

Abstract
Joubert syndrome (JS) and related disorders are a group of autosomal-recessive conditions sharing the “molar tooth sign” on axial brain MRI, together with cerebellar vermis hypoplasia, ataxia, and psychomotor delay. JS is suggested to be a disorder of cilia function and is part of a spectrum of disorders involving retinal, renal, digital, oral, hepatic, and cerebral organs. We identified mutations in ARL13B in two families with the classical form of JS. ARL13B belongs to the Ras GTPase family, and in other species is required for ciliogenesis, body axis formation, and renal function. The encoded Arl13b protein was expressed in developing murine cerebellum and localized to the cilia in primary neurons. Overexpression of human wild-type but not patient mutant ARL13B rescued the Arl13b scorpion zebrafish mutant. Thus, ARL13B has an evolutionarily conserved role mediating cilia function in multiple organs.
Introduction
Joubert syndrome (JS [MIM 213300]) is characterized by congenital cerebellar ataxia, hypotonia, oculomotor apraxia, and mental retardation. It is the most common inherited cerebellar malformation syndrome and is part of the autosomal-recessive cerebellar ataxia group of disorders.1 The neuroradiological hallmark in JS is a peculiar malformation of the midbrain-hindbrain junction known as the “molar tooth sign” (MTS), consisting of cerebellar vermis hypoplasia or dysplasia, thick and horizontally oriented superior cerebellar peduncles, and an abnormally deep interpeduncular fossa.2 The MTS has subsequently been reported in a group of syndromes termed Joubert syndrome and related disorders (JSRD). These include (1) classical JS (MIM 213300), (2) JS plus Leber congenital amaurosis (LCA, i.e., congenital retinal blindness [MIM 204000]),3 (3) JS plus nephronophthisis (NPHP [MIM 256700]), (4) JS plus LCA plus NPHP (also known as cerebello-oculo-renal syndrome [CORS (MIM 608091)]),4 (5) cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, ocular coloboma, hepatic fibrosis (COACH [MIM 216360]) syndrome,5 and (6) oral-facial-digital syndrome type VI6,7 (MIM 277170), with occurrence of other features such as encephalocele and polydactyly variably found within each subgroup. Many of these disorders have previously been linked to genes encoding cilia-localized proteins, so JSRD was considered a candidate ciliopathy disorder. The recent discoveries that JSRD can be due to mutations in CEP2908,9 (encoding a known centrosomal/cilia protein), as well as mutations in NPHP110,11 (previously implicated in the NPHP ciliopathy), RPGRIP1L12–14 (encoding a binding partner of proteins implicated in the ciliopathy retinitis pigmentosa), and TMEM67 (a.k.a. MKS3)15 (previously implicated in the Meckel syndrome ciliopathy) have further strengthened the hypothesis that JSRD is due to defective ciliary function.
Here we report the identification of the JBTS8 locus and mutations in the ARL13B gene, encoding ADP-ribosylation factor-like protein 13B. ARL13B is a small GTPase belonging to the class of Arf/Arl family within the Ras superfamily of small GTPases involved in diverse cellular functions. ARL13B is thus the first gene encoding an enzymatically active protein implicated in JS, suggesting that its study may shed light on the essential signaling pathways. Furthermore, an Arl13b null mutation was recently identified as underlying the lethal hennin mouse mutant that displays a Sonic Hedgehog-like phenotype, as well as being identified in the zebrafish scorpion mutant that displays renal cysts and curved tail, both tied to impaired cilia function. The identification of ARL13B mutations in humans with JS, together with the previous work in mouse and zebrafish, indicates an extremely broad spectrum of phenotypes across species all related to defective cilia function. The results also suggest a role for cilia-mediated Sonic Hedgehog signaling in cerebellar development and the pathogenesis of JS in humans.
Material and Methods
Genome Mapping
Family MTI-001 was recruited according to an approved Human Subjects Protocol at the University of Leeds and UCSD. DNA was extracted from peripheral blood leukocytes by salt extraction, genotyped with the Illumina Linkage IVb mapping panel,16 and analyzed with easyLINKAGE-Plus17 software, which runs Allegro version 1.2c in a PC Windows interface to calculate multipoint LOD scores. Parameters were set to autosomal recessive with full penetrance, and disease allele frequency of 0.001. Genomic regions with LOD scores over 2 were considered as candidate intervals. Linkage simulations were performed with Allegro 1.2c under the same parameters, with 500 markers at average 0.64 cM intervals, codominant allele frequencies, and parametric calculations.17 Additional subjects were enrolled according to approved protocols at University of Chicago, University of Michigan, University of Leeds, and Necker Hospital in Paris. Control samples were from healthy US adult subjects from a hypertension study, from healthy Arab controls from the United Arab Emirates, and healthy Pakistani controls from Leeds, UK.
Mutation Screening
We performed direct bidirectional sequencing of the ten coding exons and splice junction sites of ARL13B via BigDye Terminator cycle sequencing (Applied Biosystems). We screened 124 patients ascertained for the presence of nephronophthisis with or without features of Joubert syndrome (courtesy of Friedhelm Hildebrandt, U. Michigan) and 32 patients ascertained for the presence of Meckel syndrome with or without features of Joubert syndrome (courtesy of Tania Attie-Bitach, Necker Hospital, France) via bidirectional sequencing. We screened 44 patients ascertained for the presence of Meckel syndrome with or without features of Joubert syndrome (courtesy of Colin Johnson, U. Leeds) by LightScanner detection (Idaho Technologies), with subsequent bidirectional sequencing of aberrant bands. All patient samples were ascertained and managed according to approved human subjects protocols.
Candidate Genes and GenBank Accession Numbers
Human ARL6 has two GenBank numbers (NM_032146.1 and NM_177976.3) encoding the same protein, but the first encodes a longer transcript. Human ARL13B has two GenBank numbers (NM_144996.1 and NM_144996.2) differing in that the second entry is missing coding exons 2–3. Zebrafish arl13B has GenBank number NM_173272. Mouse Arl13b has GenBank number NM_026577.2.
