

The 1.95 Å resolution X-ray crystal structure of tunicamycin-resistance protein (TmrD) from D. radiodurans is described.
Keywords: tunicamycin resistance, phosphotransferases, Deinococcus radiodurans
Abstract
The open-reading frame (ORF) DR_1419 in the Deinococcus radiodurans genome is annotated as a representative of the wide family of tunicamycin-resistance proteins as identified in a range of bacterial genomes. The D. radiodurans ORF DR_1419 was cloned and expressed; the protein TmrD was crystallized and its X-ray crystal structure was determined to 1.95 Å resolution. The structure was determined using single-wavelength anomalous diffraction with selenomethionine-derivatized protein. The refined structure is the first to be reported for a member of the tunicamycin-resistance family. It reveals strong structural similarity to the family of nucleoside monophosphate kinases and to the chloramphenicol phosphotransferase of Streptomyces venezuelae, suggesting that the mode of action is possibly by phosphorylation of tunicamycin.
1. Introduction
Tunicamycin (Tn) is an antibiotic that was first discovered in the early 1970s (Takatsuki et al., 1971 ▶). The compound is composed of a uracil base, the sugar tunicamine, N-acetylglucosamine and a fatty acid (Fig. 1 ▶). The antibiotic functions by preventing protein glycosylation and specifically inhibits the formation of dolichol-linked oligosaccharides destined for eventual use in a variety of glycoconjugates. Tunicamycin-resistant strains of Bacillus subtilis have been identified (Nomura et al., 1978 ▶) and the gene, tmrB, that confers resistance has been sequenced (Harada et al., 1988 ▶) and encodes a protein of around 22 kDa. TmrB-like sequences have now been identified in a number of bacterial genomes; the sequences share a conserved Walker A ATP-binding signature, GXXXXGKT/S (Walker et al., 1982 ▶), close to the N-terminus. The exact function of the TmrB protein is unknown, although studies have shown the B. subtilis protein to be an ATP-binding membrane protein (Noda et al., 1992 ▶) using a hypothetical 18-residue C-terminal amphiphilic helix to bind to the membrane. The same team also showed the protein to directly bind tunicamycin (Noda et al., 1995 ▶) and demonstrated tunicamycin resistance to be conferred on a previously sensitive mutant of Escherichia coli when the gene was expressed in the mutant (Noda et al., 1996 ▶). Based on their results, Noda and coworkers suggested that the bacterial protein could act to lower the intracellular concentration of Tn by either blocking the uptake system or working as an efflux pump in its own right (Noda et al., 1992 ▶, 1995 ▶).
Figure 1.

The antibiotics tunicamycin (a) and chloramphenicol (b). In chloramphenicol, the red circle indicates the known phosphorylation location; in tunicamycin, as deduced from the X-ray structure presented in this paper, the red, orange and green circles indicates the most likely sites for phosphorylation.
The genome of the highly radiation-resistant bacterium Deinococcus radiodurans contains an open reading frame DR_1419 (Q9RUG7, CMR; http://cmr.jcvi.org) which codes for a protein of 194 amino acids (21.6 kDa; referred to in this paper as TmrD) excluding the initiating methionine (White et al., 1999 ▶). The TIGR automated annotation suggests that the gene product is an antibiotic resistance protein, although Makarova et al. (2001 ▶) further identify the protein as a tunicamycin-resistance protein and a predicted ATPase, sharing a high sequence identity (ClustalW score of 35 using default settings, corresponding to 38% identity over 184 aligned residues; Larkin et al., 2007 ▶) with the B. subtilis TmrB protein. As part of the the ESRF’s structural genomics initiative on D. radiodurans and to shed light on the fold and function of the family of tunicamycin-resistance proteins, we undertook the determination of its three-dimensional X-ray crystal structure. Here, we report the 1.95 Å resolution crystal structure of a truncated form of TmrD, the first for a member of the tunicamycin-resistance family of proteins.
2. Experimental
2.1. Protein purification and crystallization
The gene DR_1419 was cloned from genomic D. radiodurans DNA into the Gateway destination vector pDest17 (Gateway, Invitrogen) by the company Protein’eXpert (Grenoble, France). The correct sequence with an N-terminal hexahistidine tag (sequence MSYYHHHHHHLESTSLYKKAG) was confirmed by DNA sequencing.
