

In this paper, the 1.2 Å atomic resolution crystal structure of the 5-nitroimidazole antibiotic resistance protein NimA from Deinococcus radiodurans (DrNimA) is presented.
Keywords: NimA, Deinococcus radiodurans, antibiotic resistance proteins
Abstract
Many anaerobic human pathogenic bacteria are treated using 5-nitroimidazole-based (5-Ni) antibiotics, a class of inactive prodrugs that contain a nitro group. The nitro group must be activated in an anaerobic one-electron reduction and is therefore dependent on the redox system in the target cells. Antibiotic resistance towards 5-Ni drugs is found to be related to the nim genes (nimA, nimB, nimC, nimD, nimE and nimF), which are proposed to encode a reductase that is responsible for converting the nitro group of the antibiotic into a nonbactericidal amine. A mechanism for the Nim enzyme has been proposed in which two-electron reduction of the nitro group leads to the generation of nontoxic derivatives and confers resistance against these antibiotics. The cofactor was found to be important in the mechanism and was found to be covalently linked to the reactive His71. In this paper, the 1.2 Å atomic resolution crystal structure of the 5-nitroimidazole antibiotic resistance protein NimA from Deinococcus radiodurans (DrNimA) is presented. A planar cofactor is clearly visible and well defined in the electron-density map adjacent to His71, the identification of the cofactor and its properties are discussed.
1. Introduction
Antibiotic resistant bacteria have become a significant health problem throughout the developed world. After the treatment of bacterial infections with similar types of antibiotics for a several decades some bacteria have also become multi-drug resistant, resulting in a limited number of options for the choice of treatment drug. Knowledge of different resistance mechanisms is therefore important for efficient treatment of bacterial infections in the future as well as for the development of new drug treatments.
Many anaerobic bacterial infections are treated with 5-nitroimidazole (5-Ni) drug derivatives (Edwards, 1993a ▶,b ▶; Rafii et al., 2003 ▶; Fang et al., 2002 ▶), including metronidazole (MTZ), tinidazole (TNZ) and dimetridazole (DMZ) amongst others. 5-Ni drugs are used in multi-drug therapies against the human pathogen Helicobacter pylori (Houben et al., 1999 ▶), a spiral bacterium that colonizes the stomach; infections caused by H. pylori are associated with severe gastrointestinal disease, including peptic ulceration. Metronidazole and other 5-nitroimidazole-related drugs are also used to treat Trichomonas vaginalis, an amitochondriate flagellate which causes human urogenital trichomoniasis (Hrdy et al., 2005 ▶). Metronidazole is used for treating anaerobic infections caused by Bacteroides spp., a dominant bacterium in normal intestinal microflora. The B. fragilis group are opportunistic pathogens that are among the most clinically relevant anaerobic species. Interspecies lateral transfer of resistance genes among anaerobes have been shown, e.g. for the clinically important drugs β-lactam, clindamycin, tetracycline and metronidazole (see Löfmark et al., 2005 ▶, and references therein).
In the B. fragilis group, eight nim genes (nimA–F) have been reported to be involved in reduced susceptibility to 5-nitroimidazole antibiotics (Haggoud et al., 1994 ▶; Stubbs et al., 2000 ▶; Trinh & Reysset, 1996 ▶; Gal & Brazier, 2004 ▶) and have a sequence identity between them of 67% or greater (Fig. 1 ▶). The gene products are suggested to be 5-nitroimidazole reductases (Carlier et al., 1997 ▶). Four of the nim genes are associated with mobile insertion-sequence (IS) elements, posing a threat to the existing applications of 5-Ni drugs. Indeed, one study identified seven resistant Bacteroides strains with minimum inhibitory concentrations of >32 µg ml−1, all of which contained nim genes (Jamal et al., 2004 ▶). Even if the rate of 5-Ni resistance is still low, organisms with metronidazole resistance are beginning to emerge (Haggoud et al., 1994 ▶; Löfmark et al., 2005 ▶).
Figure 1.

Sequence alignment of DrNimA (D. radiodurans NimA; TrEMBL entry Q9RW27), BvNimA (B. vulgatus NimA; Q45801), BfNimB (B. fragilis NimB; Q45146), BtNimC (B. thetaiotaomicron NimC; Q45778), BfNimD (B. fragilis NimD; Q45150), BfNimE (B. fragilis NimE; Q9L4E6), BfNimE2 (B. fragilis NimE; Q4LAM4) and BvNimF (B. vulgatus NimF; Q8GJ54). The marked residues are the reactive His71 in addition to Pro56 and Tyr111 in the MTZ-binding site (Leiros et al., 2004 ▶) of DrNimA.
