

The three-dimensional structure of a SARS coronavirus-derived peptide, VQQESSFVM, bound to the human major histocompatibility complex (MHC) class I antigen HLA-B*1501 is presented.
Keywords: human leukocyte antigen class I, SARS coronavirus-derived peptides, HLA-B*1501
Abstract
The human leukocyte antigen (HLA) class I system comprises a highly polymorphic set of molecules that specifically bind and present peptides to cytotoxic T cells. HLA-B*1501 is a prototypical member of the HLA-B62 supertype and only two peptide–HLA-B*1501 structures have been determined. Here, the crystal structure of HLA-B*1501 in complex with a SARS coronavirus-derived nonapeptide (VQQESSFVM) has been determined at high resolution (1.87 Å). The peptide is deeply anchored in the B and F pockets, but with the Glu4 residue pointing away from the floor in the peptide-binding groove, making it available for interactions with a potential T-cell receptor.
1. Introduction
Major histocompatibility complex class I (MHC-I, or HLA-I in humans) molecules are cell-surface glycoproteins. Their function is to select peptides derived from the intracellular protein metabolism and present them to cytotoxic T cells of the adaptive immune system, thereby allowing the protein content of cells to be surveyed and controlled (reviewed in Guermonprez et al., 2002 ▶). The specificity of MHC-I molecules determines which peptides will be presented and potentially recognized by the immune system. The HLA system is highly polymorphic in a manner that effectively diversifies immune responsiveness. Almost 1600 different HLA-I alleles have been identified in human populations, making MHC-I the most polymorphic gene system known. In an attempt to reduce this complexity, it has been suggested that the majority of HLA-I molecules can be clustered into one of 12 currently defined HLA supertypes. More than 120 different HLA-I crystal structures have been reported and the majority of these belong to the HLA-A2 supertype; other supertypes are sparsely populated or not at all. Only a few structures of HLA-I alleles belonging to the HLA-B62 supertype have been reported, including two structures of HLA-B*1501 (Røder et al., 2006 ▶). HLA-B*1501 is the prototypical member of the HLA-B62 supertype. HLA-B*1501 is frequent in Southeast Asian and in European populations, where it often accounts for more than 10% of the HLA-I phenotype.
We are involved in the Human MHC Project, which aims to describe and predict the peptide-binding specificity of one or more members of each HLA supertype (Lauemøller et al., 2000 ▶). To this end, we are generating structural (Blicher et al., 2005 ▶, 2006 ▶; Røder et al., 2006 ▶) and biochemical (Buus et al., 1995 ▶; Sylvester-Hvid et al., 2002 ▶; Lauemøller et al., 2000 ▶) information on peptide–HLA interactions and using this information to generate fast computational predictions of peptide binding (Stryhn et al., 1996 ▶; Buus et al., 2003 ▶; Peters et al., 2006 ▶). A detailed understanding of peptide–MHC interactions will support a general understanding of the nature of T-cell recognition and a rational exploitation of human immune responses (Lauemøller et al., 2000 ▶). Thus, predictive algorithms allow entire genomes to be rapidly and accurately screened for potential immunogenic epitopes (Wang et al., 2007 ▶). One approach, which might be independent of large-scale biochemical experimentation, would be to use structural information to predict peptide binders (Rognan et al., 1999 ▶). To populate the ‘structure space’ of HLA molecules, we have previously reported two HLA-A*1101 (Blicher et al., 2005 ▶, 2006 ▶) and two HLA-B*1501 structures (Røder et al., 2006 ▶). Here, we report a crystal structure of HLA-B*1501 in complex with a SARS coronavirus-derived nonapeptide (VQQESSFVM).
2. Materials and methods
2.1. Cloning
A genetic construct representing the HLA-B*1501 protein, positions 1–276, was produced by site-directed mutagenesis from an HLA-A*0201 gene using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene). This procedure involved 81 nucleotide mutations, which could be accomplished using 13 DNA primers. The final HLA-B*1501 sequence was inserted into the pET28a vector (Novagen). The sequence was verified by DNA sequencing (3100 Avant, ABI).
