

Abstract
We describe a maternally transmitted genomic-imprinting syndrome of mental retardation, hypotonia, and unique dysmorphism with elongated face. We mapped the disease-associated locus to ∼7.27 Mb on chromosome 8q24 and demonstrated that the disease is caused by a missense mutation in the maternal copy of KCNK9 within this locus. KCNK9 is maternally transmitted (imprinted with paternal silencing) and encodes K2P9.1, a member of the two pore-domain potassium channel (K2P) subfamily. The mutation fully abolishes the channel's currents—both when functioning as a homodimer or as a heterodimer with K2P3.1.
Introduction
In contrast with Mendel's theory, for some autosomal human genes, only the paternal or the maternal copy of the gene is expressed. Thus, for a gene that is normally imprinted with paternal silencing, a mutation in the maternal copy of the gene will result in disease, whereas a mutation in the paternal copy will have no effect. Thus far, more than 80 genes have been demonstrated to be imprinted in humans and mice, yet only a few have been associated with a human disease.1 To date, several recognizable developmental syndromes due to abnormalities within specific domains have been described. The most studied genomic-imprinting defects in humans are Prader-Willi (PWS [MIM 176270]) and Angelman (AS [MIM 105830]) syndromes, which are due to paternal and maternal imprinting disorders of genes on chromosome 15q11-q13, and Beckwith-Wiedemann (BWS [MIM 130650]) and Russell-Silver (SRS [MIM 180860]) syndromes, which are associated with genomic imprinting of genes in two adjacent domains on chromosome 11p15.5. Other human imprinting disorders include transient neonatal diabetes (at 6q24) (MIM 601410), maternal and paternal chromosome 14 uniparental-disomy syndromes (at 14q32), and pseudohypoparathyroidism type 1b (PHP1B [MIM 603233]) (at 20q13.2).1 We now describe a human syndrome of mental retardation, hypotonia, and characteristic dysmorphism caused by a mutation in a genomically imprinted gene on chromosome 8.
Material and Methods
The study was approved by the Institutional Review Board of Soroka Medical Center.
Genome-wide linkage analysis with the 400 microsatellite markers (ABI PRISM Linkage Mapping Set MD10, Applied Biosystems) and fine mapping were done as previously described.2 Statistical analysis was done with an autosomal-dominant disease model assuming imprinting with paternal silencing. Extended two-point LOD scores were calculated on the entire kindred with SUPERLINK.3
Fine mapping of the disease-associated genomic locus was performed with 18 markers designed with Tandem Repeats Finder.4 Coding sequences of potassium channel, subfamily K, member 9 (KCNK9 [MIM 605874]) were PCR amplified from genomic DNA and sequenced. Analysis of the mutation in the entire family and controls was performed by restriction analysis of KCNK9 exon 2 with the restriction enzyme NlaIV on PCR products (the 770G→A mutation abolishes a recognition site). PCR primers used are available upon request.
Electrophysiology
Full-length cDNAs of human KCNK9 (TASK3; K2P9.1; NT_008046; NP_057685.1) and human KCNK3 (TASK1; K2P3.1; NP_002237; MIM 603220) channels were cloned into pMAX and pRAT plasmids, respectively; both plasmids include a T7 RNA polymerase promoter to enable cRNA synthesis, as well as 3′-UTR and 5′-UTR sequences of the Xenopus laevis β-actin gene to ensure efficient expression in Xenopus oocytes. The mutant was generated with the Quickchange site-directed mutagenesis technique (Stratagene, La Jolla, CA). The mutation was confirmed by DNA sequencing. cRNA was transcribed in vitro with T7 polymerase and the AmpliCap High Yield Message Maker (Epicenter, Madison, WI) kit.
Xenopus laevis oocytes were isolated and injected 7 ng of cRNA in 20 nl of RNase-free water. Whole-cell currents were measured 3–4 days after injection by the two-electrode voltage-clamp technique (GeneClamp 500B, Axon Instruments, Union City, CA). Data were sampled at 2 kHz and filtered at 0.5 kHz with Clampex 9.0 software (Axon Instruments, Union City, CA). The pipettes contained 3 M KCl, and the bath solution contained 4 mM KCl, 96 mM NaCl, 1 mM MgCl2, 0.3 mM CaCl2, and 5 mM HEPES (pH 7.4 [adjusted with NaOH]).