The 5-Ni drugs are inactive prodrugs that enter the cells by simple diffusion and are further reduced in a one-electron reduction by ferredoxin (Fd) to the toxic short-lived radical anion R—N·O2 − (Lockerby et al., 1985 ▶; Kulda, 1999 ▶; Goodwin et al., 1998 ▶). Fd obtains the relevant electron from the pyruvate:ferredoxin oxidoreductase (PFOR) complex when pyruvate and coenzyme A are converted into acetyl-coenzyme A (Ragsdale, 2003 ▶). The activation of the 5-Ni drugs is therefore dependent on the redox system in the target cell (Kwon et al., 2000 ▶), in which the toxic radicals causes DNA-helix destabilization, DNA-strand breaks, DNA unwinding and cell death (Edwards, 1993a ▶,b ▶; Declerck & De Ranter, 1986 ▶; Declerck et al., 1983 ▶) and damage to other vital cell systems is also possible (Kulda, 1999 ▶). The redox potential of MTZ is very low (E′0 = −480 mV; Sisson et al., 2002 ▶) and possible electron donors are therefore limited. In anaerobic organisms, ferredoxin-like Fe–S proteins typically have a redox potential of −430 to −460 mV, making the organisms susceptible to MTZ (Kwon et al., 2000 ▶). The lowest redox potential aerobic organisms can offer is from NAD- or NADP-containing proteins and is in the order of −320 to −324 mV; these organisms are intrinsically MTZ-resistant (Kwon et al., 2000 ▶).
Different resistance mechanisms have been reported for 5-Ni drugs, in particular MTZ, that are related, for example, to the reduced amount of available ferredoxin in T. vaginalis (Quon et al., 1992 ▶); in H. pylori the inactivation of rdxA, an oxygen-insensitive NADPH nitroreductase (Marais et al., 2003 ▶), frxA, an NAD(P)H flavin reductase, or fdxB, a ferredoxin-like protein (Kwon et al., 2000 ▶), caused resistance. H. pylori strains with an intact rdxA gene were resistant to 5-Ni drugs, suggesting that several pathways are involved (Jenks et al., 1999 ▶). In B. fragilis, 5-Ni resistance was found to be related to the nim genes (nimA–F; Gal & Brazier, 2004 ▶; Breuil et al., 1989 ▶; Haggoud et al., 1992 ▶, 1994 ▶; Trinh et al., 1995 ▶), which encode a 5-Ni reductase (Carlier et al., 1997 ▶). The structural genomics project at the European Synchrotron Radiation Facility on the extremely radiation-resistant bacterium Deinococcus radiodurans (Mattimore & Battista, 1996 ▶) has produced several crystal structures (Leiros & McSweeney, 2007 ▶; Leiros, Moe et al., 2005 ▶; Leiros, Timmins et al., 2005 ▶; Moe et al., 2006 ▶; Timmins et al., 2005 ▶, 2007 ▶; Meunier-Jamin et al., 2004 ▶; Romao et al., 2006 ▶). The crystal structure of NimA from D. radiodurans (DrNimA) has been elucidated and complexes of DrNimA with (i) pyruvate (native), (ii) pyruvate and MTZ, (iii) covalently linked pyruvate and (iv) covalently linked lactate have been obtained (Leiros et al., 2004 ▶). From these structures, a catalytic mechanism was proposed in which the cofactor played an essential role.
In the previously published 1.6 Å structure (Leiros et al., 2004 ▶) of DrNimA this cofactor was interpreted as pyruvate. In order to obtain further details and characteristics of the cofactor and its binding site, higher resolution X-ray data were collected and the atomic resolution 1.2 Å structure of native DrNimA is presented here with a particular focus on the cofactor. The structure is compared with the B. vulgatus NimA (BvNimA) sequence, which has 28% sequence identity and 54% similarity to DrNimA (Fig. 1 ▶).
