

Three conventional robots were subjected to a crystallization screening test involving 18 proteins from T. thermophilus HB8 using the sitting- and hanging-drop vapour-diffusion and microbatch methods. The number of diffraction-quality crystals and the amount of time required to obtain visible crystals depended greatly on the robots used. The combined use of different robots, especially for protein samples exhibiting low crystallization success rates, significantly increased the chance of obtaining diffraction-quality crystals.
Keywords: protein crystallization, crystallization screening, crystallization success rates
Abstract
It was essential for the structural genomics of Thermus thermophilus HB8 to efficiently crystallize a number of proteins. To this end, three conventional robots, an HTS-80 (sitting-drop vapour diffusion), a Crystal Finder (hanging-drop vapour diffusion) and a TERA (modified microbatch) robot, were subjected to a crystallization condition screening test involving 18 proteins from T. thermophilus HB8. In addition, a TOPAZ (microfluidic free-interface diffusion) designed specifically for initial screening was also briefly examined. The number of diffraction-quality crystals and the time of appearance of crystals increased in the order HTS-80, Crystal Finder, TERA. With the HTS-80 and Crystal Finder, the time of appearance was short and the rate of salt crystallization was low. With the TERA, the number of diffraction-quality crystals was high, while the time of appearance was long and the rate of salt crystallization was relatively high. For the protein samples exhibiting low crystallization success rates, there were few crystallization conditions that were common to the robots used. In some cases, the success rate depended greatly on the robot used. The TOPAZ showed the shortest time of appearance and the highest success rate, although the crystals obtained were too small for diffraction studies. These results showed that the combined use of different robots significantly increases the chance of obtaining crystals, especially for proteins exhibiting low crystallization success rates. The structures of 360 of 944 purified proteins have been successfully determined through the combined use of an HTS-80 and a TERA.
1. Introduction
The structural biological whole-cell project (http://www.thermus.org/) that is being promoted by the RIKEN SPring-8 Center aims to increase the understanding of fundamental biological phenomena in the cell system at the atomic level on the basis of the three-dimensional structures of proteins encoded by genes (Yokoyama, Matsuo et al., 2000 ▶; Yokoyama, Hirota et al., 2000 ▶; Kuramitsu et al., 1995 ▶). We selected an extreme thermophile, Thermus thermophilus HB8, as a model organism because this bacterium can grow at the highest temperature of all organisms that have a convenient gene-manipulation system and because the proteins from this bacterium are highly stable and suitable for structure–function studies. The first step in the project was the structural genomics of T. thermophilus HB8. Therefore, it was essential to efficiently crystallize a number of purified proteins and to determine their structures.
Crystallization is still one of the major bottlenecks in X-ray structure analysis (Stevens, 2000 ▶; Chayen & Saridakis, 2008 ▶), although robots have been developed for conducting a large number of crystallization trials in a short time (Saitoh et al., 2005 ▶; Miyatake et al., 2005 ▶; Hiraki et al., 2006 ▶; Sulzenbacher et al., 2002 ▶; Hui & Edwards, 2003 ▶; Shah et al., 2005 ▶). Generally, it is time-consuming to crystallize proteins. In rare cases, it takes only several hours to grow crystals if all goes smoothly. However, for 80% of the samples in our case it took more than 60 d to go from initial screening of crystallization conditions to crystals that were suitable for data collection. Efficient crystallization, especially from the viewpoint of structural genomics, requires diffraction-quality crystals shortly after screening crystallization conditions.