Bioinformatics
Predicted folding and intermolecular interactions were predicted with Swiss-Model with crystallized ARL2-GTP as a template. The resulting ARL13B structure was manipulated and rendered with PyMOL software by importing amino acid sequence for wild-type and mutant and comparing the predicted hydrogen bonds.
GTP Binding Assay
Human wild-type and mutant ARL13B cDNA were cloned separately into the pGEX 6P1 vector (GE Lifesciences), and GST-tagged purified protein was isolated according to the manufacturer's recommendations. Protein was retrieved from the soluble phase, integrity verified by SDS-PAGE analysis followed by Coomassie staining, and analyzed by circular dichroism to validate that wild-type and mutant proteins had similar secondary structure as described. GTP binding was performed with a filter-based assay as described,18 with denatured (boiled for 15 min) Arl13b protein as negative control and Rac as a positive control, and analyzed with SigmaPlot Ver 10.0, with one-way ANOVA for statistical analysis.
Zebrafish Characterization
The arl13bsco/+ line was obtained from the Zebrafish International Resource Center (ZIRC) under an approved animal protocol at UCSD. One-cell embryos from arl13bsco/+ matings were injected with 50 pg in vitro transcribed capped open reading frame RNA (Ambion Message Kit) of human wild-type or mutant transcript. After 72 hr, embryos were phenotyped by an investigator that was blinded to the genotype, and then all embryos were subject to genotyping for correlation.
Histology
Mouse tissue was fixed with 4% paraformaldehyde, then processed for 15 μm cryostat sections and immunostained as described.19 We used two different rabbit polyclonal antibodies for Arl13b, one that was recently created against a bacterially expressed GST fused to amino acids 208–428 of murine Arl13B,19 and one created against an internal polypeptide, which behaved similarly, so only the first was used for further experimentation. Neither antibody showed reactivity with the cilia in the hennin mutant19 (data not shown). Antibodies were used at the following concentrations: Arl13b, 1:2000; acetylated tubulin (Zymed 32-2700), 1:500; calbindin (Swant), 1:600. Cerebellar granule neurons were isolated with a cell-density gradient as described,20,21 fixed, and immunostained at various time points after plating.
Results
Identification of the JBTS8 Locus at the Centromeric Region of Chromosome 3
In our series, we excluded linkage to known loci in 27 consanguineous JSRD families, supporting evidence for further genetic heterogeneity. One of the largest families (MTI-001, Figure 1A), which is of Pakistani origin and the first to be recruited in this study, displayed a proven MTS on brain imaging (Table S1 available online), provided a maximum expected LOD score of 3.0, and did not display any mutations in any of the five known JSRD genes (NPHP1, AHI1, CEP290, MKS3, RPGRIP1L). We performed a 0.64 cM resolution genome-wide screen by the Illumina Whole Genome SNP Linkage IVb panel, which defined an interval of 107–112 cM (80.0–103.3 MB) on chromosome 3p12.3-q12.3, with peak multipoint LOD score of 3.0 termed JBTS8 (Figure 1B).
Figure 1.
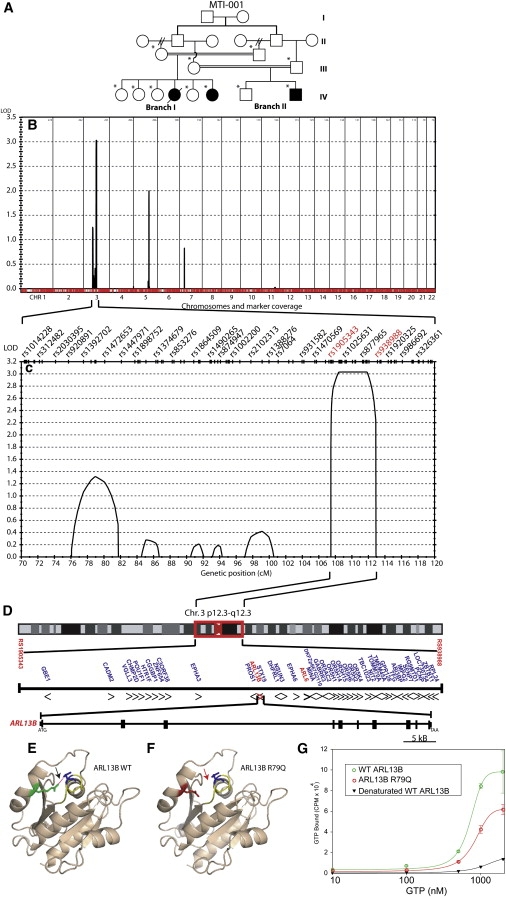
Mapping JSRD Family MTI-001 Identified the JBTS8 Locus Containing the ARL13B Gene, Mutation of which Produces Altered GTP Binding
(A) MTI-001 consists of a double first cousin marriage where each of two brothers were married twice (generation II, double hash refers to divorce); one had two half-brothers and one had two half-sisters, and each of those subsequently married his/her respective cousin (generation III). These two couples each produced an affected offspring (filled symbols in generation IV) as well as healthy siblings. One affected girl from branch 1 died from related medical complications (single hash line).
(B) Whole-genome analysis of linkage results with chromosomal position (x axis) and multipoint LOD scores (y axis) showing a significant LOD score (>3) on chr. 3. No other peaks reached this threshold for significance.
(C) Expanded view of the candidate interval showing that the linkage signal is composed of one major peak defined by recombinations at rs1005343 and rs938988 SNP markers (red), thus defining a minimal candidate interval from 107 to 112 cM.
(D) Expanded view of the candidate interval on chr. 3p12.3-q12.3, encompassing the centromere, containing 41 candidate genes, including ARL6 and ARL13B (red). Transcriptional direction indicated by arrowheads. Genomic structure of ARL13B consists of 10 coding exons extending over about 70 KB of genomic sequence.
(E) Homology modeling of wild-type ARL13B showing R (green) residue and predicted hydrogen bond (red dashed line) with D (blue) residue that engages in GTP binding.
(F) The p.R79Q mutation is a basic polar to neutral polar substitution (red) and is predicted to disrupt this hydrogen bond (absent dashes indicated by red arrow).