2. Experimental procedures
2.1. Purification of DrNimA
The DrNimA gene construct contains the Gateway destination vector pDEST17 (Invitrogen) with a 21-residue His tag (MSYYHHHHHHLESTSLYKKAG) followed by the 195-residue DrNimA protein (entry Q9RW27). The protein was recombinantly expressed in a manner similar to that described in Leiros et al. (2004 ▶), by expression in BL21 Star(DE3)pLysS cells at 310 K to an OD600 of about 1; the cells were then cooled and induced at 293 K overnight with 1 mM isopropyl β-d-1-galactopyranoside (IPTG). The harvested cells were lysed in a French press, centrifuged and the supernatant containing the soluble DrNimA protein was purified in two steps. The first step used a His-Trap Ni2+ column; the fractions containing pure DrNimA were pooled and dialysed against a buffer consisting of 25 mM Tris pH 7.5, 25 mM NaCl and 5 mM β-mercaptoethanol. In the next step, the protein was applied onto a MonoQ anion-exchange column (Amersham Bioscience) in a buffer consisting of 50 mM Tris–HCl pH 7.5, 10 mM NaCl, 5 mM β-mercaptoethanol and eluted with 1 M NaCl in the same buffer. The protein eluted in two peaks, both of which were pure according to SDS–PAGE gels, but only the main peak was used in the crystallization trials.
2.2. Crystallization, data collection and refinement
The DrNimA protein was crystallized at 277 K as rosettes of plate-shaped crystals using the hanging-drop technique with reservoir solution containing 0.6 M sodium acetate and 0.1 M MES [2-(N-morpholino)ethanesulfonic acid] pH 6. The drops were made by mixing 1 µl reservoir solution and 1 µl protein solution at 6.5 mg ml−1 and the crystals reached final dimensions of about 250 × 150 × 40 µm. These plates were substantially thicker than those previously obtained giving the 1.6 Å native data (Leiros et al., 2004 ▶), which is likely to explain their excellent diffraction properties. The thicker and better diffracting crystals might have been obtained as a consequence of the slightly lower salt concentration, the lower pH, the slightly higher protein concentration and/or the smaller drop size (2 µl compared with the previous 4 µl). The data were collected on beamline ID23-EH1 at ESRF using a MAR CCD 225 detector, with a wavelength of 0.9999 Å, 30% beam transmission of a uniform beam with maximum intensity of 200 mA, 1 s exposure time, three passes, 1° oscillation and 252° of data. The images were integrated with MOSFLM (Leslie, 1992 ▶) and scaled with SCALA and the structure factors were obtained with TRUNCATE (Collaborative Computational Project, Number 4, 1994 ▶). 3% (2004 in total) of the reflections were used in R free cross-validation. Data-collection statistics are given in Table 1 ▶.
Table 1. Data-collection statistics.
Values in parentheses are for the highest of ten resolution shells; the resolution limits for this shell are indicated.
| PDB code | 2vpa |
| Beamline | ID23-EH1 |
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 100.04, b = 38.95, c = 59.90, β = 114.36 |
| Resolution (Å) | 20–1.20 (1.26–1.20) |
| Wavelength (Å) | 0.9999 |
| No. of unique reflections | 65 888 |
| Multiplicity | 5.1 (5.0) |
| Completeness (%) | 100.0 (100.0) |
| Mean 〈I〉/〈σ(I)〉 | 10.2 (2.1) |
| Rmerge (%)† | 8.9 (61.4) |
| Wilson B factor (Å2) | 8.16 |
R
merge = 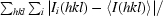
 , where I
i(hkl) is the ith measurement of reflection h and 〈I(hkl)〉 is the weighted mean of all measurements of h.
, where I
i(hkl) is the ith measurement of reflection h and 〈I(hkl)〉 is the weighted mean of all measurements of h.
The new high-resolution native DrNimA structure was refined by rigid-body optimization from the native 1.6 Å structure (PDB code 1w3o) followed by positional refinement with REFMAC5 (Murshudov et al., 1999 ▶) and manual building in O (Jones et al., 1991 ▶). The protein, the ten residues in the His tag, all water molecules, the acetate and the cofactor were all refined anisotropically throughout the refinement. Double conformations were added manually in O. Water molecules were added in REFMAC5 with the arp_waters option when above 3.5σ in the F o − F c Fourier difference map and then inspected manually in O. Refinement statistics are given in Table 2 ▶.