2.2. Protein production and purification
Escherichia coli BL21 (DE3) cells were transformed with the HLA-B*1501-pET28 vector and the protein was produced in a 2 l fermentor (Infors) by induction with IPTG as described previously (Ferré et al., 2003 ▶). Inclusion bodies were obtained by cell disruption (Constant Cell Disruption Systems), washed twice with PBS containing 0.5%(v/v) NP40 and 0.5%(w/v) deoxycholate and dissolved in 8 M urea and 25 mM Tris–HCl pH 8.0. The dissolved HLA-B*1501 protein was purified in 8 M urea at 285 K by ion-exchange chromatography (Q-Sepharose Fast Flow). Buffer A contained 8 M urea and 25 mM Tris–HCl pH 8.0 and buffer B was the same but included 1 M NaCl. A 0–50% gradient in buffer B spanning four column volumes was applied. The eluted protein was further purified by hydrophobic interaction chromatography (Phenyl Sepharose HP) to separate correctly disulfide-bonded HLA-B*1501 (buffer A, 8 M urea, 25 mM Tris–HCl pH 8.0 and 100 g l−1 ammonium sulfate; buffer B, 8 M urea and 25 mM Tris–HCl pH 8.0; gradient, 0–40% in buffer B spanning six column volumes). Finally, the HLA-B*1501 sample was purified by Sephacryl-S200 chromatography in 8 M urea, 25 mM Tris–HCl pH 8.0 and 150 mM NaCl, concentrated by ultrafiltration and subsequently stored at 253 K.
2.3. Peptide production and purification
The VQQESSFVM peptide was synthesized by conventional Fmoc chemistry and subsequently purified by reversed-phase HPLC (Schafer-N, Copenhagen). The peptide identity was verified by reversed-phase HPLC followed by ion-trap mass spectrometry (Bruker Daltonics). The purity was determined to be higher than 99%.
2.4. MHC-I complex assembly
For complex assembly, 3 mg denatured HLA-B*1501 heavy chain was rapidly diluted in 1 l 50 mM Tris–HCl pH 7.5, 3 mM EDTA and 150 mM NaCl containing 2 mg β2-microglobulin (β2m) and 1 mg peptide (the final heavy chain, β2m and peptide concentrations were 100, 170 and 1000 nM, respectively). The folding reaction was incubated at 291 K for 48 h and then concentrated to 10 ml using a pressure cell (Amicon) equipped with a 10 kDa molecular-weight cutoff filter. The highly concentrated folding reaction was allowed to settle overnight and was subsequently concentrated to 0.5 ml on a 10 kDa spin filter (Amicon). Folded MHC-I complex was separated from aggregated HLA-B*1501 heavy chain, free β2m and peptide on a Superdex-200 column (Amersham Biosciences). The fractions were analyzed (SDS–PAGE and ion-trap mass spectrometry) and those containing the two protein components, HLA-B*1501 heavy chain and β2m, were pooled and concentrated on a 10 kDa spin filter to 4.2 mg ml−1 (96 µM) in 50 mM Tris–HCl pH 7.5, 3 mM EDTA and 150 mM NaCl.
2.5. Crystallization and data collection
Crystals of the HLA-B*1501 complex were grown using the hanging-drop technique. Buffers from Crystal Screens 1 and 2 (Hampton Research) were used in screening (reservoir volume, 500 µl; drop volume, 1 µl reservoir solution and 1 µl protein solution). The experiments were set up at 277 K. No crystals appeared within the first two months, after which the crystallization trials were left at 277 K for 2 y. Single crystals appeared under initial conditions consisting of 0.2 M magnesium chloride hexahydrate, 0.1 M HEPES, 30%(v/v) PEG 400 pH 7.5. Diffraction data were collected at cryogenic temperature on the I911-5 beamline at MAX-lab, Lund, Sweden. The HLA-B*1501 complex crystallized in space group P212121, with one protein–peptide complex per asymmetric unit (see Table 1 ▶). Intensities were integrated with MOSFLM (Powell, 1999 ▶) and scaled with SCALA (Evans, 2006 ▶).