Results
Clinical Phenotype of the Syndrome
An Israeli Arab kindred presented with an apparently maternally transmitted (imprinted with paternal silencing) syndrome (Figure 1A). All affected individuals had moderate to severe mental retardation and were hyperactive. Severe feeding difficulties at infancy (requiring tube feeding in most) were followed in all patients by dysphagia of solid foods until near puberty. Generalized hypotonia at an early age was followed by weakness of proximal muscles and of the supra- and infrascapular and trapezius muscles later on. Coordination, tendon reflexes, deep and superficial sensation, and vibration were all within normal limits. Babinski sign was negative. Hearing, vision, and ophthalmologic examination were normal. As depicted in Figures 1B–1D, similar dysmorphic features were found in affected individuals, with most elements being more prominent at younger ages: The face was elongated with narrow bitemporal diameter, mild atrophy of temporalis and masseter muscles, and reduced facial movements. The eyebrows were flared, bushy, and arched upward, with downturned eyelids and sparse eyelashes in the inner third of the lower eyelids, and with congested conjunctivae in most patients. Ears were mildly protruding with a very prominent fold of the crux of the helix and a prominent antihelical fold. The nasal bridge was high and narrow with a broad nasal tip. The colummella was normal, but the philtrum was extremely short, broad, and thick in all patients. The maxillary and premaxillary regions were prominent, with hypotonia of the mandible and micrognathia—leading (in combination with the short philtrum) to an open mouth. The lips were thick, with downturned upper lips (“fish mouth”) and a lower lip that was shorter than the upper lip. Most patients had a narrow, high-arched palate with full or submucous cleft and dysphonic speech. Large and protruding incisors were seen in younger patients (Figure 1D). All patients had narrow, elongated necks, trunks, and feet. In some infants, mild joint contractures of the hips, elbows, phalanx, and feet were present and became prominent with age. A pilonidal dimple or sinus was evident in most patients. Extensive skeletal X-ray studies were normal. Thorough metabolic work-up, karyotypes, blood levels of muscle-associated enzymes, and EEG and brain CT studies were normal (mild periventricular leukomalacia was present in a single child). Muscle biopsies in two patients were compatible with spinal muscular atrophy (SMA [MIM 253300]), with normal molecular SMA tests. Mitochondria appeared normal on EM examination, and normal activity of the mitochondrial respiratory-chain enzymes was demonstrated in muscle.
Figure 1.
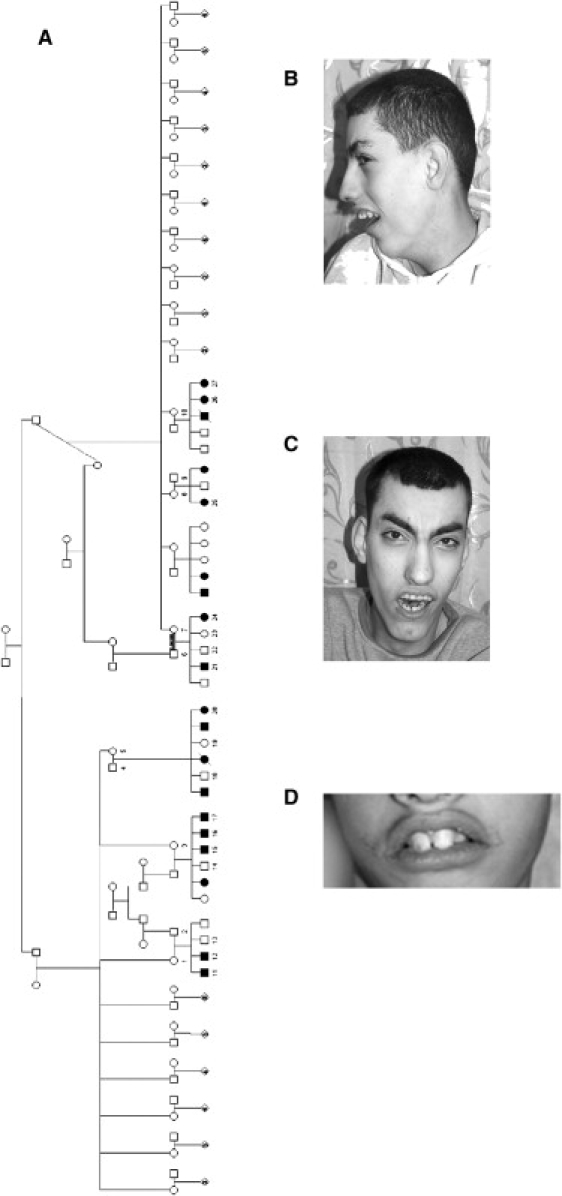
Clinical Features
(A) Pedigree of the affected Israeli Arab family. The numbers denote individuals whose DNA samples were analyzed. It can be seen that the disease is maternally transmitted (paternally imprinted).
