL-17−/−) mice and found the onset and severity of arthritis equivalent in wildtype (WT) and IL-17−/− mice. Despite evidence that IL-17 is involved in neutrophil recruitment, synovial fluid from arthritic joints showed a comparable proportion of Gr1+ neutrophils in WT and IL-17−/− mice. IL-17 is also implicated in bone destruction in autoimmune arthritis, however histological analysis of the arthritic joints from WT and IL-17−/− mice revealed a similar extent of joint cellularity, cartilage destruction and bone erosion despite significantly reduced RANKL expression. There were only subtle differences between WT and IL-17−/− in pro-inflammatory cytokine expression, T cell proliferation and autoanibody production. These data demonstrate that IL-17 is not absolutely required for autoimmune arthritis and production of other proinflammatory mediators are sufficient to compensate for the loss of IL-17 in PGIA.
Keywords: Autoimmunity, Cytokines, Inflammation, Rheumatoid Arthritis and Rodent
Introduction
Rheumatoid arthritis (RA) is a chronic systemic autoimmune disease of unknown etiology affecting ∼1% of the world population. Disease is characterized by chronic inflammation of the synovial tissues of multiple joints leading to joint destruction and loss of function (1-3). Several induced animal models have been developed to aid our understanding of the disease process and for the development of therapeutic tools for intervention (4). Two such models are collagen induced arthritis (CIA) and proteoglycan induced arthritis (PGIA); where collagen and proteoglycan are used as antigens respectively. The histological, pathological and some immune features of both CIA and PGIA closely resemble that of human disease and as such represent valuable models for the study of RA.
CD4+ T helper cells differentiate under the influence of antigen presenting cells into effector cells specializing in cytokine secretion and function. Th1-cells produce IFN-γ and are important mediators of cell immune response to infectious agents and in some autoimmune disease models (5-10). PGIA and CIA were originally designated as Th1-mediated autoimmune diseases based on robust production of IFN-γ. The importance of IFN-γ in PGIA was confirmed by the observation that arthritis onset and severity are reduced under conditions where IFN-γ is neutralized or in mice deficient in IFN-γ (11). By contrast, in CIA, lack of the IFN-γ receptor exacerbates autoimmune disease. Similar to CIA, another autoimmune model experimental autoimmune encephalomyelitis (EAE), disease is enhanced in IFN-γ deficient mice (12-14). Paradoxically CIA and EAE was associated with Th1 responses based on studies in which disease is inhibited by treatment with neutralizing antibodies to IL-12, a cytokine known to drive Th1 responses. The delineation of IL-12 into a family of cytokines including IL-23 (IL-12p40 and IL-12p19) and IL-27 (p28 and EB13) (15, 16) revealed the divergent roles for IL-12 and IL-23 in EAE and CIA (17-19). Reports showed the induction of EAE and CIA is ablated in mice lacking only IL-23 (p19−/−) and in mice deficient in IL-12p40, which lack both IL-12 and IL-23. Conversely, mice deficient in IL-12 alone (IL-12p35−/−) remain susceptible (17-19); clearly establishing a requirement for IL-23 (20, 21). Evidence that IL-12 and IFN-γ are not responsible for inflammation in CIA or EAE implied the existence of a separate population of proinflammatory effectors T cells. Studies revealed mice deficient in IL-23 have reduced IL-17 secretion (22) demonstrating a link between IL-23 and IL-17 production. A role for IL-17 in the pathogenesis of CIA and EAE was confirmed in IL-17-deficient mice, IL-17 over expression and neutralization studies (21, 23-26). These data lead to the characterization of the Th17 cell subset as a separate lineage of Th cells (27, 28). Importantly, it was found that IFN-γ regulates IL-17 activity (29) and thus provides an explanation for why a deficiency in IFN-γ or IFN-γ signaling in CIA and EAE leads to an increase in IL-17 and exacerbated disease.
There are several important functions of IL-17 that may contribute to its importance in models of arthritis and possibly rheumatoid arthritis. IL-17 acts on several cells types including macrophages, dendritic cells, T cells, endothelial cells, fibroblasts and synovial cells to upregulate chemokines, cell adhesion molecules and importantly cytokines; in particular the proinflammatory cytokines IL-1β, TNF, IL-6 (30). Neutrophils are the dominant cell population in synovial fluid of RA patients and in autoimmune models of arthritis. IL-17 is involved in the accumulation and activation of neutrophils through the induction of colony stimulating factors and C-X-C chemokines (31-33). Over expression of IL-17 in knee joints of naïve and type II collagen immunized mice results in neutrophil infiltration to the joint (24). Furthermore, an important role for IL-17 in osteoclastogenesis has been reported (24, 34-36). Both IL-1 and TNF can synergize with IL-17 to induce bone erosion in CIA. Also, IL-17 directly induces the expression of receptor activator of NK-kB ligand (RANKL). Synovial fluid of RA patients was found to contain IL-17 responsible for the activation of osteoclasts (37).
Although IFN-γ is necessary for the development of arthritis in PGIA, we do not know if IL-17 may also play a role in disease; in particular because neutrophil recruitment into synovial joint cavities and bone erosion are characteristic features of PGIA. We therefore examined the role of IL-17 in PGIA using IL-17−/− mice. Our results demonstrated that the onset and severity of arthritis was similar in IL-17−/− and WT mice. Furthermore, WT and IL-17−/− mice exhibit comparable numbers of neutrophils in synovial fluid and equivalent bone erosion in arthritic joints. In addition there was no difference between WT and IL-17−/− mice in the systemic inflammatory responses dominated by IFN-γ, TNF, IL-1β and IL-6. Our results demonstrate that any effect of IL-17 deficiency is compensated for by the production of other proinflammatory cytokines in PGIA.
Materials and Methods
Mice
IL-17-deficient mice on the BALB/c background (here designated as IL-17−/−) were generated as previously described (38). IL-17−/− mice were backcrossed to BALB/c for x generation in Dr. Iwakura's laboratory and then further backcrossed to the BALB/c (Charles Rivers, Wilmington MA) for 2 generation then intercrossed to obtain WT and IL-17−/− littermates. The BALB/c Charles Rivers colony is the most susceptible BALB/c subline for the induction of PGIA. Mice were genotype using primers specific for IL-17A. WT and IL-17−/− littermates were used in all experiments. IFNγ−/−, IL-12p40−/−, and IL-12p35−/− were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained in Rush University Medical Center facility. Female BALB/c age matched 12−14 wks of age were used in all experiments. All animal experiments were approved by the institutional Animal Care and Use Committee at Rush University Medical Center (Chicago, IL).