Table 1. Crystal data and data-collection and refinement statistics of the HLA-B*1501 structure.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Wavelength (Å) | 0.907 |
| Unit-cell parameters (Å) | a = 50.66, b = 81.71, c = 109.39 |
| Space group | P212121 |
| Molecules per ASU | 1 |
| Resolution range (Å) | 18.29–1.87 (1.97–1.87) |
| No. of unique reflections | 38220 (5506) |
| Multiplicity | 4.0 (4.1) |
| Completeness of data (%) | 99.8 (100.0) |
| Rmerge† | 0.064 (0.362) |
| 〈I/σ(I)〉 | 15.7 (3.1) |
| Model details | |
| Protein atoms | 3184 |
| Water O atoms | 357 |
| PEG (PG4) atoms | 13 |
| HEPES (EPE) atoms | 15 |
| Refinement details | |
| No. of reflections used in refinement | 36303 |
| R factor for all data‡ (%) | 18.8 |
| Rfree§ (%) | 22.7 |
| Model | |
| Mean isotropic equivalent B factor (Å2) | 25.8 |
| MHC-I heavy-chain B factor (Å2) | 22.3 |
| β2-Microglobulin B factor (Å2) | 31.3 |
| Peptide B factor (Å2) | 21.2 |
| Water B factor (Å2) | 35.1 |
| Heterocompounds B factor (Å2) | 51.0 |
| Residues in most favoured regions¶ (%) | 91.4 |
| Residues in generally disallowed regions¶ (%) | 0.0 |
R
merge = 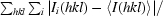
 .
.
R = 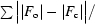
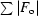 .
.
As R, but calculated on 5% of the data excluded from the refinement.
Calculated with PROCHECK (Laskowski et al., 1993 ▶).
2.6. Structure determination and refinement
The structure was solved by molecular replacement using the program Phaser (McCoy et al., 2005 ▶) with HLA-B*1501 (PDB code 1xr9) as the search model (Røder et al., 2006 ▶). An initial model consisting of the HLA-B*1501 heavy chain and β2m was automatically built with ARP/wARP (Cohen et al., 2004 ▶) and residue side chains were substituted using guiSIDE (Cohen et al., 2004 ▶). Multiple rounds of manual inspection and model building in Coot (Emsley & Cowtan, 2004 ▶) were performed, followed by restrained refinements in REFMAC5 (Murshudov et al., 1997 ▶). This included manual building of the VQQESSFVM peptide. The final structure contains HLA-B*1501 (residues 1–275), β2m (residues 1–99), the VQQESSFVM peptide, one putative PEG fragment, a putative HEPES molecule and 357 waters (Table 1 ▶). The MHC-I α-chain, the β2m chain and the bound peptide will be referred to in the following as ‘a’, ‘b’ and ‘p’, respectively. The structure has been deposited in the RCSB Protein Data Bank with code 3c9n. All figures were prepared with PyMOL (DeLano, 2002 ▶).
3. Results and discussion
This is the third crystal structure of HLA-B*1501 in complex with a peptide. The two previous structures either had Leu or Glu in position 2 of the peptide (Røder et al., 2006 ▶). The structure reported here has Gln at this position. Furthermore, the two reported structures both have Tyr as the C-terminal peptide residue, whereas this structure has a Met, which slightly alters the position of the peptide C-terminus.