(B and C) Dysmorphic features of affected siblings.
(D) Prominent incisor teeth of young affected individual.
Linkage-Analysis Studies
Genome-wide linkage analysis of seven affected individuals, one healthy individual, and four obligatory-transmitting mothers was done with 400 fluorescent end-labeled polymorphic markers as previously described.4 Assuming the disease is caused by a mutation in a paternally imprinted gene, we looked for a genomic locus with a haplotype shared by the (obligatory transmitting) healthy mothers and their affected children, but not their healthy children. Studies using polymorphic markers excluded noninformative regions as well as six of seven genomic loci identified (data not shown). The remaining single locus spanned ∼37.91 cM (corresponding to 26.34 Mb) on chromosome 8q24, between marker D8S514 and the telomeric end of 8q. Fine mapping of the locus was done, testing DNA samples of the 27 available family members (11 affected and 16 nonaffected) with additional polymorphic markers in the identified interval (details in Material and Methods). One affected child (26) in the family presented with a crossing-over event at ATCT-1389, setting the centromeric border of the common interval at ATCT-1389, delineating a ∼7.27 Mb locus (harboring ∼153 known or predicted genes) common to all affected individuals and their mothers in the family (Figure 2A). Two-point analysis with SUPERLINK3 demonstrated a LOD score above 5 at θ = 0 for two separate markers within the haplotype (Figure 2B).
Figure 2.
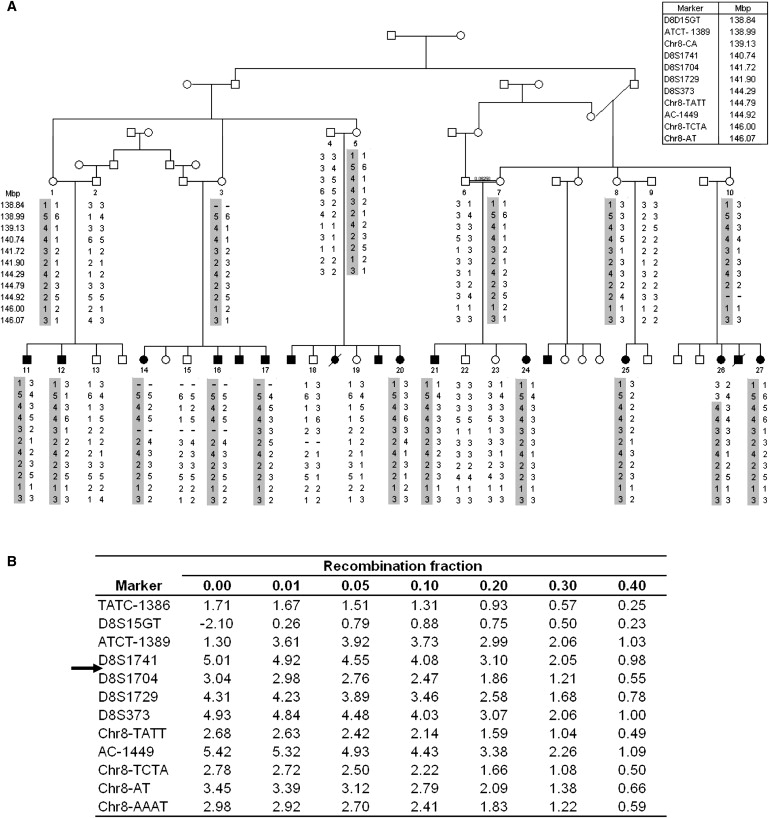
Mapping the KCNK9 Locus
(A) Partial pedigree and segregation analysis of the affected family. The numbers denote individuals whose DNA samples were analyzed. Haplotypes are represented as columns of numbers; the disease-associated haplotype is shaded. Physical distances between the markers are shown.
(B) Two-point LOD-score analysis for the polymorphic markers at the KCNK9 locus. The arrow indicates the gene location.