Table 2. Refinement statistics for the presented structures.
| R factor† (%) | 14.2 |
| Rfree† (%) | 18.0 |
| No of Rfree reflections | 2004 (3%) |
| No. of protein atoms | 1750 |
| No. of water molecules | 405 |
| No. of other molecules | 1 pyruvate, 1 acetate |
| R.m.s.d. bond lengths (Å) | 0.020 |
| R.m.s.d. bond angles (°) | 1.736 |
| Average B factors (Å2) | |
| All atoms | 13.15 |
| Protein (residues 2–195) | 10.46 |
| Water molecules | 23.69 |
| Acetate | 11.05 |
| Pyruvate | 25.22 |
| Ramachandran plot | |
| Most favoured region (%) | 93.6 |
| Additionally allowed regions (%) | 6.4 |
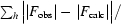
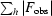 , where |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes for all reflections (R factor) or the reflections used in the test R
free set (reflections not used in the structure refinement).
, where |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes for all reflections (R factor) or the reflections used in the test R
free set (reflections not used in the structure refinement).
2.3. Mass spectrometry
The molecular weight of DrNimA was analyzed with a Q-TOF Ultima Global (Micromass) mass-spectrometry instrument. The molecular weight was predicted using the MaxEnt1 software. The soluble protein was dialyzed against 20 mM NaCl overnight and spun down and the supernatant was used. The sample was then dissolved in 50% acetonitrile with 0.1% formic acid prior to injection.
2.4. Cofactor identification
A good impression of the cofactor adjacent to His71 was seen in the OMIT density maps and several small molecules were placed in the density and run through several cycles of positional refinement. The resulting Fourier difference maps and B factor of the cofactors were then inspected and evaluated.
3. Results
3.1. Overall structure
The final 1.2 Å DrNimA model was refined anisotropically to an R factor of 14.2%, an R free of 18.0% and an overall B factor of 13.2 Å2. The final model comprises protein residues 2–195, an acetate ion, the cofactor, 405 water molecules and ten residues in the 21-residue His tag. There are 12 double conformations: residues 16, 25, 30, 80, 84, 111, 122, 138, 166, 168, 183 and 192. The root-mean-square deviation between the high-resolution (1.2 Å) and medium-resolution (1.6 Å) DrNimA structures was 0.118 Å as calculated with the Secondary Structure Matching database (http://www.ebi.ac.uk/msd-srv/ssm/); some rearrangements were found for residues Ser2, Asp3, Phe4, Asn81 and Gly83. There is one molecule in the asymmetric unit and the crystallographic twofold axis forms the functional dimer shown in Fig. 2 ▶. DrNimA was found to be a dimer in solution by performing analytical gel-filtration and cross-linking experiments (Leiros et al., 2004 ▶).
Figure 2.

Overall structure of a DrNimA dimer showing the cofactor, acetate (Act), the His tag (residues SerH1–LeuH10), His71 and some additional residues. The MTZ molecule as found in the crystal structure of DrNimA with MTZ and pyruvate (PDB code 1w3r; Leiros et al., 2004 ▶) is indicated with grey C atoms.
Inside the β-barrel one acetate ion is found to be in close contact with the guanido group of Arg79 and the side chains of His86 and Tyr70 (Fig. 2 ▶). None of these residues are conserved in other Nim proteins (see Fig. 2 in Leiros et al., 2004 ▶) or in Bacteroides sp. NimA–F as shown in Fig. 1 ▶, indicating that the acetate ion probably has no functional importance even if it is stabilizing the neighbouring residue to the reactive His71. His71 was determined to be reactive because it was found to be covalently bound to mercury when obtaining the experimental phases of DrNimA (Leiros et al., 2004 ▶) and His71 was also found to be covalently linked to both pyruvate (PDB code 1w3p) and lactate (PDB code 1w3q) as reported previously (Leiros et al., 2004 ▶). The fold and dimeric organization of DrNimA is the same as in pyridoxine 5′-phosphate oxidase (PNPO; Musayev et al., 2003 ▶), in which the active site of PNPO overlaps with the His71 and MTZ-binding site of DrNimA. His71 is suggested to be the active-site residue of DrNimA; it is conserved in several NimA sequences and its location is on the same side of the β-barrel as in other electron-transfer enzymes (Leiros et al., 2004 ▶).