3.1. Model quality
The overall structure of the present HLA-B*1501 peptide-binding groove is similar to the previously two determined HLA-B*1501 structures (Røder et al., 2006 ▶) and to other MHC-I molecules (see Fig. 1 ▶). Structural superpositioning of this and a previously published HLA-B*1501 structure (PDB code 1xr9) using the first 181 CA atoms in the α-chain yields a root-mean-square deviation of 0.34 Å. The structure consists of all of the residues of the heavy (a) and light (b) chains and the bound peptide (p) (see Table 1 ▶ for details). The model was refined to an R value of 18.8% and an R free value of 22.7%. Generally, the electron density is clearly defined and all the residues lining the peptide-binding groove and the peptide residues are completely resolved (see Fig. 2 ▶). Furthermore, the disulfide bond in the α2 domain of the peptide-binding groove is clearly defined, as depicted in the inset in Fig. 1 ▶. It has been speculated that the oxidation state of this disulfide bond is important for the loading of the peptide-binding groove with a peptide (Park et al., 2006 ▶). When this disulfide bond is oxidized and a peptide is bound, the α1 and α2 domains are thought to be more stable. Some solvent-exposed residues (including pSer5 of the peptide) are found in multiple conformations. A Ramachandran analysis estimated that 91.4% of the residues are found in the favoured regions and no residues are found in disallowed regions.
Figure 1.

Interactions in the HLA-B*1501 peptide-binding groove. The HLA-I complex is visualized from above, looking directly towards the floor of the peptide-binding groove. The peptide (VQQESSFVM) is depicted in yellow and the peptide N-terminal region is located to the left. Peptide-binding groove residues interacting with the peptide are coloured black and labelled. The insert shows the electron density (at 1σ, to a distance of 1 Å) of the characteristic disulfide bond in the α2 helix.
Figure 2.

(a) The bound peptide ligand. The 2F o − F c electron-density map (at 1σ) is shown 1 Å around the peptide. The peptide is seen from the side of the α2 helix, which positions its N-terminal end to the left. (b) Surface representation of the peptide-binding groove. The docked peptide is caged by an electron-density mesh as in (a).
3.2. Interactions with the bound peptide
The electron density of the peptide bound in the HLA-B*1501 binding groove is well defined except for the pGlu4 side chain, which is solvent-exposed and thus only partially defined (see Fig. 2 ▶).
3.2.1. Peptide residue 1
The Cγ atoms of the pVal1 side chain in the A pocket are positioned towards the aromatic ring system of aTrp167, thereby engaging in hydrophobic interactions. Furthermore, this hydrophobic p1 residue also interacts with the aTyr59 and aLeu163 residues in HLA-B*1501 as described previously (Røder et al., 2006 ▶). The N-terminal N atom is oriented towards the α2 helix. This is unusual as a water-mediated hydrogen bond is generally formed to protein residues (Ogata & Wodak, 2002 ▶). In the present structure, the corresponding water molecule (HOH287) makes hydrogen bonds to aTyr7, aTyr59 and potentially aGlu63.
3.2.2. Peptide residue 2
The pGln2 peptide residue is located in the B pocket in the same orientation as the pGlu2 in the previously described HLA-B*1501–LEKARGSTY structure (Røder et al., 2006 ▶). This residue is completely shielded from the surroundings by aArg62, thereby prohibiting any interaction with a potential T-cell receptor. The pGln2 residue makes hydrophobic interactions with aTyr7 and aIle66 that make up the bottom and top side wall of the B pocket, respectively. The N and O atoms of pGln2 interact with aTyr9, aGlu63, aSer67 and a bridging water molecule (HOH57) located at the apex of the B pocket.
3.2.3. Peptide residue 3
The pGln3 peptide residue is located in the D pocket and points towards the α2 helix. This peptide residue forms hydrophobic interactions with aTyr99, aTrp156 and aTyr159. In addition, weak hydrogen bonds to two water molecules (HOH341 and HOH539) are observed, thus bridging the peptide to the HLA-B*1501 side chains, which results in further stabilization of the bound peptide residue in this pocket.
3.2.4. Peptide residue 4
The pGlu4 peptide residue points away from the peptide-binding groove and the side chain does not interact with any HLA-B*1501 residues. This orientation of pGlu4 makes it available for potential interactions with T-cell receptors.