Determination of the Specific Gene Mutation
The next step was to identify and sequence candidate genes within the locus that are known to be imprinted in humans or mice. Using specific DNA sequences characteristic of known imprinted genes to screen for previously unreported potentially imprinted genes, Luedi et al. recently identified two human imprinted genes on chromosome 8,5 one of which lies within our identified locus: KCNK9 (also known as K2P9.1 and TASK-3). KCNK9 has been shown to be imprinted with paternal silencing in humans and mice and to be exclusively expressed from the maternal allele in the brain.6 As shown in Figure 3, sequence analysis of KCNK9 (GenBank accession number NM_016601) in affected individuals revealed a 770G→A mutation in exon 2, replacing glycine at position 236 by arginine (G236R). The mutation abolishes a recognition site of the restriction enzyme NlaIV, allowing easy analysis of the entire family and controls. Analysis of all 27 DNA samples of the kindred was compatible with the mutation being associated with the disease phenotype, implying dominant inheritance with paternal imprinting. The mutation was not found in any of 548 chromosomes from ethnically matched controls.
Figure 3.
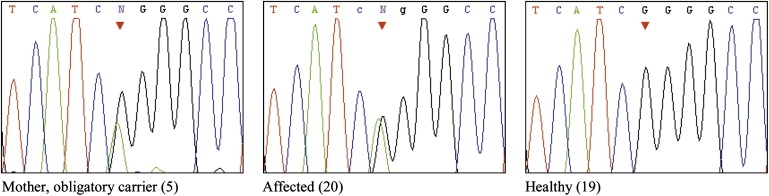
Analysis of the 770G→A Mutation in Exon 2 of KCNK9
Sequence analysis of a healthy obligatory-carrier mother and her affected and healthy offspring.
Determination of the Physiological Effect of the K2P9.1 Mutation
The biophysical implications of the K2P9.1-G236R mutation were studied with the two-electrode voltage-clamp technique (Figure 4). On the basis of the published structure of the KcsA potassium channel,7 the small G236 residue (homologous to KcsA-G104) is expected to lie within the ion-conduction pathway of the channel (Figure 4E). Hence, the introduction of the positively charged arginine residue at this position is expected to interfere with potassium conductance by forming both physical and electrostatic barriers. Wild-type and mutant channel cRNAs were transcribed in vitro and injected to Xenopus laevis oocytes, and whole-cell currents were measured 3–4 days subsequent to injection. Unlike wild-type channels, K2P9.1-G236R channels produced no measurable currents (Figure 4). Moreover, coexpression of mutant and wild-type channels resulted in a ∼4-fold decrease in wild-type currents (Figures 4B and 4C). Furthermore, a similar dominant-negative effect was observed when the K2P9.1-G236R mutant was coexpressed with human K2P3.1 (also known as TASK-1, KCNK3) (Figure 4C), a closely related K2P channel, known to form heterodimers with K2P9.1 in expression systems as well as in native tissues.8 Thus, the loss of current seen in the heterologous system is likely to occur also in vivo and cause the disease. The loss of current could be either due to expression of nonfunctional channels or due to failure to traffic to the membrane.
Figure 4.
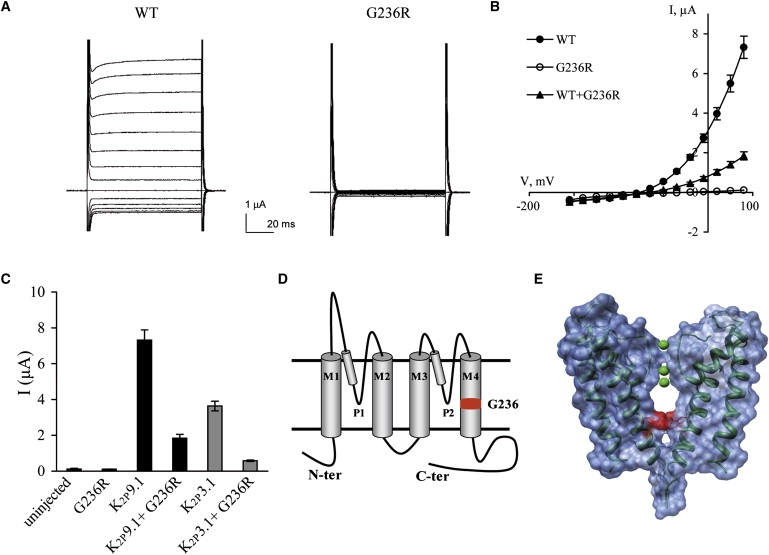
The K2P9.1-G236R Mutant Does Not Produce Functional Channels
Macroscopic currents measured by two-electrode voltage clamp.