The main peak predicted from the Q-TOF MS results as elucidated by the MaxEnt1 software was 24 279 Da, which is 139 Da lighter than the theoretical MW of DrNimA with a His tag of 24 418 Da. We then suspect that the first Met residue, with a MW of 132 Da, in the His tag is not present in the recombinantly produced protein and is possibly cleaved off during protein synthesis in Escherichia coli.
In DrNimA, ten of the residues in the His tag (SerH2–LeuH11) are visible in the electron-density map; they form an α-helix and are included in the final PDB file. After residue Ser2 in the protein the tag goes through a water channel with 12 undefined residues as indicated by the dotted lines in Fig. 2 ▶ until LeuH11; the following residues are in contact with symmetry-related molecules. The amino group of SerH2 forms ion pairs to Asp24 and Asp185 from two different symmetry-related protein molecules. This observation also supports the mass-spectrometry results that the first Met residue in the tag is absent, making SerH2 the true positively charged N-terminus. There are also several other water-mediated hydrogen bonds between the His tag and symmetry-related molecules.
3.2. The cofactor
At the end of the refinement, after adding double conformations and all the water molecules, the solvent molecules adjacent to His71 were removed from the model and the resulting OMIT electron-density maps are shown in Figs. 3 ▶(a) and 3 ▶(b). Here, it is clear that a flat cofactor is present in the structure: it seems to have a two-atom ‘base’ with four legs. Several small molecules were fitted to this OMIT electron density and refined, but the planar pyruvate seemed to be the most reasonable and was therefore kept as in the medium-resolution (1.6 Å) DrNimA structure (PDB code 1w3o). The cofactor was refined with an occupancy of 1.0, except for the O1 atom, which has an occupancy of 0.75. The mean B factor is 25.2 Å2, whereas the neighbouring residues have a mean B value for their side chains of 10.3 Å2 for His71, 7.8 Å2 for Phe140 and 11.6 Å2 for Phe98′. Since the adjacent residues have lower B values, the true occupancy of the pyruvate cofactor is probably lower than 1. Rerunning the refinement and reducing the occupancy for all atoms in the pyruvate molecule suggested an occupancy close to 0.5 in order to obtain B values more similar to those of the surrounding residues. Furthermore, O1 could also be a lighter atom than oxygen as judged from the reduced occupancy.
Figure 3.

The 1.2 Å resolution OMIT electron-density map (F o − F c) in the active site of DrNimA at 3.5σ (red) and 2.5σ (green): (a) a ‘front’ view and (b) a ‘side’ view. The model of the refined pyruvate is included in (a) and adjacent residues and water molecules are also shown. (c) Final 2F o − F c electron-density maps of His71 and pyruvate at 1.3σ (blue) and the difference maps at +3.5σ (green) and −3.5σ (red) in which His71 and pyruvate are not visible in this area of the structure. (d) Side view of (c). Adjacent residues, interatomic distances and the metronidazole (MTZ; grey C atoms) from PDB entry 1w3r are outlined in the various panels.
The assignment of a pyruvate to this moiety seems reasonable because when the O1 atom has an occupancy of 0.75 there is no difference electron density observed (Figs. 3 ▶ c and 3 ▶ d). The O1 atom of the pyruvate carbonyl group is 2.34 Å away from His71 NE2; this is a possible ion-pair interaction if both groups are charged and this might explain their short interatomic distance. All three O atoms (O1, O2, O3) are hydrogen bonded to five water molecules (W1, W2, W3, W5, W6). Finally, the hydrophobic methyl group on the pyruvate is facing Phe140 and Phe98′ and is therefore found in a hydrophobic environment.
BvNimA has been found to be a reductase (Carlier et al., 1997 ▶) and from the four previous DrNimA structures a mechanism has been proposed (Leiros et al., 2004 ▶). In the native state (PDB code 1w3o) the pyruvate ion was hydrogen bonded to the conserved His71 (Fig. 1 ▶) and upon MTZ binding (PDB code 1w3r) the cofactor–His71 hydrogen bond became shorter. A modified His71-pyruvate residue was then observed (PDB code 1w3p), with the pyruvate covalently linked to His71, a possible oxidation product of His and pyruvate, with the release of two electrons and a proton. The two released electrons are then available for reducing the antibiotic prodrug, where a two-electron reduction (R—NO2 → R—N=O) prevents the accumulation of the toxic nitro radical (R—N·O2 −).