Induction and Assessment of Arthritis
Human cartilage was obtained following joint replacement surgery and was provided through the Orthopedic Tissue, Transplant, and Implant Repository of Rush University Medical Center, with the approval of the Institutional Review Board. PG was isolated as previously described (39). Female WT and IL-17−/− mice were immunized i.p. with 150 μg human PG measured as protein in dimethyldioctadecyl ammonium bromide (DDA) (Sigma Aldrich, St. Louis, MO) as described (40). Mice received booster immunizations at weeks three and six with 100μg PG in DDA. Mice were monitored for arthritis twice weekly and scored in a blinded manner. Paw swelling was scored based on an established scoring system on a scale from one to four as follows: 0, normal; 1, mild erythema and swelling of several digits; 2, moderate erythema and swelling; 3, more diffuse erythema and swelling; and 4, severe erythema and swelling of complete paw with ankylosis. Incidence of arthritis denotes the percentage of mice that develop PGIA. Each animal received a cumulative score ranging from 0 to 16, based on individual paw scores of 0 to 4.
Detection of serum Ab titers by ELISA
Mice immunized with human PG were anesthetized and bled from the orbital plexus. Serum was obtained and examined for antibodies against mouse and human PG by ELISA. EIA tissue culture “half-area” plates (Costar Corning, Corning, NY) were coated overnight at 4°C with 0.5 μg chrondroitinase ABC-digested PG or 0.75 μg of native mouse PG in carbonate buffer. Sera were serially diluted in PBS containing 0.5% Tween-20. Samples were incubated with the immobilized PGs, and plate-bound human PG- or mouse PG-specific Ab was detected using peroxidase-conjugated rabbit IgG against mouse IgG1 and IgG2a (Zymed, San Francisco, CA), respectively; which was then detected with the substrate o- phenylenediamine (OPDA). Samples were run in duplicate. Colorimetric change in each sample was measured with a spectrophotometer at 490 nm, and compared to a standard curve of known concentrations of unlabeled murine IgG1 and IgG2a (Southern Biotechnology Associates, Birmingham, AL). Data represent the mean ±SEM of antibodies from 5−10 mice.
Assessment of T cell activation by proliferation
CD4+ T-cells from the spleens of PG immunized mice were purified by negative selection using CD4 isolation kits and AutoMACS automated separation (Miltenyi Biotech, Auburn, CA). Purified CD4+ T cells (2.5×105 cells/ml) and irradiated (2500 rad) naïve spleen cells (2.5×105 cells/ml) were cultured in 96-well Falcon plates (Fisher Scientific, Pittsburgh, PA) in 200μl serum-free medium HL-1 medium (Fisher Scientific) containing 100μg/ml penicillin, 100μg/ml streptomycin and 2mM L-glutamine (complete media) in the presence or absence of PG (10μg/ml). T cell cultures were incubated at 37°C in 5% CO2 for 5 days; the last 18h pulsed with [3H] thymidine (0.5 μCi/well). The cells were harvested using a cell harvester (Tomtec, Orange, CT) and the incorporated [3H] thymidine was measured using a scintillation counter (EC&G Wallac, Galesburg, MD). Cells were cultured in quadruplicate.
Assessment of cytokines
Single cell suspensions of splenocytes harvested from PG immunized mice were prepared as previously described (41). Splenocytes (2.0 × 106 cells/ml) were incubated in 24-well Falcon plates (Fisher Scientific) in RPMI 1640 complete medium in triplicate (41). CD4+ T cells were purified using CD4 isolation beads (Miltenyi Biotech) from spleens of immunized mice and cultured with naïve irradiated (2500 rad) spleen cells. Cells were cultured in the absence or presence of PG (20μg/ml). Cytokines were measured from supernatants harvested on day 4 by ELISA using the OPT EIA mouse IFN-γ, IL-4, IL-1β, TNF, IL-6 sets (BD PharMingen, San Diego, CA) and a mouse IL-17 ELISA kit (R&D Systems, Minneapolis, MN).
Assessment of splenic and synovial fluid cell populations
Spleen, lymph node and synovial fluid cells were extracted at the time of sacrifice and analyzed by flow cytometry. Synovial fluid was collected from ankle joints of arthritic mice by repeated flushing of the open joint cavity with PBS followed by scraping of the synovial intima with a pipet tip. Approximately 3−5 × 105 cells were obtained from a single ankle joint with an arthritis score of 3−4. Cells were immunostained using fluorescence conjugated antibodies specific for GR1 or isotype control (BD Bioscience). Spleen and lymph node cells were stained with antibodies specific for T cells (CD3, CD4, and CD8) and B cells (CD19). Stained cells were acquired using a FACScanto II (BD Pharmingen) and data were analyzed with FACSDiva software (BD Pharmingen).
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Hind paws were minced and joint tissue RNA isolated using Tri-Reagent (Molecular Research Center, Cincinnati, OH). Reverse transcription was performed with random hexamers for priming and Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA). Gene specific amplification was performed using iQ SYBR Green Supermix (BioRad, Herclules, CA) and normalized to β-actin levels for each sample. All samples were run in triplicate on a BioRad iQ5 machine using BioRad proprietary iQ5 software. To confirm that the same amount of RNA was added to each PCR reaction, murine β actin amplification was performed on each sample. Relative fold induction was calculated using the formula 2–(ΔΔCt), where ΔΔCT is ΔCT(treatment) – ΔCT(control), ΔCT is CT(target gene) – CT(β actin), and CT is the cycle at which the threshold is crossed. PCR product quality was monitored using post-PCR melt curve analysis. Controls were from naïve non-immunized and target genes from PG-immunized WT and IL-17−/− joint tissue.
Histology
Hind ankle joints of immunized mice were isolated at week 13 after initial immunization. Joints were fixed in formalin, decalcified in 5% formic acid, embedded in paraffin and H&E. Bone erosion was measured as follows: at least 5 non-articulating bone surface areas, approximately 1mm length were measured at the distal tibia, calcaneus and 1 or 2 metatarsal bones. Erosive changes were scored on three semi-serial sections of the joint spaced 50 μm apart. Results were expressed as a percentage of the eroded length compared to the total length examined. Mean values were obtained for individual mice and then a mean was obtained for each group (n=10). Data represent the mean ± SEM percent bone erosion Cellular infiltration was measured on a scale of 0−4 by a blinded observer and values represent mean ± SEM of (n=11) sections from 2 independent experiments.