Proteins are usually crystallized by mixing a drop of a protein solution with a drop of a crystallization solution containing precipitating reagents such as salts and polyethylene glycol. The mixed drop is then driven to sufficient supersaturation for nucleation (McPherson, 1999 ▶). The most commonly used crystallization methods are the sitting- and hanging-drop vapour-diffusion (VD) and microbatch (MB) methods, although various other crystallization methods have been developed (DeLucas et al., 2003 ▶; McPherson, 2004 ▶; Chayen, 1998 ▶). The sitting- and hanging-drop VD methods are most frequently used for high-throughput crystallization by most crystallization robots (Hiraki et al., 2006 ▶; Sulzenbacher et al., 2002 ▶; Hosfield et al., 2003 ▶; Miyatake et al., 2005 ▶; Santarsiero et al., 2002 ▶). The MB method is also popular for large-scale screening of crystallization conditions (Luft et al., 2001 ▶; Adams et al., 2003 ▶; Saitoh et al., 2005 ▶). With the modified MB method, the droplet is allowed to gradually become concentrated using a water-permeable oil system (D’Arcy et al., 2004 ▶). Recently, the microfluidic free-interface diffusion (FID) method was developed for the rapid screening of initial crystallization conditions with the use of only several microlitre aliquots of the protein solutions (Hansen et al., 2002 ▶; Segelke, 2005 ▶).
Three conventional robots were subjected to a crystallization condition screening test using 18 proteins (Table 1 ▶). This test prior to large-scale crystallization for structural genomics was used to determine which crystallization screening protocol is best suited to the project. The robots used were an HTS-80 for sitting-drop VD (RIKEN and Panasonic Factory Solutions Co. Ltd; Miyatake et al., 2005 ▶), a Crystal Finder for hanging-drop VD (RIKEN and Ishikawajima Inspection and Instrumentation Co. Ltd) and a TERA for modified MB (RIKEN; Saitoh et al., 2005 ▶) (Table 2 ▶). These robots, which were designed for microlitre-scale experiments, are rather conventional compared with those used for high-throughput nanolitre methods (Santarsiero et al., 2002 ▶; DeLucas et al., 2003 ▶; Au et al., 2006 ▶; Walter et al., 2005 ▶; Chayen & Saridakis, 2008 ▶), indicating that the results obtained here should be of some help for the manual screening of crystallization conditions in small-scale laboratories. In addition to microlitre-scale investigations, a TOPAZ for nanoscale FID (Fluidigm Corp.; Segelke, 2005 ▶) was also subjected to preliminary tests in the initial condition search. The screening data were analyzed and compared among the robots and TOPAZ. We then started crystallization experiments for the structural genomics work with the combined use of the HTS-80 and TERA and succeeded in determining the structures of 360 of 944 purified proteins. In this paper, we report the results of the crystallization screening tests as they relate to the crystallization success rate, the time of appearance of crystals and the number of successful trials common to the two robots and specific to each robot.
Table 1. 18 protein samples selected for crystallization condition screening.
| Locus tag† | Function | PDB code | MW (kDa) | pI | Concentration (mgml1) | Buffer (protein solution)‡ | |
|---|---|---|---|---|---|---|---|
| A | TTHA1797 | Probable amidase | 2dc0 | 46520 | 5.91 | 10.82 | (i) |
| B | TTHA0859 | Uridylate kinase | 25274 | 7.30 | 13.10 | (i) | |
| C | TTHA1431 | Conserved hypothetical protein | 2cz8 | 7748 | 5.77 | 38.58 | (i) |
| D | TTHA1437 | Transcription regulator, Crp family | 23818 | 5.69 | 10.32 | (ii) | |
| E | TTHA0789 | Putative glutaryl-CoA dehydrogenase | 2eba | 42780 | 6.31 | 32.02 | (i) |
| F | TTHA0122 | Manganese-containing pseudocatalase | 2cwl | 33333 | 5.34 | 10.68 | (i) |
| G | TTHA1671 | Adenylate kinase | 20754 | 5.05 | 10.24 | (i) | |
| H | TTHB192 | Hypothetical protein | 1wj9 | 23724 | 10.79 | 5.27 | (i) |
| I | TTHB029 | Conserved hypothetical protein | 2e67 | 29598 | 5.30 | 7.42 | (i) |
| J | TTHA0735 | Cytidine deaminase | 13280 | 7.16 | 11.11 | (i) | |
| K | TTHA1056 | Putative phosphoglucosamine mutase | 46968 | 6.04 | 19.25 | (i) | |
| L | TTHA0895 | Universal stress-protein family | 1wjg | 14756 | 5.14 | 20.02 | (i) |
| M | TTHA1699 | Conserved hypothetical protein | 2cx5 | 16555 | 9.51 | 10.99 | (i) |
| N | TTHA1281 | Conserved hypothetical protein | 2e6x | 8137 | 5.26 | 11.77 | (i) |
| O | TTHA0338 | Conserved hypothetical protein | 2cw5 | 26917 | 5.97 | 7.04 | (i) |
| P | TTHA0890 | Putative 3-hydroxyacyl-CoA dehydrogenase | 84291 | 5.56 | 37.26 | (i) | |
| Q | TTHA1969 | Chromosome-partitioning protein, ParB family | 29741 | 10.15 | 14.54 | (iii) | |
| R | TTHA1623 | Metallo--lactamase superfamily protein | 2z1n | 22299 | 5.03 | 9.44 | (i) |
Protein samples are arranged in decreasing order of crystallization success rate with the HTS-80 and sitting-drop VD (see Fig. 1 ▶).