(G) GTP binding assay via recombinant wild-type ARL13B (green) or p.R79Q mutant (red) showing altered kinetics and binding capacity. Experiment performed in triplicate. Error bars represent SEM. Negative control was denatured wild-type ARL13B (black). Statistical differences observed between wild-type and mutant at 500, 1000, and 2000 nM GTP concentration (∗p < 0.05, one-way ANOVA).
Patients from MTI-001 Have a Homozygous p.R79Q Change in the ARL13B Gene
This interval encompassed the centromere, with 41 genes, and markers rs1905343 and rs938988 formed the p- and q-arm breakpoints, respectively (Figure 1C). Four genes within this interval were listed within the cilia proteome databases,22,23 based upon the original work that identified these proteins as localized to or predicted to function at the cilium,24–30 including CEP97, MINA, ARL6, and ARL13B, and thus were considered as strong candidates (Figure 1D). Of these, only ARL6 (ADP-ribosylation factor-like 6) had been implicated in a human disease, where mutations are the cause of Bardet-Biedl syndrome 3 (BBS3).31,32 Because predominantly missense mutations were identified in ARL6, and because JSRD is a phenotypically more severe disorder than BBS, we considered that more severe mutations (such as stop codon or splice site mutations) might be found in MTI-001. Therefore, the seven coding exons of ARL6 were sequenced in affecteds from this family, but no sequence changes were encountered. The ARL13B gene (also known as ARL2L1, or Y37E3.5 in C. elegans) had previously been demonstrated to be expressed in ciliated neurons and specifically found to localize to the ciliary axoneme in C. elegans sensory neurons,25,31 so we considered this the next-most-likely candidate gene. Sequence analysis identified a homozygous c.G236A mutation in exon 3 that is predicted to lead to a p.R79Q (positively charged to noncharged) amino acid substitution (Figure S1). This mutation was not encountered in 288 individuals (96 of Pakistani origin, 96 individuals of Arab origin, 96 of mixed US origin), providing >80% power to distinguish this from a normal sequence variant,33 and suggesting that it does not represent a common allele. Genotyping of all members of MTI-001 showed that this homozygous mutation segregated with the phenotype in affecteds but not in unaffecteds or obligate carriers.
Impaired GTP Binding Associated with the R79Q Mutation
The p.R79Q mutation identified in family MTI-001 occurred within the highly conserved GTP-binding domain.34,35 Therefore, we questioned whether this mutation might affect GTP binding. Because ARL13B is 36% identical and 61% similar in amino acid sequence with ARL2, we used the crystal structure of ARL2 to model the effect of the p.R79Q mutation on intramolecular forces. This modeling showed that R79 (R74 in ARL2) of the switch II domain establishes a hydrogen bond with D30 (D25 in ARL2) of the P loop domain. This interaction is conserved in other ARF family proteins36 and is predicted to be disrupted with alteration in amino acid charge because of the p.R79Q substitution (Figures 1E and 1F), and as a result might disrupt GTP binding. To test this hypothesis, we cloned both wild-type and p.R79Q ARL13B human cDNAs into bacterial expression vectors (Figure S2), isolated recombinant proteins (Figure S3), verified that both were soluble based upon their isolation from the liquid phase and that they had similar secondary structure based upon similar circular dichroism analysis37 (not shown), and then tested their ability to bind GTP in an established in vitro binding assay. We found that the concentration for half-maximal GTP binding by ARL13B was ∼7.2 × 10−7 M. At this concentration, the amount of GTP bound to the mutant protein was about half of the amount bound to the wild-type protein (p < 0.05 for each of three comparisons), whereas negative control was just above background (Figure 1G). We conclude that the p.R79Q mutation interferes with GTPase binding activity of ARL13B and that this mutation could lead to impairment of its cilia function.
Mutation Analysis in a JS Cohort
We tested for mutations in ARL13B within several cohorts of JSRD. By using bidirectional sequencing, we screened 182 patients from our cohort diagnosed with JSRD and identified two additional mutations in one family. Family MTI-423 has one affected female displaying a proven MTS on brain imaging (Figure S4). The affected female displayed a compound heterozygous mutation c.G246A (exon 3) resulting from a stop codon p.W82X from the mother and a c.C598T (exon 5) from the father that results in a positive charged to uncharged amino acid p.R200C missense mutation within the coiled-coil domain (Figure 2A; Figure S1). This family did not display mutations in any of the known JSRD genes (NPHP1, AHI1, CEP290, TMEM67, RPGRIP1L). Each of these ARL13B substitution mutations was fully conserved across evolution in all mammals as well as Xenopus tropicalis, Danio rerio, and Tetraodon Nigroviridis (Figure 2B), and none of these mutations or any other predicted functional changes were identified by direct bidirectional sequencing of genomic DNA in a cohort of 182 healthy individuals (364 chromosomes). The early lethality of the hennin mouse suggests a critical role for ARL13B in early embryonic development and may provide one explanation for the low percentage of ARL13B mutations in the JSRD cohort and the low representation of truncating mutations among the ARL13B patients.
Figure 2.
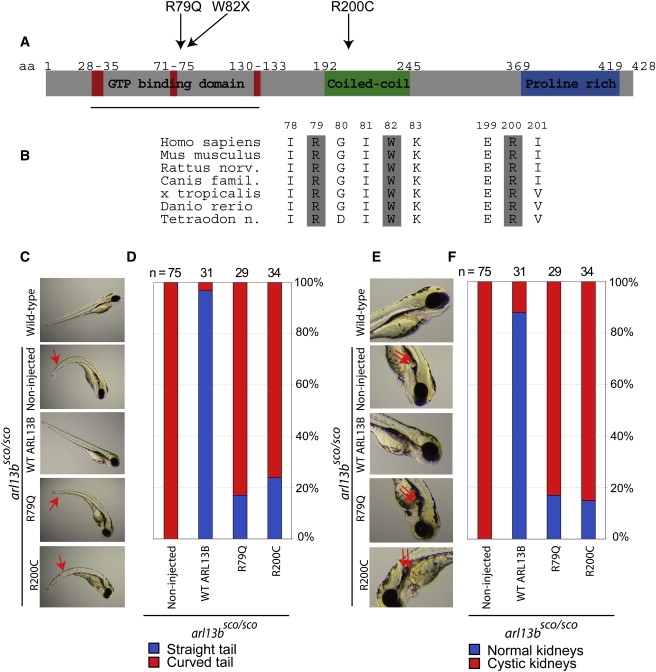
Human Mutations in ARL13B Occur in Conserved Residues and Interfere with Evolutionarily Conserved Functions
(A) Domain structure of ARL13B protein, with GTP binding, coiled-coil, and proline-rich domains. Red indicates conserved regions involved in GTP binding. Mutations are indicated along the top.