Statistical analysis
The Mann-Whitney U test was used to compare non-parametric data for statistical significance. P < 0.05 was considered significant.
Results
IL-12p35, p40 and IFN-γ essential for PGIA severity
The role of IL-12 in autoimmune disease models has been reexamined in light of the delineation of the IL-12 family of cytokines. Here we used mice deficient in either IL-12p35 or IL-12p40 to examine the role of IL-12 in PGIA. IL-12p35−/− and IL-12p40−/− mice were immunized with PG and the arthritis onset and severity were compared to WT mice. The incidence of disease was not significantly reduced in IL-12p35−/− and IL-12p40−/− mice (Fig.1A); however, a significant and sustained reduction in disease severity was observed in both the IL-12p35−/− and IL12p40−/− mice in comparison to WT mice (Fig.1B). The similarity in arthritis onset and severity in the IL-12p35−/− and IL-12p40−/− mice demonstrated that IL-12 is critical for the development of PGIA. However, we cannot eliminate a role for IL-23 in PGIA without investigating the development of arthritis in IL-23p19−/− mice.
Figure 1.

PGIA is dependant on IL-12p35, IL-12p40 and IFN-γ. Groups of mice age matched female mice were immunized i.p. with human PG in adjuvant three times at 3-wk intervals and monitored for arthritis onset and severity by a blinded observer. WT (n=10), IL-12p35−/− (n=8), IL-12p40−/− (n=8) incidence (A) is expressed as the percentage of mice that developed arthritis. Disease severity (B) is the sum of paw inflammation scores divided by the number of arthritic mice. WT (n=17) and IFNγ−/− (n=17) mice were immunized as described. Incidence (C) and severity (D) are shown. Results are shown as the mean scores ±SEM for week after the initial immunization. Asterisks (*) denote significant differences (p≤0.05). Data are representative of 2−3 experiments performed.
We have previously demonstrated a requirement for IFN-γ in PGIA (11). Here we confirm these findings to show despite early resistance to disease, IFN-γ−/− mice eventually succumb to disease, albeit at a reduced incidence and severity in comparison to WT mice (Fig. 1C and D). IFN-γ deficiency does not prevent the development of arthritis; however, it is an important factor in governing the severity of disease. The difference in disease onset and the reduction in the incidence of arthritis in IFN-γ−/− mice when compared to either IL-12p35−/− or IL-12p40−/− mice suggest IFN-γ−/− and IL-12 contribute different signals in the induction of arthritis. Together these results demonstrate development of PGIA is dependent upon IL-12 and IFN-γ; indicating the Th1 pathway is operative in this autoimmune disease.
Systemic and local joint expression of inflammatory cytokines in PGIA
The emerging function of IL-17 in several models of inflammation (21, 42-46) suggested a possible role for IL-17 in PGIA. We first examined the concentration of IFNγ and IL-17 and several known proinflammatory cytokines TNF, IL-6 and IL-1β in synovial fluid from inflamed ankle joints of WT mice (Fig. 2A). Pooled samples of synovial fluid from arthritic mice were examined by ELISA and standardized to total protein. The T cell derived cytokines IFN-γ and IL-17 were present at low concentrations; conversely, TNF and particularly IL-6 and IL-1β were highly expressed.
Figure 2.

Inflammatory cytokines expressed systemically and in joints of arthritic mice. (A) IFN-γ, IL-17, IL-4, TNF, IL-6 and IL-1β cytokine concentrations in synovial fluid of arthritic WT mice standardized to total protein levels (n=8). (B) RNA was harvested from inflamed joint tissues and analyzed by qRT-PCR. The relative increase in RNA expression to non-immune joint tissue was determined. Asterisks (*) denotes significant differences between p≤0.05 IFN-γ and IL-17 transcripts. (C) Spleen cells from immunized WT mice were cultured in the presence and absence of PG for 4 days (n=5). Supernatants were harvested and assayed by ELISA for IFN-γ and IL-17. Values represent the mean ± SEM of (quadruplicate cultures of individual mice). Results are representative of at least 2 experiments. Asterisks (*) denote significant differences (p≤0.05) between IFN-γ and IL-17 cytokine production..
We next assessed the level of IFNγ and IL-17 transcripts in arthritic joint tissue. IFN-γ expression was modestly elevated in the joint tissue from arthritic mice while IL-17 transcripts were elevated over 3 fold compared to control non-immune joint tissue (Figure 2B). The reduced levels of IL-17 protein in comparison to RNA transcripts may be due to the utilization of IL-17 protein by synovial tissue cells. To determine if IL-17 was produced systemically in PGIA, spleen cells from arthritic mice were re-stimulated in vitro with PG and supernatants were examined for IFN-γ and IL-17 production by ELISA (Figure 2C). IFN-γ and IL-17 were produced in spleen cell cultures; however, significantly more IFN-γ was produced in comparison to IL-17. The predominance of IFN-γ in the spleen and IL-17 mRNA in the joint suggested both cytokines may play a role in the generation of a robust inflammatory response in PGIA.
IL-17 is not required for development of PGIA
To assess the role of IL-17 in the development of PGIA we used IL-17−/− mice. WT and IL-17−/− were initially tested for expression of IL-17 by ELISA and the level of IL-17 protein in the supernatant from IL-17−/− T cells was below the limits of detection (Fig.3A). WT and IL-17−/− mice were immunized with PG and the development of arthritis monitored over time. IL-17−/− mice succumbed to disease with similar kinetics and degree of severity in comparison to WT mice (Fig. 3A and B). These data demonstrate that IL-17 is not critical for the development of PGIA. Together with our previous data showing an important role for IFN-γ in PGIA, these data support an inflammatory role for IFN-γ (11) and not IL-17 in this model indicating that in different models of arthritis IFN-γ and IL-17 may function differently.
Figure 3.

IL-17−/− mice succumb to PGIA with similar onset and severity as WT mice. WT (n=7) and IL-17−/− (n=17) age matched female mice were immunized i.p. with human PG in adjuvant three times at 3-wk intervals and monitored for arthritis onset and severity by a blinded observer. (A) Incidence is expressed as the percentage of mice that developed arthritis. (B) Disease severity is the sum of paw inflammation scores for each mouse divided by the number of arthritic mice. Results are shown as the mean scores ±SEM for week after the initial immunization. Asterisks (*) denote significant differences (p≤0.05). Data are representative of 3 experiments performed.