(i) 20mM TrisHCl pH 8.0, 1mM dithiothreitol (DTT); (ii) 20mM TrisHCl pH 8.0, 1mM DTT, 150mM NaCl; (iii) 20mM TrisHCl pH 8.0, 1mM DTT, 500mM NaCl.
Table 2. Robots, methods and conditions for crystallization condition screening.
| HTS-80 sitting-drop VD | Crystal Finder hanging-drop VD | TERA modified MB | TOPAZ microfluidic FID | |
|---|---|---|---|---|
| Protein solution | 1.0l | 1.0l | 0.5l | 0.7nl |
| Crystallization solution | 1.0l | 1.0l | 0.5l | 2.0nl |
| Reservoir (l) | 100 | 200 | ||
| Oil (l) | 15† | |||
| Cell (m) | 700 100 10 | |||
| Temperature (K) | 293 | 293 | 291 | 293 |
| Humidity (%) | 60 | 90 | ||
| Crystallization plate | 96-well sitting-drop plate (Corning, 3773) | 50-well custom-made plate (Ishikawajima Inspection and Instrumentation) | Microwell minitray, 72 wells (Nunc, 438733) | Topaz 4.96 screening chip (Fluidigm, TPZ-M-4.96) |
7:3 paraffin oil:silicone oil.
2. Experimental methods
2.1. Protein samples
18 proteins which were overexpressed in Escherichia coli and purified in good yields were selected for crystallization condition screening from among T. thermophilus HB8 proteins (Table 1 ▶). Desalting and buffer exchange of protein samples were performed on a HiLoad 16/60 Superdex 75pg column (GE Healthcare Biosciences) equilibrated with 20 mM Tris–HCl pH 8.0. If a protein precipitated from this low ionic strength solution, 150 or 500 mM NaCl was added to improve the solubility. Finally, dithiothreitol was added to the protein solution to prevent protein degradation. The protein concentration was determined by measuring the absorbance at 280 nm (Kuramitsu et al., 1990 ▶). The buffer conditions used for each protein solution are shown in Table 1 ▶. The three-dimensional structures of 11 of the 18 proteins have been determined and submitted to the Protein Data Bank (PDB).
2.2. Crystallization condition screening
Each protein sample was screened against 384 cocktails from commercial crystallization screening kits (OptiMix-1, OptiMix-2, OptiMix-3 and OptiMix-PEG; Fluidigm Corp.) listed in Table S11. The screening period was set at nine weeks (63 d) for the robots (HTS-80, Crystal Finder and TERA). The protein drops were monitored six times, i.e. one day, one week, three weeks, five weeks, seven weeks and nine weeks after crystallization screening was started. The concentration rate for a protein drop (microbatch) with a TERA robot depends on the composition of the oil and the surrounding humidity. The conditions selected as the most suitable for crystallization condition screening were a 7:3 ratio of paraffin and silicone oil and 60% humidity (RIKEN, unpublished results). The screening period for the TOPAZ was two weeks because it was designed for rapid screening of initial crystallization conditions (Fluidigm Corp). The appearance of crystals was checked seven times, i.e. at the beginning and then 1, 2, 4, 7, 10 and 14 d after crystallization screening was started.