(A) Raw current traces for representative oocytes expressing WT-K2P9.1 and K2P9.1-G236R channels and bathed in 5 mM potassium solution. Oocyte membrane potential was held at −80 mV and pulsed from −155 to +40 mV in 15 mV voltage steps for 70 ms with 1 s interpulse intervals.
(B) K2P9.1 steady-state current-voltage relationships for WT-K2P9.1 (full circles), K2P9.1-G236R mutated channels (open circles), and coexpression of WT-K2P9.1 + K2P9.1-G236R (triangles) (mean ± SEM, n = 6-9 cells).
(C) Currents at +40 mV of oocytes injected with equal cRNA amounts of the different channels, as indicated (mean ± standard error of the mean, n = 6–9 cells).
(D) Predicted membrane topology of a K2P9.1 subunit indicating the two P domains and four transmembrane segments (M1–4). The predicted position of G236 is highlighted in red.
(E) Surface representation of two opposing KcsA subunits.7 G104 was replaced by an arginine residue (highlighted in red), as in the K2P9.1-G236 mutant. Ribbon representation of the carbon backbone is in dark green, and potassium ions are in light green. Molecular graphics images were produced with the UCSF Chimera (an extensible molecular-modeling system) package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
Discussion
Genomic imprinting has been associated with very few human phenotypes, caused by defects in specific imprinting domains on human autosomes. Each domain is under the regulatory control of an “imprinting center” that harbors a differentially methylated region. A number of molecular mechanisms result in differential silencing of some genes within these domains, making them vulnerable to naturally occurring genetic and epigenetic aberrations.1
We clearly demonstrate that the disease is caused by a mutation in the maternal copy of KCNK9 and that the mutation fully abolishes the activity of this potassium channel both when acting as a homodimer and when functioning as a heterodimer. KCNK9 was recently shown to be imprinted with paternal silencing, well in line with the form of heredity demonstrated in the kindred presented here. KCNK9 encodes K2P9.1 potassium channel, a member of the two-pore domain (K2P) family.9 K2P potassium channels comprise the newest branch of the potassium-channel superfamily serving to carry leak or “background” currents that are mostly time- and voltage-independent. K2P channels operate as dimers with four transmembrane segments and two pore-forming domains in each subunit (Figure 4D). Leak currents shape the duration, frequency, and amplitude of action potentials and regulate cell responsiveness and excitability8 by modulating membrane resting potential and resistance. The 15 human members of the K2P channel family have a wide and diverse anatomical distribution pattern and are involved in numerous physiological processes. To our knowledge, this is the first report of a disease-causing mutation in a member of this gene family. Mammalian K2P9.1 channels are involved in various physiological functions, including adjustment of neuronal excitability10–13 and regulation of aldosterone secretion.14 Human K2P9.1 channels are expressed primarily in the central nervous system and particularly in the cerebellum.15 K2P9.1 activity is modulated by extracellular pH, neurotransmitters, volatile anesthetics, and more.9
KCNK9 resides within 6.2 Mb of the marker D8S256, linked with bipolar disorder,16 and has been suggested to be a candidate for idiopathic-absence epilepsies.17 K2P9.1 channels were suggested to play an important role in K+-dependent apoptosis of cerebellar granule neurons in culture18 and in maturation of neurons in the cerebellum during neuronal development.19 Indeed, through the use of knockout mice, K2P9.1 was shown to modulate action-potential firing in cerebellar granule neurons,20 circadian rhythms, cognitive functions, and specific pharmacological effects.20,21 Moreover, we expect that the physiological impact of a dominant loss-of-function mutation might exceed that of a complete null mutation, because nonfunctional K2P9.1 protein might affect the function of associated proteins such as K2P3.1 (Figure 4C) and hyperpolarization-activated cyclic-nucleotide-gated (HCN2) channels.22 In cerebellar granule neurons, the standing outward K+ current (IKso) corresponds to either K2P9.1 homomeric or K2P9.1/K2P3.1 heteromeric channels. Because K2P3.1 knockout mice exhibited compromised motor performance consistent with altered cerebellar function,10 a mutated K2P9.1 channel is expected to further diminish IKso and perturb neuronal activity. In addition, membrane resting potential of thalamocortical relay neurons is shaped by the interaction of K2P9.1 and HCN2 channels.22 HCN channels that conduct Ih, a hyperpolarization-triggered cationic current that contributes to the maintenance of neuronal membrane potential, were recently proposed to take part in febrile seizures and epilepsy.23 We thus hypothesize that damaging the possible K2P9.1-HCN2 association might also alter the Ih current and potentially contribute to the phenotype described here. Moreover, we expect K2P9.1 to have a more pivotal role in human cerebellum than in mice because of its high relative expression levels.15 Taken together, the K2P9.1 loss of function during brain development specifically disrupts the efficiency of the cerebro-cerebellar pathways, resulting in cognitive deficits and deterioration in muscle strength and function.