(B) Evolutionary conservation of ARL13B, showing that each mutated residues is conserved at least to Danio rerio (zebrafish).
(C–F) Wild-type zebrafish have straight tails and absence of cystic kidney. Noninjected arl13bsco/sco embryos have curved tails (red arrow) and cystic kidneys (double red arrow). The arl13bsco/sco embryos injected with human wild-type ARL13B RNA show rescue of the curved tail and absence of cystic kidney. The human p.R79Q and p.R200C mutant ARL13B RNA failed to rescue either phenotype in the majority of embryos, although about 15%–24% showed evidence of rescued phenotype, suggesting a hypomorphic allele. A control amino acid substitution (p.S371I) in ARL13B showed a rescue effect similar to wild-type (not shown).
Delineation of the Phenotypic Spectrum Associated with ARL13B Mutations
The phenotype observed in patients with identified mutations consists predominantly of classical Joubert syndrome. All patients displayed the MTS (Table S1). In MTI-001, two affecteds each displayed a small occipital encephalocele. In the other family, there was no occipital encephalocele, and there were no other supratentorial cerebral abnormalities and no renal abnormalities upon diagnostic ultrasound (performed on both families). Study of urinary concentration defects may be a more sensitive measure of early kidney involvement and was not available on any of these patients, so it is possible that renal symptoms might develop in the future. One affected from MTI-001 (now deceased) displayed evidence of mild nonspecific pigmentary retinopathy on clinical examination, although electroretinogram was normal on the affected siblings, so the involvement of ARL13B in retinal function is not yet clear. We also screened a cohort of 124 patients with JS with nephronophthisis and a cohort of 76 patients with overlapping Meckel syndrome with JS features. No mutation in either of these cohorts was encountered, suggesting that mutations in ARL13B are restricted to the classical form of JSRD (which can include encephalocele and retinopathy)38 and are not identifiable in other related ciliopathies, at least at this level of detection.
Human Wild-Type but Not Mutated ARL13B Rescue the Zebrafish Scorpion Mutant
To test the evolutionary conservation of ARL13B, we utilized zebrafish (Danio rerio). Comparison of the human and zebrafish ARL13B shows 75% similarity and 59% identity in more than 90% of the predicted amino acid sequence. We took advantage of the scorpion zebrafish mutant in the arl13b locus.39 The arl13bsco allele is due to a retroviral insertion into the first exon, which inactivates the gene, as evidenced by the finding that morpholino antisense treatment against arl13b produced a phenocopy of the arl13bsco/sco mutant.40 These mutants display fully penetrant curved tail and cystic kidney phenotypes, which are evident by 72 hr post fertilization (hpf) (n = 75, Figures 2C and 2E). We tested for rescue of the phenotype upon injection of RNA into 1-cell stage embryos from arl13bsco/+ × arl13bsco/+ matings, which produce 1/4 arl13bsco/sco embryos, by phenotyping the fish at 72 hpf by investigators blinded to the genotype. Injection of RNA corresponding to the zebrafish arl13b cDNA caused complete rescue of the phenotype, as evidenced by the fact that no mutants were identified by this blinded genotyping protocol (n = 20). Injection of the human ARL13B RNA similarly rescued the phenotype in the majority of embryos (Figures 2D and 2F). Among these mutants, the curved tail phenotype was rescued in 97% and the cystic kidney was rescued in 88% of embryos (n = 31). There was no overt overexpression phenotype that resulted from injection of either RNA into wild-type sibling embryos. Thus, the ARL13B gene displays evolutionarily conserved functions at the genetic level in embryogenesis.
In order to test the functional impairment of the patient missense mutations, we injected RNA corresponding to the cDNA for the human open reading frame of ARL13B, with each of these mutations introduced into the coding region in a similar assay. We opted to use the human rather than the zebrafish cDNA for this work, because it represented a more stringent test of function of the human mutations. Injection of each of these mutant RNAs resulted in a marked reduction in rescue when compared with the wild-type human RNA or a control nonpathogenic amino acid substitution-containing RNA. For this control, we chose the p.S371I transversion, because S371 is an evolutionarily conserved amino acid (similar to R79 and R200), it occurs in a part of the protein without known secondary structure, and the p.S371I alteration does not change the charge of the residue. The curved tail phenotype was rescued in 17% and 24%, and the cystic kidney phenotype was rescued in 17% and 15% of the arl13bsco/sco mutants for the p.R79Q and p.R200C mutations, respectively (n = 29 for p.R79Q, 34 for p.R200C, Figures 2C–2F). Contrarily, for the p.S371I control substitution, the curved tail phenotype was rescued in 100% and the cystic kidney phenotype was rescued in 91% of the arl13bsco/sco mutants (n = 11 for p.S371I, not shown). These data indicate that the human RNA with these human pathogenic mutations fail to function as well as the wild-type or control mutagenized human RNA and suggests that these pathogenic mutations at least partially impair the function of the protein during embryogenesis, leading to the JSRD phenotype.
Arl13b Is Expressed in Organs Affected in JS, with Protein Localized to Cilia
We next tested for expression of Arl13b in mouse tissues that are involved in JSRD patients at the presumed time points of affection to determine whether expression of the protein might account for the human phenotype. Arl13b protein was localized to cilia of all organs examined where they are known to be present, including the developing cerebellum (Figure 3). The epithelial cells lining the distal renal collecting ducts have well-described cilia, and we observed Arl13b staining of cilia structures within these tubules (Figure 3A). This staining might predict a role for Arl13b in kidney function, although none of our patients with ARL13B mutations displayed evidence of impaired kidney function. The retinal photoreceptor inner and outer segments, corresponding to the cell body and rhodopsin-containing disks, are bridged by a connecting cilium. We observed robust Arl13b staining of the connecting cilium at P10 (Figure 3B), similar to many of the proteins involved in LCA.41,42 It is interesting to note that one of the patients with an ARL13B mutation displayed evidence of retinopathy (Table S1). It will be important to follow the remaining patients to determine whether they develop progressive involvement of these organs in the future.