When a protein drop contains a high concentration of a salt, it is possible that not protein crystals but salt crystals grow in the drop. We thus examined salt crystallization for each of the three sets of buffer conditions, (i)–(iii), listed in Table 1 ▶. The examination method was the same as that used for protein crystallization condition screening except that the drops did not contain protein (Table 3 ▶).
Table 3. Salt crystallization in protein-free drops.
| Buffer (i) | Buffer (ii) | Buffer (iii) | |
|---|---|---|---|
| HTS-80 | |||
| No. of trials with salt crystal(s) | 1 | 4 | 1 |
| Average appearance time (d) | 1.0 | 9.5 | 1.0 |
| Crystal Finder | |||
| No. of trials with salt crystal(s) | 1 | 0 | 1 |
| Average appearance time (d) | 50.0 | 8.0 | |
| TERA | |||
| No. of trials with salt crystal(s) | 13 | 10 | 12 |
| Average appearance time (d) | 53.3 | 58.8 | 45.0 |
| TOPAZ | |||
| No. of trials with salt crystal(s) | 3 | 0 | 1 |
| Average of appearance time (d) | 14.0 | 0.0 |
2.3. Evaluation method for crystallization condition screening
Microscopic images of protein drops were automatically obtained and stored on a hard disk. The images were scored by eye with a four-point scale of 0–3. Score 0 indicates that no precipitate appeared in the protein drop. Score 1 indicates that a noncrystalline precipitate was observed in the drop. Score 2 indicates that crystals of less than 50 × 50 × 50 µm, needle crystals with a cross-section of less than 50 × 50 µm or plate-like crystals with a thickness of less than 50 µm were observed in the drop. Score 3 indicates that crystals of larger than 50 × 50 × 50 µm were observed in the drop. Crystals evaluated as score 3 would be suitable for X-ray diffraction studies and were tentatively defined as diffraction-quality crystals. The TOPAZ (microfluidic FID) with a cell volume of 700 × 100 × 10 µm is not able to produce score 3 crystals. A three-point scale, 0–2, was therefore used.
The crystallization success rate was defined as the ratio of the number of successful trials with score 2 or 3 crystals to the total number of trials (384). As an example, consider a screening that resulted in 20 drops with score 2 and ten with score 3 crystals. The crystallization success rate (%) was therefore (20 + 10)/384 × 100 = 7.8. The time of appearance of crystals was defined as the time it took to obtain score 2 crystals. As an example, in one screen it took 7 d to detect score 2 crystals and another 7 d to obtain crystals large enough to be score 3 crystals. The time of appearance in this case was 7 d.
3. Results and discussion
3.1. Crystallization success rate
Table 3 ▶ shows the salt-crystal growth in protein-free drops. Salt crystallization was examined by the same method as used for the screening of protein crystallization conditions, except that protein-free drops were used in place of protein drops (see §2.2). The numbers of trials with salt crystals for buffers (i)–(iii) were very low for the HTS-80 and Crystal Finder. The maximum number was four out of 384 trials (1.04%) for buffer (ii) and the HTS-80 (Table 1 ▶). Salt crystals with score 3 were not observed in any buffer. On the other hand, the rate of salt crystallization with the TERA was high compared with the other robots. The numbers of trials with salt crystals ranged from ten to 13, of which 7–10 were observed 7–9 weeks after crystallization screening was started.