Joint tissue histology is similar in WT and IL-17−/− mice
To determine if the similarity in paw erythema and swelling in WT and IL-17−/− mice corresponded to comparable cellar infiltration and joint damage we examined joint histology from hind limbs. The histological picture in WT and IL-17−/− mice was characteristic of acute arthritis (Figure 4A-D). Infiltration of mononuclear and polymorphonuclear cells in the synovial cavity and adjacent tissues, edema of the synovial and periarticular tissues and synovial hyperplasia was similar in WT and IL-17−/− mice.
Figure 4.
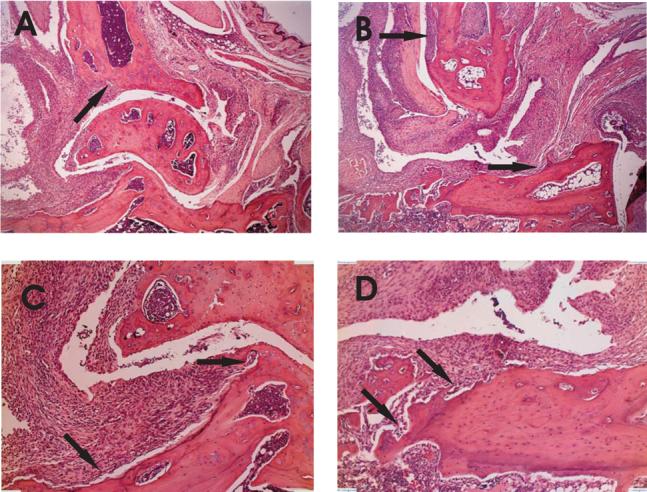

Similar histopathology of WT and IL-17−/− arthritic ankle joints. Hind limbs of immunized mice were dissected, decalcified and embedded in paraffin. Tissue sections were stained with H&E. Representative sections from WT (A and C) and IL-17−/− (B and D) are shown. Magnification objective X4 (A and B) and X10 (C and D). Arrows indicate areas of bone erosion. Cellular infiltration (E) was measured on a scale of 0−4 (n=11). (F) FACS analysis of pooled synovial fluid from WT (n=7) and IL-17−/− mice (n=17) stained for Gr1+ and analyzed by FACS. Values are the mean ±SEM. Data are representative of two separate experiments.
We further characterized the synovial infiltrating cells because IL-17 is known to play an important role in the recruitment of neutrophils (24, 47). Neutrophils were necessary to maintain chronic inflammation in PGIA as depletion of neutrophils with anti-Gr1 mAb suppressed chronic inflammation (data not shown). If IL-17 was necessary for neutrophil recruitment in PGIA we would anticipate that IL-17 deficiency would lead to a reduction of neutrophils recruited to the synovial cavity. We isolated the synovial fluid from ankle joints of WT and IL-17−/− arthritic mice and stained for Gr1+ cells (Figure 4F). Gr1+ cells comprised 80−90% of the cell population in the synovial fluid. There was no significant difference in the proportion of Gr1+ neutrophils or the number (data not shown) in the synovial fluid of WT and IL-17−/− arthritic mice. These data demonstrate that in PGIA, IL-17 deficiency does not affect inflammation or infiltration of neutrophils.
Expression of RANKL transcripts does not correlate with bone erosion in IL-17−/− mice
IL-17 induces the expression of RANKL in osteoblasts and RANKL is an important regulator of osteoclastogenesis (37, 48). To investigate the possibility that RANKL expression may be reduced as consequence of ablated IL-17, we compared the expression of RANKL transcripts in inflamed joint tissues in WT and IL-17−/− mice (Figure 5A). We observed a reduction in RANKL expression of more than 50% in IL-17−/− mice. To determine if the reduction in RANKL expression resulted in decreased bone erosion, we examined the degree of bone erosion in WT and IL-17−/− arthritic mice. Despite the substantial reduction in RANKL expression in the joint tissues we observed similar degree of bone erosion in WT and IL-17−/− mice visualized as cellular infiltration into non-articulating bone surface areas (Figure 5B) suggesting the level of RANKL expression was sufficient for osteoclast activation and bone erosion. Thus, the extent of cellular infiltration, the similarity in the neutrophil composition, and the level of bone erosion in WT and IL-17−/− mice suggest that in the absence of IL-17 a redundant pathway exists for inflammation.
Figure 5.

RANKL mRNA reduced, bone erosion unaffected by IL-17 deficiency. (A) qRT-PCR analysis of RANKL mRNA transcripts from inflamed hind limbs of arthritic WT (n=14) and IL-17−/− (n=9) mice. Each sample was standardized to ß-actin levels and run in triplicate. Date represents RANKL expression in IL-17−/− joint tissue relative to WT. (B) Bone erosion was measured as described in material and methods from arthritic WT (n=10) and IL-17−/− (n=10). Values are the mean ±SEM. Asterisks (*) denote significant differences (p≤0.05).
Inflammatory cytokine expression in spleen and joint
We have seen robust inflammation in IL-17−/− mice similar to WT mice (Figure 3A, B); demonstrating effective homing of inflammatory cells to the joint. To determine if there were any differences in the downstream effects of IL-17 deficiency we measured the ability of ex vivo spleen cells from arthritic WT and IL-17−/− mice to generate cytokines associated with inflammation. Concentrations of TNF, IL-1ß and IL-6 were similar in WT and IL-17−/− splenocyte culture supernatants (Fig. 6A-C). We also examined the expression of cytokine transcripts from joint tissues of arthritic mice (Fig. 6D-F). Similar to results from spleen cultures, TNF expression in WT and IL-17−/− was comparable. However, IL-1ß was expressed significantly more in IL-17−/− joints while conversely, IL-6 expression was significantly less in IL-17−/− mice compared to WT. Despite differences in mRNA expression, inflammation was indistinguishable between IL-17−/− and WT mice (Figure 3A, B) demonstrating the complex and redundant pathways utilized in mediating inflammation.
Figure 6.

Systemic inflammatory cytokines unaffected by IL-17 deficiency whereas joint tissue mRNA transcripts altered. Spleen cells from immunized mice (n=5) were cultured in the presence or absence of PG for 4 days. Supernatants were harvested and assayed by ELISA for (A) TNF, (B) IL-1β, (C) IL-6. Values represent the mean ±SEM. (D) TNF, (E) IL-1β, (F) IL-6 transcripts were harvested from inflamed ankle joints of arthritic and WT and IL-17−/− and analyzed by qRT-PCR. Each sample was standardized to β-actin expression and run in triplicate. Date represents cytokine transcript expression in IL-17−/− joint tissue relative to WT. Values are the mean ±SEM. Asterisks (*) denote significant differences (p≤0.05). Results are representative of 2 experiments.