3.2.5. Peptide residue 5
The pSer5 peptide residue is found in two conformations, both of which point towards the α1 helix of the C pocket. The Oγ atom in the A conformation makes a hydrogen bond to the Nδ2 atom of aAsn70 and the backbone O atom of aIle66, both of which are located in the α1 helix, and forms a weak hydrogen bond to a water molecule located at the surface (HOH327). In the B conformation, a hydrogen bond is formed to the Nδ2 atom of aAsn70 and two water molecules (HOH79 and HOH364).
3.2.6. Peptide residue 6
The pSer6 peptide residue points down towards the β-sheet floor of the peptide-binding groove. The conformation observed does not interact with any HLA-B*1501 residues, but makes a hydrogen bond to two water molecules (HOH67 and HOH348) present between the peptide and the floor of the peptide-binding groove, thereby forming water-mediated contacts with the HLA-B*1501 protein. Therefore, only larger peptide residues at this position are likely to contribute significantly to the overall MHC-I binding.
3.2.7. Peptide residue 7
The pPhe7 peptide residue is solvent-exposed and points directly away from the floor of the peptide-binding groove. The electron density of this residue is clearly defined, revealing that pPhe7 is tilted slightly towards the α2 helix. The conformation of pPhe7 allows contacts with aAla150 located on the α2 helix segment. The backbone N atom of this residue also makes a hydrogen bond to the O∊1 atom on the aGlu152 residue. The large aTrp147 also excludes the possibility of the pPhe7 residue side chain packing into the peptide-binding groove, which forces the pPhe7 residue to be solvent-exposed. The orientation of pPhe7 makes it available for interactions with a potential T-cell receptor.
3.2.8. Peptide residue 8
The side chain of this peptide residue does not contact any HLA-B*1501 residues. The pVal8 residue protrudes from the binding pocket. However, owing to the small size of pVal8, it is unlikely to be a dominating peptide residue that plays any significant role in the T-cell receptor docking and recognition.
3.2.9. Peptide residue 9
The last pocket of the peptide-binding groove of HLA-I is the F pocket. As previously described for HLA-B*1501, this pocket is lined by hydrophobic residues and can accommodate large aromatic residues such as Tyr and Phe (Røder et al., 2006 ▶). The present HLA-B*1501–peptide structure contains a C-terminal methionine residue, pMet9, which is found in an extended conformation reaching the apex of the F pocket. The F pocket is lined by aLeu81, aLeu95, aTyr123 and aTrp147, rendering this environment mostly hydrophobic. aSer116 is located at the bottom of the F pocket and points towards the α1 domain of the peptide-binding groove. Hence, aSer116 points away from the docked pMet9 side chain and is unable to interact with this C-terminal peptide residue. pMet9 along with a114 have been shown to not only have a direct influence on the F-pocket specificity but also to influence the interaction of HLA-I with tapasin (Park et al., 2003 ▶; Thammavongsa et al., 2006 ▶). The peptide C-terminal O atoms make one hydrogen bond to the Nδ2 atom of aAsn80 and one to the Nζ atom of aLys146, which stabilizes peptide binding and at the same time shields the peptide C-terminus from interactions with a T-cell receptor.
Supplementary Material
PDB reference: HLA-B*1501–VQQESSFVM complex, 3c9n, r3c9nsf
Acknowledgments
This work was funded by the Danish Centre for Synchrotron Radiation (DANSYNC), EU 6FP 503231 and NIH HHSN266200400025C.