The protein crystallization condition screening data obtained seven and nine weeks after crystallization screening was started were removed because salt crystallization in the drops could not be neglected. Fig. 1 ▶ shows the crystallization success rates for 18 proteins for 384 crystallization trials. The average crystallization success rates for the 18 protein samples for the HTS-80 (sitting-drop VD), Crystal Finder (hanging-drop VD) and TERA (modified MB) were 7.0%, 7.0% and 6.8%, respectively, i.e. they were nearly equal. With the TERA, the number of diffraction-quality crystals was three to four times higher than with the HTS-80 and Crystal Finder. The ratios of the number of trials with score 3 crystals (diffraction-quality crystals) to the number of successful trials with score 2 or 3 crystals were 13.7%, 16.9% and 53.5% for the HTS-80, Crystal Finder and TERA, respectively. In terms of diffraction-quality crystals, at least in this experiment, the TERA was the best, followed by the HTS-80 and Crystal Finder. It is important for X-ray studies that diffraction-quality crystals are easily obtainable in the initial screening, because score 2 crystals cannot always be optimized to diffraction-quality crystals. With the TOPAZ (microfluidic FID), the average crystallization success rate (11.3%) was 1.6 times higher than with the HTS-80, Crystal Finder and TERA. Therefore, the TOPAZ is most suitable for screening the initial crystallization conditions.
Figure 1.

Number of successful crystallization trials for 18 protein samples for an HTS-80 (sitting-drop VD), a Crystal Finder (hanging-drop VD), a TERA (modified MB) and a TOPAZ (microfluidic FID). Crystallization conditions were screened against 384 trials. Protein samples are arranged in decreasing order of successful crystallization trials with the HTS-80 in this figure.
3.2. Time crystals appeared
Fig. 2 ▶ shows the distribution of the average time of appearance for crystals. Comparison of Fig. 1 ▶ with Fig. 2 ▶ reveals that a protein sample exhibiting a high crystallization success rate tended to crystallize rapidly. The average time of appearance increased in the order HTS-80, Crystal Finder, TERA. The HTS-80 can reduce the time spent on crystallization condition optimization, because the time required for one cycle of optimization is considered to be short compared with Crystal Finder or TERA. The concentration speed for the TERA can be increased by using a high proportion of silicone oil and/or low-humidity conditions. However, this enhances the possibility of salt crystallization, making the conditions unsuitable for crystallization screening (RIKEN, unpublished results). Development of nanolitre crystallization techniques might make the time of appearance for the modified MB shorter without enhancing the salt crystallization in the protein drops (DeLucas et al., 2003 ▶; Cherezov et al., 2004 ▶). Crystals were produced most rapidly using the TOPAZ system, making it well suited for rapid screening of the initial crystallization conditions.
Figure 2.

Distribution of the average time of appearance of crystals for each of 18 protein samples. Protein samples are arranged in decreasing order of successful crystallization trials with the HTS-80 (see Fig. 1 ▶).
3.3. Proteins exhibiting low crystallization success rates
In the case of protein samples exhibiting low crystallization success rates, the number of crystallization conditions common to any two robots was extremely small compared with the total number of crystallization conditions specific to each robot (Fig. 3 ▶). The numbers of crystallization conditions common to the HTS-80 and Crystal Finder, common to the HTS-80 and TERA, common to the Crystal Finder and TERA, specific to the HTS-80, specific to the Crystal Finder and specific to the TERA were five, one, zero, 16, 19 and ten, respectively, when six protein samples exhibiting crystallization success rates of less than 1.6% were taken into consideration. The TOPAZ had no crystallization condition in common with the HTS-80, Crystal Finder or TERA.
Figure 3.