Imprinting of both the human and mouse orthologs of this gene does not seem to be regulated by short-range cis-acting differential methylation at their CpG-island promoter.6 In the mouse, Kcnk9 is located on chromosome 15, 260 kb downstream of the imprinted Peg13 gene, a non-protein-coding RNA gene of unknown function. Similar to Kcnk9, Peg13 is expressed predominantly in brain, but, in contrast, the maternal allele is silenced and marked by DNA methylation in somatic tissues.24 Nearly full methylation of the Peg13 differentially methylated region (DMR) was shown in mature oocytes with near complete absence of methylation in sperm DNA. It was thus hypothesized that the nearby murine Peg13-DMR acts as a long-range acting imprinting control element controlling the imprinting of the distal murine Kcnk9 gene. To date, there is no known Peg13 or Peg13-DMR human homolog,6,24,25 and the mechanism of imprinting of human KCNK9 remains to be elucidated.
To our knowledge, the syndrome we describe is the first human syndrome caused by a defect in a specific imprinted gene on chromosome 8. Imprinting syndromes can evolve from a mutation or deletion of the “active” copy of an imprinted gene, but they can also be caused by defects in an adjacent imprinting center or by uniparental disomy of the “inactive” copy of the gene. A single previous report suggested possible association of maternal uniprental disomy of chromosome 8 with early-onset ileal carcinoid tumor.26 This finding does not conflict with our data, and it remains to be seen whether this single case is an incidental finding or whether it reflects a true phenotypic effect of unimaternal disomy of chromosome 8. Of more relevance to our findings is a single case report27 demonstrating that paternal uniparental disomy of chromosome 8 found in lymphocytes is not associated with a human clinical phenotype. This latter report in conjunction with our data can be explained in several ways. One option is that the previously reported case of unipaternal disomy of chromosome 8 was a mosaic. Thus, for instance, it might reflect a trisomy rescue that occurred differently in various tissues, resulting in unipaternal disomy in blood cells, yet in one maternal and one parental copy of chromosome 8 in all or most cells of neuronal tissues. However, because there is no evidence of any moasicism in the blood of the individual reported, the above suggestion is possible yet not very likely. Alternatively and perhaps more likely, the previously reported case of paternal uniparental disomy is indeed such in all tissues. In such a case, it might well be that complete null mutation and loss of function of human K2P9.1 is compensated for by other molecules and has no phenotypic effect. This would then suggest that the missense mutation we describe might exert a dominant-negative effect on other molecules, as suggested by the dominant-negative effect that was observed when the K2P9.1-G236R mutant was coexpressed with human K2P3.1 (Figure 4C). If this is the case, this would be to the best of our knowledge the first report of a mutation in a genomically imprinted gene causing a human phenotype through a dominant-negative effect on other (nonimprinted) molecules. If this interpretation is true, null mutations (or deletions) of KCNK9 would have no phenotypic effect in humans. Future identification and characterization of more cases of human unipaternal disomy of chromosome 8 will allow better understanding of the mechanism through which the phenotype we describe evolves and will determine whether unipaternal disomy of human chromsome 8 (or null mutations or deletions of KCNK9) are of clinical significance.
Supplemental Data
Supplemental Data include a genome scan and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSF Chimera, http://www.cgl.ucsf.edu/chimera
Acknowledgments
We deeply thank the Morris Kahn Family Foundation for making this study possible. This work was also supported by grants from the Israel Science Foundation (431/03) and the Zlotowski Center for Neuroscience to N.Z. We thank Professor Hanna Mandel for the metabolic work-up; Professor Shapira for the analysis of muscle biopsies; Dr. Dan Reich and the team at the Department of Neonatology, Tal Shoshani, for technical assistance; Professor Juan (Moshe) Chemke, Professor Dvora Abeliovich, Dr. Morkos Siman, and Dr. Ahmed Haj-Daud for clinical insights; the families and patients; and the devoted family physicians and pediatricians responsible for the routine medical care of the family members in their communities.