PG-specific T cell and B cell responses were similar in WT and IL-17−/− mice
The demonstrated inflammatory nature of both IFN-γ and IL-17 in autoimmune diseases suggests both cytokines may contribute to inflammation in PGIA. IL-17 production is regulated by IFN-γ (29) and some data suggests that IL-17 may regulate IFN-γ expression (49). To determine if IFN-γ was up-regulated in IL-17−/− mice we cultured CD4+ T cells from arthritic WT and IL-17−/− mice in the presence and absence of PG. There was no significant difference in IFN-γ production by CD4+ T cells or whole spleen cultures from WT and IL-17−/− mice (Figure 7A and data not shown).
Figure 7.

Cyokines, PG-specific CD4+ T cell proliferation enhanced and PG-specific antibody isotopes minimally affected by IL-17 deficiency. Spleen cells from PG immunized WT (n=8) and IL-17−/− mice (n=8). CD4+ T cells were isolated and stimulated in the presence of absence of PG with irradiated naïve spleen cells. Supernatants were harvested and assayed by ELISA for IFN-γ (A) or IL-4 (B). For proliferation T cells were pulsed with [3H]-thymidine (C). Serum concentrations of anti-human and anti-mouse PG IgG1 (D) and IgG2a (E). Values are the mean ±SEM. Asterisks (*) denote significant differences (p≤0.05).
We have shown that IL-4 functions to suppress the severity of PGIA via STAT6 (11, 41, 50). Similarly IL-4 inhibits the IL-17-induced inhibition of chondrocyte proteoglycan synthesis in intact murine articular cartilage (34). To examine the possible role of IL-4 in IL-17−/− mice we assessed IL-4 production from arthritic WT and IL-17−/− mice (Figure 7B). In accord with our earlier studies we found minimal IL-4 production in WT or IL-17−/− mice. We were unable to identify an inhibitory role for IL-17 in IL-4 production in PGIA.
In order to determine if CD4+ T cells were similarly activated we examined the antigen-specific proliferation of CD4+ T cells from immunized WT and IL-17−/− mice. We found proliferation to be significantly higher in the IL-17 deficient CD4+ T cells compared to WT (Fig. 7C). This is in accord with a previously described anti-proliferative role for IL-17 (51). However, we were unable to identify a significant difference in either the number of total lymphocytes in the spleen or lymph nodes or the percent CD3, CD4, CD8, and CD19 cells in WT and IL-17−/− mice (data not shown).
We have previously shown PG specific T cells and B cells to be critical for the development of PGIA (52, 53). Recently, IL-17 deficiency has been shown to inhibit IgG2a production in CIA (42), and to play a role in germinal center development in autoimmune mice (54). To determine if B cell responses were intact in IL-17−/− mice we examined PG specific antibody isotype concentrations from the serum of immunized arthritic mice. We found anti-mouse and anti- human PG IgG1and IgG2a antibody isotypes to be comparable in WT and IL-17−/− mice (Fig. 7D and E).
Discussion
PGIA is critically dependant on CD4+ T cells and their production of IFN-γ (41, 55). The recent identification of the new members of the IL-12 family and the discovery of the pathogenic Th17 subset (15, 26, 28, 56) suggested the possibility that IL-17 may be involved in the pathogenesis of PGIA. We first determined that IL-12 (p40/p35)-deficient mice developed significantly less severe arthritis than WT mice. Although we cannot rule out a role for IL-23 (p40/p19) or IL-35 (p35/EbI3) the reduction in arthritis correlates a similar suppression of arthritis in IFN-γ−/− and STAT4−/− mice further establishing PGIA as a Th1-mediated disease. We next assessed whether IL-17 was produced in PGIA. We found IL-17 transcripts to be expressed in arthritic joint tissues but only minimally in the spleen; the converse of the IFN-γ expression pattern. To demonstrate a contribution of IL-17 in development of PGIA, we used IL-17-deficient mice. There was no apparent consequence of a loss of IL-17 expression on the onset or the severity of arthritis. Assessment of the inflamed joints histologically, showed that the cellular infiltration, cartilage destruction and bone erosion were similar in IL-17−/− and WT mice.
In PGIA, the synovial fluid is comprised mainly of neutrophils. The role of IL-17 in granulopoesis and the recruitment of neutrophils (23) suggested that the primary cell population infiltrating the inflamed joint may be altered in IL-17−/− mice. However, the numbers and percentages of Gr1+ neutrophils in the synovial fluid of arthritic joints were similar in WT and IL-17−/− mice. These data reveal a redundant pathway for neutrophil recruitment in PGIA. It is likely that IFN-γ or downstream effector cytokines such as TNF, IL-1β and IL-6, which are present in IL-17−/− mice, mediate the recruitment of neutrophils in the absence of IL-17. Sun et al., demonstrated that IL-12 is capable of promoting IFN-γ dependant recruitment of neutrophils (57) and augmentation of TNF, which is itself capable of recruiting neutrophils (58). In addition, IL-6 has been shown in a model of the rheumatoid synovitis to directly mediate neutrophil recruitment (59). Thus, the ability of IL-17 to augment neutrophil recruitment in the presence of other proinflammatory cytokine is not required in PGIA.
In several models of arthritis IL-17 plays a role in stimulating osteoclastogenesis. We quantified the degree of bone erosion in serial sections of arthritic hind limbs from WT and IL-17−/− mice. We were unable to identify any difference in the extent of bone erosion between WT and IL-17−/− mice. IL-17 induces fibroblasts to produce TNF and IL-6 and promotes bone erosion in CIA via RANKL and IL-1β (47). However, our data indicate that IFN-γ, TNF, IL-1β, and IL-6 were sufficient to induce bone erosion in the absence of IL-17. We observed an approximate 50% reduction in RANKL expression in IL-17−/− mice confirming the importance of IL-17 in RANKL expression. Nevertheless, the degree of inhibition of RANKL was insufficient to prevent destruction of bone. Studies looking at bone erosion in 3 separate models, ovariectomized mice, LPS stimulation and inflammation models reveal IFN-γ stimulates osteoclast activation indirectly through T cell secretion of RANKL (60). There are several overlapping effects of IFN-γ, IL-1β, IL-6, and TNF with IL-17 suggesting that in PGIA the lack of one of these cytokines might be easily compensated for by the presence of the others.