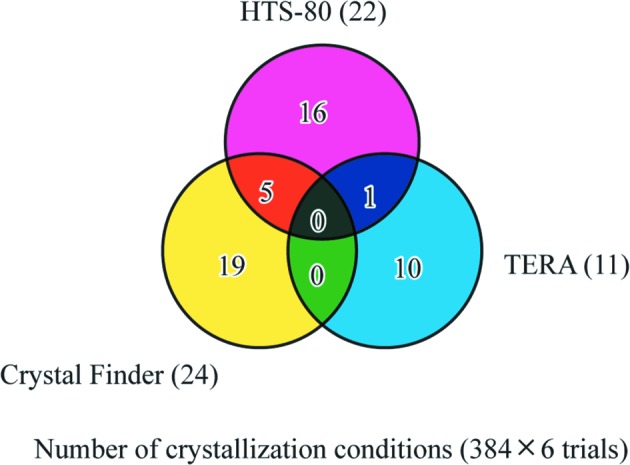
The number of successful crystallization trials common to robots and specific to each robot for proteins with low crystallization success rates. Six protein samples exhibiting crystallization success rates of less than 1.6% were selected for analysis of the successful trials. The numbers of successful trials specific to the HTS-80, Crystal Finder and TERA are shown in magenta, yellow and blue, respectively. Those common to the HTS-80 and Crystal Finder, to the HTS-80 and TERA and to the Crystal Finder and TERA are shown in orange, violet and green, respectively. Those common to the HTS-80, TERA and Crystal Finder are shown in black.
There were some cases in which the number of successful trials for each sample differed greatly depending on the robot or crystallization methods used. For example, the numbers of successful trials for TTHA0890 were four and zero for the HTS-80 (sitting-drop vapour diffusion) and Crystal Finder (hanging-drop vapour diffusion), respectively, while those for TTHA1969 were one and nine. These results indicated that the crystallization conditions were liable to be affected by the robots or crystallization methods used. The combined use of several robots or crystallization methods was thus considered to be favourable for crystallization condition screening in structural genomics work on T. thermophilus HB8.
4. Conclusion
We performed crystallization condition screening of 18 protein samples from T. thermophilus HB8 using conventional crystallization robots: an HTS-80 (sitting-drop VD), a Crystal Finder (hanging-drop VD) and a TERA (modified MB). A TOPAZ (microfluidic FID) was also subjected to a brief screening test. The number of successful crystallization trials with diffraction-quality crystals and the time of appearance of crystals increased in the order HTS-80, Crystal Finder, TERA. With the TERA, the number of diffraction-quality crystals was three to four times higher than with the HTS-80 or Crystal Finder, although it took a longer time for crystals to grow compared with the other robots. With the HTS-80 and Crystal Finder, most of the crystallization conditions were determined within 24 and 38 d, respectively, and the rate of salt crystallization in protein drops was less than 2%. For the protein samples exhibiting low crystallization success rates, there were few crystallization conditions that were common to the robots used. In some cases, the level of difficulty in crystallization depended greatly on the robot used. The TOPAZ, which is specialized for initial crystallization screening, is characterized by rapid crystallization of protein solutions and a significantly higher crystallization success rate than those of the conventional robots.
Based on the results obtained from this crystallization screening test, we conclude the following. The combined use of conventional robots employing different crystallization methods significantly increases the likelihood of identifying crystallization conditions, especially with samples that are difficult to crystallize. We have achieved a high success rate (38% of the purified proteins producing structures) by combining the HTS-80 and the TERA robots for crystallization screening.
Supplementary Material
Supporting information file. DOI: 10.1107/S1744309108013572/tt5010sup1.pdf
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: TT5010).