Figure 3.
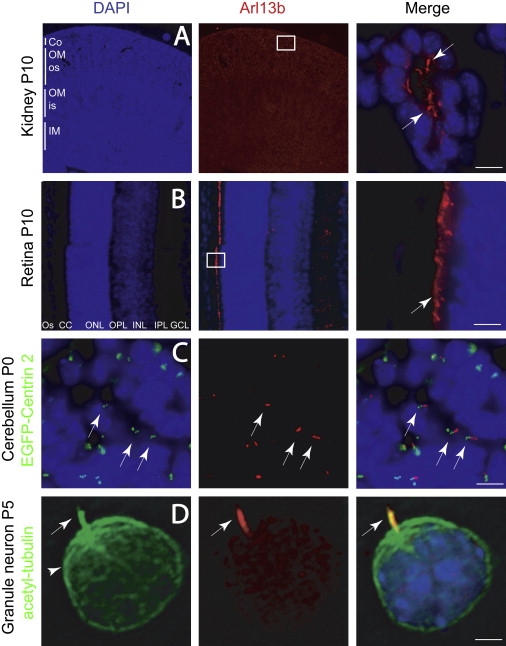
Arl13b Localization in Cilia within Kidney, Retina, and Developing Cerebellar Granule Neurons
Boxed region indicates location of high-power field shown in the third panel in (A) and (B).
(A) Kidney tubule shows Arl13b-positive cilia protruding into the lumen (arrows) in close approximation to the epithelial nuclei (blue, DAPI).
(B) Retina shows intense Arl13b staining in photoreceptor connecting cilia layer (CC, arrow).
(C) External granule layer (EGL) of P0 cerebellum from EGFP-Centrin 2 transgenic mouse stained with Arl13b. EGFP-labeled centrioles (green dots, arrow, left) are evident in many cells, defining the position of the basal body. An Arl13b-positive cilium projects adjacent to the basal body in many of these cells (arrows, right).
(D) Acutely dissociated CGN showing cilia (arrow) stained with acetylated tubulin (green) costains with Arl13b (red). Acetylated tubulin also stains the microtubule cytoskeleton (arrowhead), which is negative for Arl13b.
Abbreviations: Co, cortex; os, outer stripe of medulla; is, inner stripe of medulla; OM, outer medulla; IM, inner medulla; GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; CC, connecting cilia; OS, outer segment; P, postnatal day. Scale bar represents 20 μm in (A)–(C) and 5 μm in (D).
To test for expression in the developing cerebellum, the major site of involvement in JSRD, we examined Arl13b localization in postnatal mice. At the P0 time point, the cerebellum displays well-developed folia and consists of three well-defined layers: the external granule layer (EGL), Purkinje cell layer (PCL), and internal granule layer (IGL). Granule neuron precursors proliferate and migrate parallel to the surface of the cerebellum within the EGL. Postmitotic granule neurons then migrate radially into the cerebellar parenchyma and past the PCL to settle in the IGL. We detected strong staining of cilia-like structures on granule neurons both in the EGL and IGL (Figure 3C), which is consistent with previous serial electron microscopic sectioning of the cerebellum,43,44 as well as recent studies that have used staining for adenylate cyclase III to label cilia.45,46 Staining of Arl13b across the spectrum of cerebellar development in mouse demonstrated cilia-like structures in granule neuron populations, both in the EGL and IGL (Figure S5). There was a time dependence to the staining of cilia-like structures during cerebellar development, with about 40% of cells showing an immunopositive cilia at E16, dropping to less than 1% of cells by P21.
In order to be certain that these ARL13B-immunopositive structures represented cilia, we obtained sections from the cerebellum at P0 in a transgenic mouse that expresses EGFP fused to Centrin 2, a constitutive centrosome/basal body marker that localizes to centrioles.47 As expected, we found that most cells in the EGL contained a readily identifiable pair of centrioles adjacent to the nucleus, representing the basal body. We found an immunopositive ARL13B structure emanating from the region surrounding the basal body in many of these cells (Figure 3C), thereby demonstrating the spatial relationship between the two structures and suggesting that ARL13B marks the cilium. However, in many cells, ARL13B was not identified near a pair of centrioles, suggesting the possibility that ARL13B stains the cilia in only a fraction of granule neurons or that only a fraction of granule neurons contain a cilium.
In order to differentiate between these possibilities, we isolated cerebellar granule neuron precursors from P5 mouse by using a cell-density gradient, cultured and fixed at various time points. We found that a fraction of these primary cultured neurons display a cilia, as evidenced by labeling of a structure protruding from the cell body that was positive for acetylated tubulin (Figure 3D). Although the acetylated tubulin staining was strongly positive in the cilia, it extended into many of the microtubules within the cell soma. All cells with a cilium based upon this staining also displayed immunopositivity for Arl13b, and Arl13b staining was largely restricted to the cilia. Thus, Arl13b appears to be a sensitive and specific marker for the cilium in many cells including in the developing cerebellum. The data also support a role for Arl13b in the pathogenesis of JSRD, and thus strongly links JSRD to other human ciliopathies.
Discussion
ARL13B encodes ADP-ribosylation factor-like protein 13B (previously known as ADP-ribosylation factor-like protein 2-like 1) and belongs to the small Ras GTPase superfamily. ADP-ribosylation factor (ARF) GTP-binding proteins are among the best-characterized members of the Ras superfamily of GTPases, with well-established roles in membrane trafficking pathways. However, the ARL proteins appear to be functionally distinct, and many have roles in microtubule function.48,49 A mutation in Arl13b was very recently identified as the cause for the murine hennin phenotype that displays a disrupted neural tube during embryogenesis.19 The encoded Arl13b protein is localized to the primary cilia where it presumably plays a role in microtubule assembly or transport akin to the roles of other Arl proteins. However, the Arl13bhnn/hnn mutant is lethal at embryonic day 13.5–14.5 because of the severe cilia phenotype, thereby precluding a more detailed analysis of later functions.