It is now apparent that IFN-γ regulates the development of Th17 cells (26, 28) and disease severity in a number of autoimmune models (21, 42, 61-63). Likewise, there is some evidence that IL-17 may regulate IFN-γ production. Komiyama et al report that MOG-specific IFN-γ producing T cells are increased in IL-17−/− mice (12). IL-4 also inhibits Th17 production (26, 28), however at present there is no evidence that IL-17 regulates IL-4 production. Our results do not support a role for IL-17 in regulation of either IFN-γ or IL4 production in PGIA as PG-specific CD4+ T cells from WT and IL-17−/− mice produce equivalent amounts of IFN-γ and IL-4. As a measure of the efficacy of T cell responses in IL-17−/− mice, we examined the proliferation of CD4+ T cells in response to PG. We found the IL-17−/− CD4+ cells to proliferate significantly more than WT cells. This data is in accord with the documented anti-proliferative effects of IL-17 seen in intestinal epithelial cells (51) and could contribute to the robust inflammation seen in the IL-17−/− mice.
Several models of induced and spontaneous arthritis and other autoimmune diseases are dependent on IL-17 (21, 42-46) whereas several are dependent on IFN-γ (5-11, 41, 64). There are a number of possible mechanisms to explain the lack of reliance on IL-17. First, the dominant IFN-γ response in PGIA appears sufficient to drive the production of proinflammatory cytokines such as TNF, IL-1β and IL-6 in the absence of IL-17. Secondly, the consequence of a strong IFN-γ response is the inhibition of IL-17 making it less important in inflammation. Thus, we would predict that if the IFN-γ response was reduced, a role of IL-17 might be uncovered. It is presently unclear what drives the IFN-γ response in PGIA but since Th1 and Th17 cells differentiate under the influence of different cytokines it is likely that the cytokine milieu in which disease is initiated determines the T cell phenotype. It is also possible that IFN-γ and IL-17 act at different phases of disease. For example, in CIA early blockade of IFN-γ inhibits disease while late blockade exasperates disease (65). Similarly, in a model of uveoretinitis IL-17 was important for disease severity only late in the maintenance phase (45).
Despite strong evidence for IL-17 in some autoimmune disease models, data supporting a role for IL-17 in RA is inconclusive. Recent reports show increased IL-17 in RA synovial fluids and in the T cell areas of RA synovial tissue (66, 67). IL-17 was also over expressed in serum and activated PBMC cultures of patients with RA (68). However, another study found the frequency of Th17 cells in the joints of RA patients was significantly decreased compared to peripheral blood of the same RA patients (69). IFN-γ and its receptor IFN-γR are significantly upregulated in the joints of RA patients compared to patients with osteoarthritis (70). Also a genetic linkage of RA to IFN-γ is detected in affected sibling paired families (71). The role of IFN-γ and IL-17 in RA remains the subject of intense research. Moreover, as the differences between mouse and human Th subset biology become more evident (72-74) we should be cautious assuming physiological relevance to any one model particularly in light of the heterogeneity of human disease (75-77).
Here, using an established murine model of RA, we have shown IL-17 is unnecessary for the onset or severity of inflammation in PGIA. We further show that neutrophil recruitment to inflamed joints and bone erosion are unaffected by a deficiency in IL-17.
Acknowledgments
This research was supported by grants from the National Institutes of Health (NIH) AR47652 to A. Finnegan.
Abbreviations used in this manuscript
- RA
rheumatoid arthritis
- PG
human proteoglycan (aggregan)
- PGIA
PG-induced arthritis
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2−3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Goronzy JJ, Weyand CM. Rheumatoid arthritis. Immunol Rev. 2005;204:55–73. doi: 10.1111/j.0105-2896.2005.00245.x. [DOI] [PubMed] [Google Scholar]
- 2.Firestein GS. Evolving concepts in rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 3.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 4.Joe B, Griffiths MM, Remmers EF, Wilder RL. Animal models of rheumatoid arthritis and related inflammation. Curr Rheumatol Rep. 1999;1:139–148. doi: 10.1007/s11926-999-0011-7. [DOI] [PubMed] [Google Scholar]
- 5.Schwarting A, Tesch G, Kinoshita K, Maron R, Weiner HL, Kelley VR. IL-12 drives IFN-gamma-dependent autoimmune kidney disease in MRL-Fas(lpr) mice. J Immunol. 1999;163:6884–6891. [PubMed] [Google Scholar]
- 6.Schwarting A, Wada T, Kinoshita K, Tesch G, Kelley VR. IFN-gamma receptor signaling is essential for the initiation, acceleration, and destruction of autoimmune kidney disease in MRL-Fas(lpr) mice. J Immunol. 1998;161:494–503. [PubMed] [Google Scholar]
- 7.Zhang GX, Xiao BG, Bai XF, van der Meide PH, Orn A, Link H. Mice with IFN-gamma receptor deficiency are less susceptible to experimental autoimmune myasthenia gravis. J Immunol. 1999;162:3775–3781. [PubMed] [Google Scholar]
- 8.Yu S, Medling B, Yagita H, Braley-Mullen H. Characteristics of inflammatory cells in spontaneous autoimmune thyroiditis of NOD.H-2h4 mice. J Autoimmun. 2001;16:37–46. doi: 10.1006/jaut.2000.0458. [DOI] [PubMed] [Google Scholar]
- 9.Haas C, Ryffel B, Le Hir M. IFN-gamma is essential for the development of autoimmune glomerulonephritis in MRL/Ipr mice. J Immunol. 1997;158:5484–5491. [PubMed] [Google Scholar]
- 10.Egwuagu CE, Sztein J, Mahdi RM, Li W, Chao-Chan C, Smith JA, Charukamnoetkanok P, Chepelinsky AB. IFN-gamma increases the severity and accelerates the onset of experimental autoimmune uveitis in transgenic rats. J Immunol. 1999;162:510–517. [PubMed] [Google Scholar]
- 11.Finnegan A, Grusby MJ, Kaplan CD, O'Neill SK, Eibel H, Koreny T, Czipri M, Mikecz K, Zhang J. IL-4 and IL-12 regulate proteoglycan-induced arthritis through Stat- dependent mechanisms. J Immunol. 2002;169:3345–3352. doi: 10.4049/jimmunol.169.6.3345. [DOI] [PubMed] [Google Scholar]
- 12.Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- 13.Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26:1641–1646. doi: 10.1002/eji.1830260735. [DOI] [PubMed] [Google Scholar]