References
- 1.Temple I.K. Imprinting in human disease with special reference to transient neonatal diabetes and Beckwith-Wiedemann syndrome. Endocr. Dev. 2007;12:113–123. doi: 10.1159/000109638. [DOI] [PubMed] [Google Scholar]
- 2.Harel T., Goldberg Y., Shalev S.A., Chervinski I., Ofir R., Birk O.S. Limb-girdle muscular dystrophy 2I: Phenotypic variability within a large consanguineous Bedouin family associated with a novel FKRP mutation. Eur. J. Hum. Genet. 2004;12:38–43. doi: 10.1038/sj.ejhg.5201087. [DOI] [PubMed] [Google Scholar]
- 3.Fishelson M., Geiger D. Exact genetic linkage computations for general pedigrees. Bioinformatics. 2002;18:S189–S198. doi: 10.1093/bioinformatics/18.suppl_1.s189. [DOI] [PubMed] [Google Scholar]
- 4.Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luedi P.P., Dietrich F.S., Weidman J.R., Bosko J.M., Jirtle R.L., Hartemink A.J. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007;17:1723–1730. doi: 10.1101/gr.6584707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruf N., Bähring S., Galetzka D., Pliushch G., Luft F.C., Nürnberg P., Haaf T., Kelsey G., Zechner U. Sequence-based bioinformatic prediction and QUASEP identify genomic imprinting of the KCNK9 potassium channel gene in mouse and human. Hum. Mol. Genet. 2007;16:2591–2599. doi: 10.1093/hmg/ddm216. [DOI] [PubMed] [Google Scholar]
- 7.Doyle D.A., Morais Cabral J., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., MacKinnon R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 8.Goldstein S.A., Bockenhauer D., O'Kelly I., Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat. Rev. Neurosci. 2001;2:175–184. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- 9.Kim Y., Bang H., Kim D. TASK-3, a new member of the tandem pore K+ channel family. J. Biol. Chem. 2000;275:9340–9347. doi: 10.1074/jbc.275.13.9340. [DOI] [PubMed] [Google Scholar]
- 10.Aller M.I., Veale E.L., Linden A.M., Sandu C., Schwaninger M., Evans L.J., Korpi E.R., Mathie A., Wisden W., Brickley S.G. Modifying the subunit composition of TASK channels alters the modulation of a leak conductance in cerebellar granule neurons. J. Neurosci. 2005;25:11455–11467. doi: 10.1523/JNEUROSCI.3153-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang D., Han J., Talley E.M., Bayliss D.A., Kim D. Functional expression of TASK-1/TASK-3 heteromers in cerebellar granule cells. J. Physiol. 2004;554:64–77. doi: 10.1113/jphysiol.2003.054387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berg A.P., Talley E.M., Manger J.P., Bayliss D.A. Motoneurons express heteromeric TWIK-related acid-sensitive K+ (TASK) channels containing TASK-1 (KCNK3) and TASK-3 (KCNK9) subunits. J. Neurosci. 2004;24:6693–6702. doi: 10.1523/JNEUROSCI.1408-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meuth S.G., Budde T., Kanyshkova T., Broicher T., Munsch T., Pape H.C. Contribution of TWIK-related acid-sensitive K+ channel 1 (TASK1) and TASK3 channels to the control of activity modes in thalamocortical neurons. J. Neurosci. 2003;23:6460–6469. doi: 10.1523/JNEUROSCI.23-16-06460.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czirják G., Enyedi P. TASK-3 dominates the background potassium conductance in rat adrenal glomerulosa cells. Mol. Endocrinol. 2002;16:621–629. doi: 10.1210/mend.16.3.0788. [DOI] [PubMed] [Google Scholar]