References
- Blicher, T., Kastrup, J. S., Buus, S. & Gajhede, M. (2005). Acta Cryst. D61, 1031–1040. [DOI] [PMC free article] [PubMed]
- Blicher, T., Kastrup, J. S., Pedersen, L. Ø., Buus, S. & Gajhede, M. (2006). Acta Cryst. F62, 1179–1184. [DOI] [PMC free article] [PubMed]
- Buus, S., Lauemøller, S. L., Worning, P., Kesmir, C., Frimurer, T., Corbet, S., Fomsgaard, A., Hilden, J., Holm, A. & Brunak, S. (2003). Tissue Antigens, 62, 378–384. [DOI] [PubMed]
- Buus, S., Stryhn, A., Winther, K., Kirkby, N. & Pedersen, L. O. (1995). Biochim. Biophys. Acta, 1243, 453–460. [DOI] [PubMed]
- Cohen, S. X., Morris, R. J., Fernandez, F. J., Ben Jelloul, M., Kakaris, M., Parthasarathy, V., Lamzin, V. S., Kleywegt, G. J. & Perrakis, A. (2004). Acta Cryst. D60, 2222–2229. [DOI] [PubMed]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics system. http://www.pymol.org.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Ferré, H., Ruffet, E., Blicher, T., Sylvester-Hvid, C., Nielsen, L. L. B., Hobley, T. J., Thomas, O. R. T. & Buus, S. (2003). Protein Sci.12, 551–559. [DOI] [PMC free article] [PubMed]
- Guermonprez, P., Valladeau, J., Zitvogel, L., Théry, C. & Amigorena, S. (2002). Annu. Rev. Immunol.20, 621–667. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Lauemøller, S. L., Kesmir, C., Corbet, S. L., Fomsgaard, A., Holm, A., Claesson, M. H., Brunak, S. & Buus, S. (2000). Rev. Immunogenet.2, 477–491. [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Ogata, K. & Wodak, S. J. (2002). Protein Eng.15, 697–705. [DOI] [PubMed]
- Park, B., Lee, S., Kim, E. & Ahn, K. (2003). J. Immunol.170, 961–968. [DOI] [PubMed]
- Park, B., Lee, S., Kim, E., Cho, K., Riddell, S. R., Cho, S. & Ahn, K. (2006). Cell, 127, 369–382. [DOI] [PubMed]
- Peters, B., Bui, H., Frankild, S., Nielson, M., Lundegaard, C., Kostem, E., Basch, D., Lamberth, K., Harndahl, M., Fleri, W., Wilson, S. S., Sidney, J., Lund, O., Buus, S. & Sette, A. (2006). PLoS Comput. Biol.2, e65. [DOI] [PMC free article] [PubMed]
- Powell, H. R. (1999). Acta Cryst. D55, 1690–1695. [DOI] [PubMed]
- Røder, G., Blicher, T., Justesen, S., Johannesen, B., Kristensen, O., Kastrup, J., Buus, S. & Gajhede, M. (2006). Acta Cryst. D62, 1300–1310. [DOI] [PubMed]
- Rognan, D., Lauemøller, S. L., Holm, A., Buus, S. & Tschinke, V. (1999). J. Med. Chem.42, 4650–4658. [DOI] [PubMed]
- Stryhn, A., Pedersen, L. Ø., Romme, T., Holm, C. B., Holm, A. & Buus, S. (1996). Eur. J. Immunol.26, 1911–1918. [DOI] [PubMed]
- Sylvester-Hvid, C., Kristensen, N., Blicher, T., Ferré, H., Lauemøller, S. L., Wolf, X. A., Lamberth, K., Nissen, M. H., Pedersen, L. Ø. & Buus, S. (2002). Tissue Antigens, 59, 251–258. [DOI] [PubMed]
- Thammavongsa, V., Raghuraman, G., Filzen, T. M., Collins, K. L. & Raghavan, M. (2006). J. Immunol.177, 3150–3161. [DOI] [PubMed]
- Wang, M., Lamberth, K., Harndahl, M., Røder, G., Stryhn, A., Larsen, M. V., Nielsen, M., Lundegaard, C., Tang, S. T., Dziegiel, M. H., Rosenkvist, J., Pedersen, A. E., Buus, S., Claesson, M. H. & Lund, O. (2007). Vaccine, 25, 2823–2831. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: HLA-B*1501–VQQESSFVM complex, 3c9n, r3c9nsf