- 14.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 15.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- 16.Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science. 2005;307:430–433. doi: 10.1126/science.1103336. [DOI] [PubMed] [Google Scholar]
- 17.Robinson DS, Lloyd CM. Asthma: T-bet--a master controller? Curr Biol. 2002;12:R322–324. doi: 10.1016/s0960-9822(02)00830-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 20.Mandi B, Hadhazy C, Rethy A, Kiss EK, Glant T. Studies on cartilage formation. XVI. Chemical and histochemical assay of lipids in the regenerating articular cartilage. Acta Biol Acad Sci Hung. 1975;26:115–133. [PubMed] [Google Scholar]
- 21.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 22.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 23.Lund RJ, Chen Z, Scheinin J, Lahesmaa R. Early target genes of IL-12 and STAT4 signaling in th cells. J Immunol. 2004;172:6775–6782. doi: 10.4049/jimmunol.172.11.6775. [DOI] [PubMed] [Google Scholar]
- 24.Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, van den Berg WB. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–659. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- 25.Lubberts E, Joosten LA, van de Loo FA, Schwarzenberger P, Kolls J, van den Berg WB. Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm Res. 2002;51:102–104. doi: 10.1007/BF02684010. [DOI] [PubMed] [Google Scholar]
- 26.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 28.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 29.Cruz A, Khader SA, Torrado E, Fraga A, Pearl JE, Pedrosa J, Cooper AM, Castro AG. Cutting edge: IFN-gamma regulates the induction and expansion of IL-17-producing CD4 T cells during mycobacterial infection. J Immunol. 2006;177:1416–1420. doi: 10.4049/jimmunol.177.3.1416. [DOI] [PubMed] [Google Scholar]
- 30.Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 31.Laan M, Cui ZH, Hoshino H, Lotvall J, Sjostrand M, Gruenert DC, Skoogh BE, Linden A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- 32.Witowski J, Pawlaczyk K, Breborowicz A, Scheuren A, Kuzlan-Pawlaczyk M, Wisniewska J, Polubinska A, Friess H, Gahl GM, Frei U, Jorres A. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J Immunol. 2000;165:5814–5821. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]
- 33.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 34.Lubberts E, Joosten LA, Chabaud M, van Den Bersselaar L, Oppers B, Coenen-De Roo CJ, Richards CD, Miossec P, van Den Berg WB. IL-4 gene therapy for collagen arthritis suppresses synovial IL-17 and osteoprotegerin ligand and prevents bone erosion. J Clin Invest. 2000;105:1697–1710. doi: 10.1172/JCI7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lubberts E, Joosten LA, van de Loo FA, van den Gersselaar LA, van den Berg WB. Reduction of interleukin-17-induced inhibition of chondrocyte proteoglycan synthesis in intact murine articular cartilage by interleukin-4. Arthritis Rheum. 2000;43:1300–1306. doi: 10.1002/1529-0131(200006)43:6<1300::AID-ANR12>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 36.Lubberts E, van den Bersselaar L, Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJ, Kolls JK, Joosten LA, van den Berg WB. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-kappa B ligand/osteoprotegerin balance. J Immunol. 2003;170:2655–2662. doi: 10.4049/jimmunol.170.5.2655. [DOI] [PubMed] [Google Scholar]
- 37.Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 39.Glant TT, Finnegan A, Mikecz K. Proteoglycan-induced arthritis: immune regulation, cellular mechanisms, and genetics. Crit Rev Immunol. 2003;23:199–250. doi: 10.1615/critrevimmunol.v23.i3.20. [DOI] [PubMed] [Google Scholar]
- 40.Hanyecz A, Berlo SE, Szanto S, Broeren CP, Mikecz K, Glant TT. Achievement of a synergistic adjuvant effect on arthritis induction by activation of innate immunity and forcing the immune response toward the Th1 phenotype. Arthritis Rheum. 2004;50:1665–1676. doi: 10.1002/art.20180. [DOI] [PubMed] [Google Scholar]
- 41.Finnegan A, Mikecz K, Tao P, Glant TT. Proteoglycan (aggrecan)-induced arthritis in BALB/c mice is a Th1-type disease regulated by Th2 cytokines. J Immunol. 1999;163:5383–5390. [PubMed] [Google Scholar]
- 42.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 43.Cho ML, Kang JW, Moon YM, Nam HJ, Jhun JY, Heo SB, Jin HT, Min SY, Ju JH, Park KS, Cho YG, Yoon CH, Park SH, Sung YC, Kim HY. STAT3 and NF-kappaB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J Immunol. 2006;176:5652–5661. doi: 10.4049/jimmunol.176.9.5652. [DOI] [PubMed] [Google Scholar]
- 44.Hirota K, Hashimoto M, Yoshitomi H, Tanaka S, Nomura T, Yamaguchi T, Iwakura Y, Sakaguchi N, Sakaguchi S. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J Exp Med. 2007;204:41–47. doi: 10.1084/jem.20062259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshimura T, Sonoda KH, Miyazaki Y, Iwakura Y, Ishibashi T, Yoshimura A, Yoshida H. Differential roles for IFN-{gamma} and IL-17 in experimental autoimmune uveoretinitis. Int Immunol. 2008;20:209–214. doi: 10.1093/intimm/dxm135. [DOI] [PubMed] [Google Scholar]
- 46.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koenders MI, Lubberts E, Oppers-Walgreen B, van den Bersselaar L, Helsen MM, Di Padova FE, Boots AM, Gram H, Joosten LA, van den Berg WB. Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. 2005;167:141–149. doi: 10.1016/S0002-9440(10)62961-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymphnode organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 49.Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol. 2007;81:1258–1268. doi: 10.1189/jlb.1006610. [DOI] [PubMed] [Google Scholar]
- 50.Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, Mizoguchi E, Mudter J, Galle PR, Bhan A, Autschbach F, Sullivan BM, Szabo SJ, Glimcher LH, Blumberg RS. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J Exp Med. 2002;195:1129–1143. doi: 10.1084/jem.20011956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Awane M, Andres PG, Li DJ, Reinecker HC. NF-kappa B-inducing kinase is a common mediator of IL-17-, TNF-alpha-, and IL-1 beta-induced chemokine promoter activation in intestinal epithelial cells. J Immunol. 1999;162:5337–5344. [PubMed] [Google Scholar]