Currently, mutations are identified in CEP290 in about 40% of patients that also display NPHP and LCA,50 in AHI1 in 10%–20% of patients mostly without NPHP,51,52 and in probably lower percentages in TMEM67, NPHP1, and RPGRIP1L genes. Although there are two additional JS loci (JBTS) that have been described where the causative gene has not yet been reported,53–55 fewer than 10% of families map to these loci.56 Thus, fewer than 50% of patients can receive a molecular diagnosis, and genotype-phenotype correlations subsequently are not yet clarified. Our preliminary data indicate that mutations in ARL13B are identified probably in less than 1% of JS, akin to the mutation frequency of ARL6 in BBS, although it will be interesting to determine the frequency of mutations in ARL13B among larger cohorts of JSRD patients.
Akin to the findings in BBS3, which is the only other disease to be linked to mutations in an ARL protein, the majority of mutations identified in ARL13B to date are missense.31,32 Additionally, our zebrafish data indicate that the missense mutations are probably hypomorphic alleles, because expression of each missense mutation rescues the phenotype with intermediate potency in the arl13bsco/sco mutant. This most likely reflects a requirement for the protein during embryogenesis, because both Arl3 and Arl13b mutant mice are often lethal prior to birth.19,57 Because Arl13b has been implicated in Sonic hedgehog (Shh) signaling,19 and because Shh58,59 as well as genes critical in cilia function have been implicated in proliferation of cerebellar granule neuron precursors,46,60 our data suggest that Joubert syndrome may represent a defect in cilia function mediating Shh signaling in this proliferation, although the exact signaling mechanisms and disrupted developmental pathways remain to be discovered.
Supplemental Data
Supplemental Data include a list of other members of the International JSRD Study Group, five figures, and one table and are available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BLAST, http://www.ncbi.nlm.nih.gov/
Pfam database, http://www.sanger.ac.uk/Software/Pfam/
Human Genome Browser, http://www.genome.ucsc.edu/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Cilia Proteome, http://www.ciliaproteome.org/ and http://www.bcgsc.ca/
Zebrafish database, http://zfin.org/
Zebrafish International Resource Center, http://zebrafish.org/zirc/home/guide.php/
Pymol, http://www.pymol.org
Swiss-Model, http://swissmodel.expasy.org/
Acknowledgments
We thank the Marshfield Clinic Research Foundation and the US National Heart, Lung, and Blood Institute for genotyping support; the UCSD Neurosciences Microscopy Core for imaging (P30 NS047101); Jeong-Soo Lee (Dana Farber Cancer Institute) for the zebrafish arl13b clone; Mark Lawson (UCSD) for help with SigmaPlot and statistical analysis; Daniel O'Connor (UCSD) for control DNA samples; and members of the Gleeson laboratory for helpful discussions. Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number N01-HG-65403. F.H. is supported by NIH grants R01-DK068306, R01-DK064614, and R01-DK069272 and is a Doris Duke Distinguished Clinical Scientist. V.C. was supported by a French Foundation for Medical Research (FRM) fellowship. S.L.B. was supported by the Neuroplasticity of Aging Postdoctoral Training Grant (T32 AG00216). J.Y.B. was supported by the Irvington Institute Fellowship Program of the Cancer Research Institute. This work was supported by grants from the US National Institute of Neurological Disease and Stroke, the Simons Foundation, and the Burroughs Wellcome Fund Award in Translational Research (to J.G.G.). The authors report no conflict of interest.
References
- 1.Palau F., Espinos C. Autosomal recessive cerebellar ataxias. Orphanet J. Rare Dis. 2006;1:47. doi: 10.1186/1750-1172-1-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maria B.L., Hoang K.B., Tusa R.J., Mancuso A.A., Hamed L.M., Quisling R.G., Hove M.T., Fennell E.B., Booth-Jones M., Ringdahl D.M. “Joubert syndrome” revisited: Key ocular motor signs with magnetic resonance imaging correlation. J. Child Neurol. 1997;12:423–430. doi: 10.1177/088307389701200703. [DOI] [PubMed] [Google Scholar]
- 3.King M.D., Dudgeon J., Stephenson J.B. Joubert's syndrome with retinal dysplasia: neonatal tachypnoea as the clue to a genetic brain-eye malformation. Arch. Dis. Child. 1984;59:709–718. doi: 10.1136/adc.59.8.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Satran D., Pierpont M.E., Dobyns W.B. Cerebello-oculo-renal syndromes including Arima, Senior-Loken and COACH syndromes: more than just variants of Joubert syndrome. Am. J. Med. Genet. 1999;86:459–469. [PubMed] [Google Scholar]
- 5.Verloes A., Lambotte C. Further delineation of a syndrome of cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma, and hepatic fibrosis. Am. J. Med. Genet. 1989;32:227–232. doi: 10.1002/ajmg.1320320217. [DOI] [PubMed] [Google Scholar]
- 6.Haug K., Khan S., Fuchs S., Konig R. OFD II, OFD VI, and Joubert syndrome manifestations in 2 sibs. Am. J. Med. Genet. 2000;91:135–137. doi: 10.1002/(sici)1096-8628(20000313)91:2<135::aid-ajmg11>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 7.Gleeson J.G., Keeler L.C., Parisi M.A., Marsh S.E., Chance P.F., Glass I.A., Graham J.M., Maria B.L., Barkovich A.J., Dobyns W.B. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am. J. Med. Genet. 2004;125A:125–134. doi: 10.1002/ajmg.a.20437. [DOI] [PubMed] [Google Scholar]