In the fourth structure lactate was found to be covalently linked to His71 (PDB code 1w3q), where lactate can be formed by reducing pyruvate, also consuming two protons and two electrons. For the latter reaction possible electron donors were discussed but not identified. DrNimA was found to perform both an oxidation and a reduction of the cofactor, both of which are needed for a reductase enzyme to perform consecutive rounds of catalysis. His71 was proved to be reactive and the cofactor was important in the proposed mechanism and was therefore studied further in this paper. In the target cell the inactive prodrugs are activated in a one-electron reduction, but in the proposed two-electron reduction by the Nim enzyme the drugs become nontoxic and therefore confirm antibiotic resistance in these enzymes. Pyruvate is used by other enzymes; it can be synthesized by anaerobic bacteria (Ragsdale, 2003 ▶) and might then also be available for the Nim enzymes.
4. Conclusions
The 1.2 Å structure of DrNimA clearly shows OMIT electron density corresponding to a planar cofactor closely linked to the reactive His71. The crystallographic refinement supported pyruvate as being compatible with this cofactor, with satisfactory polar environments for the three O atoms and hydrophobic environments for the methyl group. The potentially negatively charged carboxyl group of pyruvate is bound to the side chain of His71 with a short interatomic distance of 2.34 Å. Further biochemical analysis will be necessary to confirm whether pyruvate is the true cofactor; however, in our hands DrNimA batches prepared under different conditions all contain a cofactor with similar geometry and chemical coordination, suggesting that the protein has a strong cofactor preference. Further characteristics of the cofactor are being elucidated by more extensive electrospray mass spectrometry and biophysical experiments on DrNimA.
Supplementary Material
PDB reference: NimA, 2vpa, r2vpasf
Acknowledgments
CT is grateful to the ESRF for providing financial support as a visiting scientist in the Macromolecular Crystallography Group. Dr Jack-Ansgar Bruun is gratefully acknowledged for help in performing the mass spectrometry experiments. This work was made possible by the facilities offered by the Partnership for Structural Biology in Grenoble.
References
- Breuil, J., Dublanchet, A., Truffaut, N. & Sebald, M. (1989). Plasmid, 21, 151–154. [DOI] [PubMed]
- Carlier, J. P., Sellier, N., Rager, M. N. & Reysset, G. (1997). Antimicrob. Agents Chemother.41, 1495–1499. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Declerck, P. J. & De Ranter, C. J. (1986). Biochem. Pharmacol.35, 59–61. [DOI] [PubMed]
- Declerck, P. J., De Ranter, C. J. & Volckaert, G. (1983). FEBS Lett.164, 145–148. [DOI] [PubMed]
- Edwards, D. I. (1993a). J. Antimicrob. Chemother.31, 9–20. [DOI] [PubMed]
- Edwards, D. I. (1993b). J. Antimicrob. Chemother.31, 201–210. [DOI] [PubMed]
- Fang, H., Edlund, C., Hedberg, M. & Nord, C. E. (2002). Int. J. Antimicrob. Agents.19, 361–370. [DOI] [PubMed]
- Gal, M. & Brazier, J. S. (2004). J. Antimicrob. Chemother.54, 109–116. [DOI] [PubMed]
- Goodwin, A., Kersulyte, D., Sisson, G., Veldhuyzen van Zanten, S. J., Berg, D. E. & Hoffman, P. S. (1998). Mol. Microbiol.28, 383–393. [DOI] [PubMed]
- Haggoud, A., Reysset, G., Azeddoug, H. & Sebald, M. (1994). Antimicrob. Agents Chemother.38, 1047–1051. [DOI] [PMC free article] [PubMed]
- Haggoud, A., Reysset, G. & Sebald, M. (1992). FEMS Microbiol. Lett.74, 1–5. [DOI] [PubMed]