- 52.Hollo K, Glant TT, Garzo M, Finnegan A, Mikecz K, Buzas E. Complex pattern of Th1 and Th2 activation with a preferential increase of autoreactive Th1 cells in BALB/c mice with proteoglycan (aggrecan)-induced arthritis. Clin Exp Immunol. 2000;120:167–173. doi: 10.1046/j.1365-2249.2000.01174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.O'Neill SK, Shlomchik MJ, Glant TT, Cao Y, Doodes PD, Finnegan A. Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe autoimmune arthritis. J Immunol. 2005;174:3781–3788. doi: 10.4049/jimmunol.174.6.3781. [DOI] [PubMed] [Google Scholar]
- 54.Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, Le TV, Lorenz RG, Xu H, Kolls JK, Carter RH, Chaplin DD, Williams RW, Mountz JD. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 55.Banerjee S, Webber C, Poole AR. The induction of arthritis in mice by the cartilage proteoglycan aggrecan: roles of CD4+ and CD8+ T cells. Cell Immunol. 1992;144:347–357. doi: 10.1016/0008-8749(92)90250-s. [DOI] [PubMed] [Google Scholar]
- 56.Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol. 2006;18:349–356. doi: 10.1016/j.coi.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 57.Sun K, Salmon SL, Lotz SA, Metzger DW. Interleukin-12 promotes gamma interferon-dependent neutrophil recruitment in the lung and improves protection against respiratory Streptococcus pneumoniae infection. Infect Immun. 2007;75:1196–1202. doi: 10.1128/IAI.01403-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Ramos BF, Jakschik BA. Neutrophil recruitment by tumor necrosis factor from mast cells in immune complex peritonitis. Science. 1992;258:1957–1959. doi: 10.1126/science.1470922. [DOI] [PubMed] [Google Scholar]
- 59.Lally F, Smith E, Filer A, Stone MA, Shaw JS, Nash GB, Buckley CD, Rainger GE. A novel mechanism of neutrophil recruitment in a coculture model of the rheumatoid synovium. Arthritis Rheum. 2005;52:3460–3469. doi: 10.1002/art.21394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, Weitzmann MN, Pacifici R. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. 2007;117:122–132. doi: 10.1172/JCI30074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Irmler IM, Gajda M, Brauer R. Exacerbation of antigen-induced arthritis in IFN-gamma-deficient mice as a result of unrestricted IL-17 response. J Immunol. 2007;179:6228–6236. doi: 10.4049/jimmunol.179.9.6228. [DOI] [PubMed] [Google Scholar]
- 62.Rohn TA, Jennings GT, Hernandez M, Grest P, Beck M, Zou Y, Kopf M, Bachmann MF. Vaccination against IL-17 suppresses autoimmune arthritis and encephalomyelitis. Eur J Immunol. 2006;36:2857–2867. doi: 10.1002/eji.200636658. [DOI] [PubMed] [Google Scholar]
- 63.Hofstetter HH, Ibrahim SM, Koczan D, Kruse N, Weishaupt A, Toyka KV, Gold R. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell Immunol. 2005;237:123–130. doi: 10.1016/j.cellimm.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 64.Wang B, Andre I, Gonzalez A, Katz JD, Aguet M, Benoist C, Mathis D. Interferon-gamma impacts at multiple points during the progression of autoimmune diabetes. Proc Natl Acad Sci U S A. 1997;94:13844–13849. doi: 10.1073/pnas.94.25.13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosloniec EF, Latham K, Guedez YB. Paradoxical roles of IFN-gamma in models of Th1-mediated autoimmunity. Arthritis Res. 2002;4:333–336. doi: 10.1186/ar432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lubberts E, Koenders MI, van den Berg WB. The role of T-cell interleukin-17 in conducting destructive arthritis: lessons from animal models. Arthritis Res Ther. 2005;7:29–37. doi: 10.1186/ar1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stamp LK, James MJ, Cleland LG. Interleukin-17: the missing link between T-cell accumulation and effector cell actions in rheumatoid arthritis? Immunol Cell Biol. 2004;82:1–9. doi: 10.1111/j.1440-1711.2004.01212.x. [DOI] [PubMed] [Google Scholar]
- 68.Kim KW, Cho ML, Park MK, Yoon CH, Park SH, Lee SH, Kim HY. Increased interleukin-17 production via a phosphoinositide 3-kinase/Akt and nuclear factor kappaB-dependent pathway in patients with rheumatoid arthritis. Arthritis Res Ther. 2005;7:R139–148. doi: 10.1186/ar1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi JI, Kaibara N, Hori A, Iwamoto Y, Yoshikai Y. Th1 but not Th17 cells predominate in the joints of patients with rheumatoid arthritis. Ann Rheum Dis. 2007 doi: 10.1136/ard.2007.080341. [DOI] [PubMed] [Google Scholar]
- 70.Dolhain RJ, ter Haar NT, Hoefakker S, Tak PP, de Ley M, Claassen E, Breedveld FC, Miltenburg AM. Increased expression of interferon (IFN)-gamma together with IFN-gamma receptor in the rheumatoid synovial membrane compared with synovium of patients with osteoarthritis. Br J Rheumatol. 1996;35:24–32. doi: 10.1093/rheumatology/35.1.24. [DOI] [PubMed] [Google Scholar]
- 71.John S, Myerscough A, Marlow A, Hajeer A, Silman A, Ollier W, Worthington J. Linkage of cytokine genes to rheumatoid arthritis. Evidence of genetic heterogeneity. Ann Rheum Dis. 1998;57:361–365. doi: 10.1136/ard.57.6.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- 73.Chen Z, Tato CM, Muul L, Laurence A, O'Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. 2007;56:2936–2946. doi: 10.1002/art.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 75.Hueber W, Kidd BA, Tomooka BH, Lee BJ, Bruce B, Fries JF, Sonderstrup G, Monach P, Drijfhout JW, van Venrooij WJ, Utz PJ, Genovese MC, Robinson WH. Antigen microarray profiling of autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2005;52:2645–2655. doi: 10.1002/art.21269. [DOI] [PubMed] [Google Scholar]
- 76.Zendman AJ, van Venrooij WJ, Pruijn GJ. Use and significance of anti-CCP autoantibodies in rheumatoid arthritis. Rheumatology (Oxford) 2006;45:20–25. doi: 10.1093/rheumatology/kei111. [DOI] [PubMed] [Google Scholar]
- 77.van der Pouw Kraan TC, Wijbrandts CA, van Baarsen LG, Voskuyl AE, Rustenburg F, Baggen JM, Ibrahim SM, Fero M, Dijkmans BA, Tak PP, Verweij CL. Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: assignment of a type I interferon signature in a subpopulation of patients. Ann Rheum Dis. 2007;66:1008–1014. doi: 10.1136/ard.2006.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]