- 15.Rusznák Z., Pocsai K., Kovács I., Pór A., Pál B., Bíró T., Szücs G. Differential distribution of TASK-1, TASK-2 and TASK-3 immunoreactivities in the rat and human cerebellum. Cell. Mol. Life Sci. 2004;61:1532–1542. doi: 10.1007/s00018-004-4082-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McInnis M.G., Lan T.H., Willour V.L., McMahon F.J., Simpson S.G., Addington A.M., MacKinnon D.F., Potash J.B., Mahoney A.T., Chellis J. Genome-wide scan of bipolar disorder in 65 pedigrees: Supportive evidence for linkage at 8q24, 18q22, 4q32, 2p12, and 13q12. Mol. Psychiatry. 2003;8:288–298. doi: 10.1038/sj.mp.4001277. [DOI] [PubMed] [Google Scholar]
- 17.Zara F., Bianchi A., Avanzini G., Di Donato S., Castellotti B., Patel P.I., Pandolfo M. Mapping of genes predisposing to idiopathic generalized epilepsy. Hum. Mol. Genet. 1995;4:1201–1207. doi: 10.1093/hmg/4.7.1201. [DOI] [PubMed] [Google Scholar]
- 18.Lauritzen I., Zanzouri M., Honoré E., Duprat F., Ehrengruber M.U., Lazdunski M., Patel A.J. K+-dependent cerebellar granule neuron apoptosis. Role of TASK leak K+ channels. J. Biol. Chem. 2003;278:32068–32076. doi: 10.1074/jbc.M302631200. [DOI] [PubMed] [Google Scholar]
- 19.Zanzouri M., Lauritzen I., Duprat F., Mazzuca M., Lesage F., Lazdunski M., Patel A. Membrane potential-regulated transcription of the resting K+ conductance TASK-3 via the calcineurin pathway. J. Biol. Chem. 2006;281:28910–28918. doi: 10.1074/jbc.M606092200. [DOI] [PubMed] [Google Scholar]
- 20.Brickley S.G., Aller M.I., Sandu C., Veale E.L., Alder F.G., Sambi H., Mathie A., Wisden W. TASK-3 two-pore domain potassium channels enable sustained high-frequency firing in cerebellar granule neurons. J. Neurosci. 2007;27:9329–9340. doi: 10.1523/JNEUROSCI.1427-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linden A.M., Sandu C., Aller M.I., Vekovischeva O.Y., Rosenberg P.H., Wisden W., Korpi E.R. TASK-3 knockout mice exhibit exaggerated nocturnal activity, impairments in cognitive functions, and reduced sensitivity to inhalation anesthetics. J. Pharmacol. Exp. Ther. 2007;323:924–934. doi: 10.1124/jpet.107.129544. [DOI] [PubMed] [Google Scholar]
- 22.Meuth S.G., Kanyshkova T., Meuth P., Landgraf P., Munsch T., Ludwig A., Hofmann F., Pape H.C., Budde T. Membrane resting potential of thalamocortical relay neurons is shaped by the interaction among TASK3 and HCN2 channels. J. Neurophysiol. 2006;96:1517–1529. doi: 10.1152/jn.01212.2005. [DOI] [PubMed] [Google Scholar]
- 23.Dubé C.M., Brewster A.L., Richichi C., Zha Q., Baram T.Z. Fever, febrile seizures and epilepsy. Trends Neurosci. 2007;30:490–496. doi: 10.1016/j.tins.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith R.J., Dean W., Konfortova G., Kelsey G. Identification of novel imprinted genes in a genome-wide screen for maternal methylation. Genome Res. 2003;13:558–569. doi: 10.1101/gr.781503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies W., Smith R.J., Kelsey G., Wilkinson L.S. Expression patterns of the novel imprinted genes Nap1l5 and Peg13 and their non-imprinted host genes in the adult mouse brain. Gene Expr. Patterns. 2004;4:741–747. doi: 10.1016/j.modgep.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 26.Karanjawala Z.E., Kääriäinen H., Ghosh S., Tannenbaum J., Martin C., Ally D., Tuomilehto J., Valle T., Collins F.S. Complete maternal isodisomy of chromosome 8 in an individual with an early-onset ileal carcinoid tumor. Am. J. Med. Genet. 2000;93:207–210. doi: 10.1002/1096-8628(20000731)93:3<207::aid-ajmg9>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 27.Benlian P., Foubert L., Gagné E., Bernard L., De Gennes J.L., Langlois S., Robinson W., Hayden M. Complete paternal isodisomy for chromosome 8 unmasked by lipoprotein lipase deficiency. Am. J. Hum. Genet. 1996;59:431–436. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.