References
- Adams, M. W., Dailey, H. A., DeLucas, L. J., Luo, M., Prestegard, J. H., Rose, J. P. & Wang, B.-C. (2003). Acc. Chem. Res. 36, 191–198. [DOI] [PubMed]
- Au, K. et al. (2006). Acta Cryst. D62, 1267–1275. [DOI] [PubMed]
- Chayen, N. E. (1998). Acta Cryst. D54, 8–15. [DOI] [PubMed]
- Chayen, N. E. & Saridakis, E. (2008). Nature Methods, 5, 147–153. [DOI] [PubMed]
- Cherezov, V., Peddi, A., Muthusubramaniam, L., Zheng, Y. F. & Caffrey, M. (2004). Acta Cryst. D60, 1795–1807. [DOI] [PubMed]
- D’Arcy, A., Sweeney, A. M. & Haber, A. (2004). Methods, 34, 323–328. [DOI] [PubMed]
- DeLucas, L. J., Bray, T. L., Nagy, L., McCombs, D., Chernov, N., Hamrick, D., Cosenza, L., Belgovskiy, A., Stoops, B. & Chait, A. (2003). J. Struct. Biol. 142, 188–206. [DOI] [PubMed]
- Hansen, C. L., Skordalakes, E., Berger, J. M. & Quake, S. R. (2002). Proc. Natl Acad. Sci. USA, 99, 16531–16536. [DOI] [PMC free article] [PubMed]
- Hiraki, M. et al. (2006). Acta Cryst. D62, 1058–1065. [DOI] [PubMed]
- Hosfield, D., Palan, J., Hilgers, M., Scheibe, D., McRee, D. E. & Stevens, R. C. (2003). J. Struct. Biol. 142, 207–217. [DOI] [PubMed]
- Hui, R. & Edwards, A. (2003). J. Struct. Biol. 142, 154–161. [DOI] [PubMed]
- Kuramitsu, S., Hiromi, K., Hayashi, H., Morino, Y. & Kagamiyama, H. (1990). Biochemistry, 29, 5469–5476. [DOI] [PubMed]
- Kuramitsu, S., Kawaguchi, S. & Hiramatsu, Y. (1995). Protein Eng. 8, 964.
- Luft, J. R., Wolfley, J., Jurisica, I., Glasgow, J., Fortier, S. & DeTitta, G. T. (2001). J. Cryst. Growth, 232, 591–595.
- McPherson, A. (1999). Crystallization of Biological Macromolecules. New York: Cold Spring Harbor Laboratory Press.
- McPherson, A. (2004). Methods, 34, 254–265. [DOI] [PubMed]
- Miyatake, H., Kim, S.-H., Motegi, I., Matsuzaki, H., Kitahara, H., Higuchi, A. & Miki, K. (2005). Acta Cryst. D61, 658–663. [DOI] [PubMed]
- Saitoh, K., Kawabata, K., Asama, H., Mishima, T., Sugahara, M. & Miyano, M. (2005). Acta Cryst. D61, 873–880. [DOI] [PubMed]
- Santarsiero, B. D., Yegian, D. T., Lee, C. C., Spraggon, G., Gu, J., Scheibe, D., Uber, D. C., Cornell, E. W., Nordmeyer, R. A., Kolbe, W. F., Jin, J., Jones, A. L., Jaklevic, J. M., Schultz, P. G. & Stevens, R. C. (2002). J. Appl. Cryst. 35, 278–281.
- Segelke, B. (2005). Expert Rev. Proteomics, 2, 165–172. [DOI] [PubMed]
- Shah, A. K., Liu, Z.-J., Stewart, P. D., Schubot, F. D., Rose, J. P., Newton, M. G. & Wang, B.-C. (2005). Acta Cryst. D61, 123–129. [DOI] [PubMed]
- Stevens, R. C. (2000). Curr. Opin. Struct. Biol. 10, 558–563. [DOI] [PubMed]
- Sulzenbacher, G. et al. (2002). Acta Cryst. D58, 2109–2115. [DOI] [PubMed]
- Walter, T. S. et al. (2005). Acta Cryst. D61, 651–657. [DOI] [PMC free article] [PubMed]
- Yokoyama, S., Hirota, H., Kigawa, T., Yabuki, T., Shirouzu, M., Terada, T., Ito, Y., Matsuo, Y., Kuroda, Y., Nishimura, Y., Kyogoku, Y., Miki, K., Masui, R. & Kuramitsu, S. (2000). Nature Struct. Biol. 7, Suppl, 943–945. [DOI] [PubMed]
- Yokoyama, S., Matsuo, Y., Hirota, H., Kigawa, T., Shirouzu, M., Kuroda, Y., Kurumizaka, H., Kawaguchi, S., Ito, Y., Shibata, T., Kainosho, M., Nishimura, Y., Inoue, Y. & Kuramitsu, S. (2000). Prog. Biophys. Mol. Biol. 73, 363–376. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information file. DOI: 10.1107/S1744309108013572/tt5010sup1.pdf