- 8.Valente E.M., Silhavy J.L., Brancati F., Barrano G., Krishnaswami S.R., Castori M., Lancaster M.A., Boltshauser E., Boccone L., Al-Gazali L. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat. Genet. 2006;38:623–625. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 9.Sayer J.A., Otto E.A., O'Toole J.F., Nurnberg G., Kennedy M.A., Becker C., Hennies H.C., Helou J., Attanasio M., Fausett B.V. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat. Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 10.Castori M., Valente E.M., Donati M.A., Salvi S., Fazzi E., Procopio E., Galluccio T., Emma F., Dallapiccola B., Bertini E. NPHP1 gene deletion is a rare cause of Joubert syndrome related disorders. J. Med. Genet. 2005;42:e9. doi: 10.1136/jmg.2004.027375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parisi M.A., Bennett C.L., Eckert M.L., Dobyns W.B., Gleeson J.G., Shaw D.W., McDonald R., Eddy A., Chance P.F., Glass I.A. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am. J. Hum. Genet. 2004;75:82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arts H.H., Doherty D., van Beersum S.E., Parisi M.A., Letteboer S.J., Gorden N.T., Peters T.A., Marker T., Voesenek K., Kartono A. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat. Genet. 2007;39:882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- 13.Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 14.Vierkotten J., Dildrop R., Peters T., Wang B., Ruther U. Ftm is a novel basal body protein of cilia involved in Shh signalling. Development. 2007;134:2569–2577. doi: 10.1242/dev.003715. [DOI] [PubMed] [Google Scholar]
- 15.Baala L., Romano S., Khaddour R., Saunier S., Smith U.M., Audollent S., Ozilou C., Faivre L., Laurent N., Foliguet B. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am. J. Hum. Genet. 2007;80:186–194. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murray S.S., Oliphant A., Shen R., McBride C., Steeke R.J., Shannon S.G., Rubano T., Kermani B.G., Fan J.B., Chee M.S. A highly informative SNP linkage panel for human genetic studies. Nat. Methods. 2004;1:113–117. doi: 10.1038/nmeth712. [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann K., Lindner T.H. easyLINKAGE-Plus—automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- 18.Tisdale E.J. A Rab2 mutant with impaired GTPase activity stimulates vesicle formation from pre-Golgi intermediates. Mol. Biol. Cell. 1999;10:1837–1849. doi: 10.1091/mbc.10.6.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caspary T., Larkins C.E., Anderson K.V. The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka T., Serneo F.F., Higgins C., Gambello M.J., Wynshaw-Boris A., Gleeson J.G. Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J. Cell Biol. 2004;165:709–721. doi: 10.1083/jcb.200309025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatten M.E. Neuronal regulation of astroglial morphology and proliferation in vitro. J. Cell Biol. 1985;100:384–396. doi: 10.1083/jcb.100.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inglis P.N., Boroevich K.A., Leroux M.R. Piecing together a ciliome. Trends Genet. 2006;22:491–500. doi: 10.1016/j.tig.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 23.Gherman A., Davis E.E., Katsanis N. The ciliary proteome database: An integrated community resource for the genetic and functional dissection of cilia. Nat. Genet. 2006;38:961–962. doi: 10.1038/ng0906-961. [DOI] [PubMed] [Google Scholar]
- 24.Efimenko E., Bubb K., Mak H.Y., Holzman T., Leroux M.R., Ruvkun G., Thomas J.H., Swoboda P. Analysis of xbx genes in C. elegans. Development. 2005;132:1923–1934. doi: 10.1242/dev.01775. [DOI] [PubMed] [Google Scholar]
- 25.Blacque O.E., Perens E.A., Boroevich K.A., Inglis P.N., Li C., Warner A., Khattra J., Holt R.A., Ou G., Mah A.K. Functional genomics of the cilium, a sensory organelle. Curr. Biol. 2005;15:935–941. doi: 10.1016/j.cub.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 26.Broadhead R., Dawe H.R., Farr H., Griffiths S., Hart S.R., Portman N., Shaw M.K., Ginger M.L., Gaskell S.J., McKean P.G. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature. 2006;440:224–227. doi: 10.1038/nature04541. [DOI] [PubMed] [Google Scholar]
- 27.Andersen J.S., Wilkinson C.J., Mayor T., Mortensen P., Nigg E.A., Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 28.Li J.B., Gerdes J.M., Haycraft C.J., Fan Y., Teslovich T.M., May-Simera H., Li H., Blacque O.E., Li L., Leitch C.C. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117:541–552. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 29.Avidor-Reiss T., Maer A.M., Koundakjian E., Polyanovsky A., Keil T., Subramaniam S., Zuker C.S. Decoding cilia function: Defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117:527–539. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- 30.Stolc V., Samanta M.P., Tongprasit W., Marshall W.F. Genome-wide transcriptional analysis of flagellar regeneration in Chlamydomonas reinhardtii identifies orthologs of ciliary disease genes. Proc. Natl. Acad. Sci. USA. 2005;102:3703–3707. doi: 10.1073/pnas.0408358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan Y., Esmail M.A., Ansley S.J., Blacque O.E., Boroevich K., Ross A.J., Moore S.J., Badano J.L., May-Simera H., Compton D.S. Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat. Genet. 2004;36:989–993. doi: 10.1038/ng1414. [DOI] [PubMed] [Google Scholar]
- 32.Chiang A.P., Nishimura D., Searby C., Elbedour K., Carmi R., Ferguson A.L., Secrist J., Braun T., Casavant T., Stone E.M. Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3) Am. J. Hum. Genet. 2004;75:475–484. doi: 10.1086/423903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collins J.S., Schwartz C.E. Detecting polymorphisms and mutations in candidate genes. Am. J. Hum. Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanzal-Bayer M., Renault L., Roversi P., Wittinghofer A., Hillig R.C. The complex of Arl2-GTP and PDE delta: from structure to function. EMBO J. 2002;21:2095–2106. doi: 10.1093/emboj/21.9.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vetter I.R., Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 36.Pasqualato S., Menetrey J., Franco M., Cherfils J. The structural GDP/GTP cycle of human Arf6. EMBO Rep. 2001;2:234–238. doi: 10.1093/embo-reports/kve043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly S.M., Jess T.J., Price N.C. How to study proteins by circular dichroism. Biochim. Biophys. Acta. 2005;1751:119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 38.Zaki M.S., Abdel-Aleem A., Abdel-Salam G., Marsh S.E., Silhavy J.L., Barkovich A.J., Ross M.E., Saleem S.N., Dobyns W.B., Gleeson J.G. The molar tooth sign: A new Joubert syndrome and related cerebellar disorders classification system tested in Egyptian families. Neurology. 2008;70:556–565. doi: 10.1212/01.wnl.0000277644.12087.fd. [DOI] [PubMed] [Google Scholar]