- Houben, M. H., van de Beek, D., Hensen, E. F., Craen, A. J., Rauws, E. A. & Tytgat, G. N. (1999). Aliment. Pharmacol. Ther.13, 1047–1055. [DOI] [PubMed]
- Hrdy, I., Cammack, R., Stopka, P., Kulda, J. & Tachezy, J. (2005). Antimicrob. Agents Chemother.49, 5033–5036. [DOI] [PMC free article] [PubMed]
- Jamal, W. Y., Rotimi, V. O., Brazier, J. S., Johny, M., Wetieh, W. M. & Duerden, B. I. (2004). Med. Princ. Pract.13, 147–152. [DOI] [PubMed]
- Jenks, P. J., Ferrero, R. L. & Labigne, A. (1999). J. Antimicrob. Chemother.43, 753–758. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Kulda, J. (1999). Int. J. Parasitol.29, 199–212. [DOI] [PubMed]
- Kwon, D. H., El-Zaatari, F. A., Kato, M., Osato, M. S., Reddy, R., Yamaoka, Y. & Graham, D. Y. (2000). Antimicrob. Agents Chemother.44, 2133–2142. [DOI] [PMC free article] [PubMed]
- Leiros, H.-K. S., Kozielski-Stuhrmann, S., Kapp, U., Terradot, L., Leonard, G. A. & McSweeney, S. M. (2004). J. Biol. Chem.279, 55840–55849. [DOI] [PubMed]
- Leiros, H.-K. S. & McSweeney, S. M. (2007). J. Struct. Biol.159, 92–102. [DOI] [PubMed]
- Leiros, I., Moe, E., Smalås, A. O. & McSweeney, S. (2005). Acta Cryst. D61, 1049–1056. [DOI] [PubMed]
- Leiros, I., Timmins, J., Hall, D. R. & McSweeney, S. (2005). EMBO J.24, 906–918. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Lockerby, D. L., Rabin, H. R. & Laishley, E. J. (1985). Antimicrob. Agents Chemother.27, 863–867. [DOI] [PMC free article] [PubMed]
- Löfmark, S., Fang, H., Hedberg, M. & Edlund, C. (2005). Antimicrob. Agents. Chemother.49, 1253–1256. [DOI] [PMC free article] [PubMed]
- Marais, A., Bilardi, C., Cantet, F., Mendz, G. L. & Megraud, F. (2003). Res. Microbiol.154, 137–144. [DOI] [PubMed]
- Mattimore, V. & Battista, J. R. (1996). J. Bacteriol.178, 633–637. [DOI] [PMC free article] [PubMed]
- Meunier-Jamin, C., Kapp, U., Leonard, G. A. & McSweeney, S. (2004). J. Biol. Chem.279, 25830–25837. [DOI] [PubMed]
- Moe, E., Leiros, I., Smalås, A. O. & McSweeney, S. (2006). J. Biol. Chem.281, 569–577. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. (1999). Acta Cryst. D55, 247–255. [DOI] [PubMed]
- Musayev, F. N., Di Salvo, M. L., Ko, T.-P., Schirch, V. & Safo, M. K. (2003). Protein Sci.12, 1455–1463. [DOI] [PMC free article] [PubMed]
- Quon, D. V., d’Oliveira, C. E. & Johnson, P. J. (1992). Proc. Natl Acad. Sci. USA, 89, 4402–4406. [DOI] [PMC free article] [PubMed]
- Rafii, F., Wynne, R., Heinze, T. M. & Paine, D. D. (2003). FEMS Microbiol. Lett.225, 195–200. [DOI] [PubMed]
- Ragsdale, S. W. (2003). Chem. Rev.103, 2333–2346. [DOI] [PubMed]
- Romao, C. V., Mitchell, E. P. & McSweeney, S. (2006). J. Biol. Inorg. Chem.11, 891–902. [DOI] [PubMed]
- Sisson, G., Goodwin, A., Raudonikiene, A., Hughes, N. J., Mukhopadhyay, A. K., Berg, D. E. & Hoffman, P. S. (2002). Antimicrob. Agents Chemother.46, 2116–2123. [DOI] [PMC free article] [PubMed]
- Stubbs, S. L., Brazier, J. S., Talbot, P. R. & Duerden, B. I. (2000). J. Clin. Microbiol.38, 3209–3213. [DOI] [PMC free article] [PubMed]
- Timmins, J., Leiros, H.-K. S., Leonard, G., Leiros, I. & McSweeney, S. (2005). J. Mol. Biol.347, 949–963. [DOI] [PubMed]
- Timmins, J., Leiros, I. & McSweeney, S. (2007). EMBO J.26, 3260–3271. [DOI] [PMC free article] [PubMed]
- Trinh, S., Haggoud, A., Reysset, G. & Sebald, M. (1995). Microbiology, 141, 927–935. [DOI] [PubMed]
- Trinh, S. & Reysset, G. (1996). J. Clin. Microbiol.34, 2078–2084. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: NimA, 2vpa, r2vpasf