- 39.Sun Z., Amsterdam A., Pazour G.J., Cole D.G., Miller M.S., Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131:4085–4093. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- 40.Amsterdam A., Burgess S., Golling G., Chen W., Sun Z., Townsend K., Farrington S., Haldi M., Hopkins N. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 1999;13:2713–2724. doi: 10.1101/gad.13.20.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.den Hollander A.I., Koenekoop R.K., Mohamed M.D., Arts H.H., Boldt K., Towns K.V., Sedmak T., Beer M., Nagel-Wolfrum K., McKibbin M. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat. Genet. 2007;39:889–895. doi: 10.1038/ng2066. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Y., Hong D.H., Pawlyk B., Yue G., Adamian M., Grynberg M., Godzik A., Li T. The retinitis pigmentosa GTPase regulator (RPGR)- interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc. Natl. Acad. Sci. USA. 2003;100:3965–3970. doi: 10.1073/pnas.0637349100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Del Cerro M.P., Snider R.S. The Purkinje cell cilium. Anat. Rec. 1969;165:127–130. doi: 10.1002/ar.1091650202. [DOI] [PubMed] [Google Scholar]
- 44.Del Cerro M.P., Snider R.S. Studies on the developing cerebellum. II. The ultrastructure of the external granular layer. J. Comp. Neurol. 1972;144:131–164. doi: 10.1002/cne.901440202. [DOI] [PubMed] [Google Scholar]
- 45.Bishop G.A., Berbari N.F., Lewis J., Mykytyn K. Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J. Comp. Neurol. 2007;505:562–571. doi: 10.1002/cne.21510. [DOI] [PubMed] [Google Scholar]
- 46.Chizhikov V.V., Davenport J., Zhang Q., Shih E.K., Cabello O.A., Fuchs J.L., Yoder B.K., Millen K.J. Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J. Neurosci. 2007;27:9780–9789. doi: 10.1523/JNEUROSCI.5586-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Higginbotham H., Bielas S., Tanaka T., Gleeson J.G. Transgenic mouse line with green-fluorescent protein-labeled Centrin 2 allows visualization of the centrosome in living cells. Transgenic Res. 2004;13:155–164. doi: 10.1023/b:trag.0000026071.41735.8e. [DOI] [PubMed] [Google Scholar]
- 48.Burd C.G., Strochlic T.I., Gangi Setty S.R. Arf-like GTPases: Not so Arf-like after all. Trends Cell Biol. 2004;14:687–694. doi: 10.1016/j.tcb.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Gillingham A.K., Munro S. The small G proteins of the Arf family and their regulators. Annu. Rev. Cell Dev. Biol. 2007;23:579–611. doi: 10.1146/annurev.cellbio.23.090506.123209. [DOI] [PubMed] [Google Scholar]
- 50.Brancati F., Barrano G., Silhavy J.L., Marsh S.E., Travaglini L., Bielas S.L., Amorini M., Zablocka D., Kayserili H., Al-Gazali L. CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am. J. Hum. Genet. 2007;81:104–113. doi: 10.1086/519026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parisi M.A., Doherty D., Eckert M.L., Shaw D.W., Ozyurek H., Aysun S., Giray O., Al Swaid A., Al Shahwan S., Dohayan N. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J. Med. Genet. 2006;43:334–339. doi: 10.1136/jmg.2005.036608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Valente E.M., Brancati F., Silhavy J.L., Castori M., Marsh S.E., Barrano G., Bertini E., Boltshauser E., Zaki M.S., Abdel-Aleem A. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann. Neurol. 2006;59:527–534. doi: 10.1002/ana.20749. [DOI] [PubMed] [Google Scholar]
- 53.Keeler L.C., Marsh S.E., Leeflang E.P., Woods C.G., Sztriha L., Al-Gazali L., Gururaj A., Gleeson J.G. Linkage analysis in families with Joubert syndrome plus oculo-renal involvement identifies the CORS2 locus on chromosome 11p12-q13.3. Am. J. Hum. Genet. 2003;73:656–662. doi: 10.1086/378206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valente E.M., Salpietro D.C., Brancati F., Bertini E., Galluccio T., Tortorella G., Briuglia S., Dallapiccola B. Description, nomenclature, and mapping of a novel cerebello-renal syndrome with the molar tooth malformation. Am. J. Hum. Genet. 2003;73:663–670. doi: 10.1086/378241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saar K., Al-Gazali L., Sztriha L., Rueschendorf F., Nur E.K.M., Reis A., Bayoumi R. Homozygosity mapping in families with Joubert syndrome identifies a locus on chromosome 9q34.3 and evidence for genetic heterogeneity. Am. J. Hum. Genet. 1999;65:1666–1671. doi: 10.1086/302655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valente E.M., Marsh S.E., Castori M., Dixon-Salazar T., Bertini E., Al-Gazali L., Messer J., Barbot C., Woods C.G., Boltshauser E. Distinguishing the four genetic causes of Joubert syndrome-related disorders. Ann. Neurol. 2005;57:513–519. doi: 10.1002/ana.20422. [DOI] [PubMed] [Google Scholar]
- 57.Schrick J.J., Vogel P., Abuin A., Hampton B., Rice D.S. ADP-ribosylation factor-like 3 is involved in kidney and photoreceptor development. Am. J. Pathol. 2006;168:1288–1298. doi: 10.2353/ajpath.2006.050941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wechsler-Reya R.J., Scott M.P. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 59.Lewis P.M., Gritli-Linde A., Smeyne R., Kottmann A., McMahon A.P. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev. Biol. 2004;270:393–410. doi: 10.1016/j.ydbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 60.Spassky N., Han Y.G., Aguilar A., Strehl L., Besse L., Laclef C., Romaguera Ros M., Garcia-Verdugo J.M., Alvarez-Buylla A. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev. Biol. 2008;317:246–259. doi: 10.1016/j.ydbio.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.