E. coli BL21(DE3)pLysS cells (Novagen) transformed with pDEST17-DR_1419 were grown at 310 K and 200 rev min−1 in LB medium with 50 mg l−1 ampicillin and 34 mg l−1 chloramphenicol. At an OD600 of 0.6, the cells were induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 3 h at 310 K. The harvested cells were resuspended in lysis buffer (10 mM sodium phosphate pH 7.5, 500 mM NaCl, 10 mM imidazole pH 7.5, 10 mM MgCl2, DNaseI, lysozyme, one Mini Complete EDTA-free protease-inhibitor cocktail tablet; Roche) and lysed by sonication. The soluble lysate was applied onto a 5 ml nickel-loaded HisTrap column (GE Healthcare). The protein was eluted with a linear gradient of imidazole from 10 to 500 mM, dialysed against 50 mM imidazole pH 7.5 and concentrated to no greater than 3 mg ml−1. Initial screening for suitable crystallization conditions was carried out by the hanging-drop method using standard commercial screening solutions both manually and using the Cartesian robot of the High Throughput Crystallization Laboratory of the EMBL Grenoble Outstation. In these initial trials, clusters of thin plate-like crystals were obtained in 10–14% PEG 4000, 0.8 M sodium formate, 0.1 M sodium acetate pH 4.5.
In order to improve crystal quality and because degradation of the protein to a stable fragment of weight ∼20 kDa was observed over two weeks whilst stored at 277 K limited proteolysis of the protein with α-chymotrypsin (Sigma, 5 mg ml−1 stock solution, 1/500 ratio) was performed. The stable fragment after 30 min digestion was purified and identified by molecular-weight determination using electrospray-mass spectrometry and N-terminal sequencing (performed by the Institute Biologie Structurale, Grenoble, France) as the fragment corresponding to residues 18–205 of the His-tagged DR_1419 protein. This corresponds to a truncation of most of the N-terminal His tag and 18 C-terminal residues,
This fragment was cloned into the pDEST17 vector (RoBioMol platform, IBS, Grenoble) with a cleavable His-TEV tag (sequence MAHHHHHHGHHHQLENLYFQGKKAGL, including the five residues of the original hexahistidine tag remaining after proteolysis), transformed into E. coli BL21(DE3)pLysS cells (Novagen) and the cells were grown and the protein was purified as for the full-length protein. The yield (70 mg l−1 of culture) and the solubility (20 mg ml−1) of the truncated protein were significantly increased compared with the full-length protein (15 mg l−1 of culture and 4 mg ml−1, respectively). The selenomethionine-substituted protein, the structure of which is described here, was then produced using established protocols (Van Duyne et al., 1993 ▶) and purified in a similar manner, with the exception that 10 mM β-mercaptoethanol was added to the buffers that were used in the procedure. The yield was slightly less than for the truncated native protein. The pure protein fraction, with uncleaved tag, was then concentrated to 5 mg ml−1 and stored at 253 K for use in crystallization.
The best crystals were obtained at 293 K after 3–6 d using hanging drops containing 2 µl protein solution, 0.4–0.8 µl 0.1 M CdCl2 and 1.6–1.2 µl of a reservoir solution containing 11–13% PEG 4000, 0.8 M sodium formate and 0.1 M sodium acetate pH 5.0.
To determine the nature of the protein in solution, size-exclusion analysis of the tagged protein was performed using a Superdex200 column (calibrated with known protein standards) in 10 mM Tris pH 7.5, 200 mM NaCl.
Henceforth, in this paper all residue numbering refers to the native TmrD sequence without the initiating methionine residue.
2.2. X-ray data acquisition, structure determination and refinement
Crystals were immersed for less than 5 s in cryoprotectant buffer consisting of reservoir solution and 25%(v/v) glycerol and cryocooled in a nitrogen-gas stream at 100 K. All data were recorded on beamline ID23-1 (Nurizzo et al., 2006 ▶) at the ESRF (Grenoble, France) using an ADSC 315 (Area Detector Systems Corporation, USA) CCD-based detector whilst the storage ring was operated in uniform fill mode (maximum 200 mA stored current). Following a fluorescence scan, single-wavelength anomalous diffraction (SAD) data for phasing were collected from a single crystal of selenomethionine-substituted protein at the peak of the Se K absorption edge (‘peak’ data set; wavelength 0.979 Å). Higher resolution data for refinement were then collected from a second crystal of selenomethionine-substituted protein (‘refinement’ data set). Crystals of the full-length protein with His tag gave diffraction data to no better than 2.8 Å resolution and eventual molecular replacement of the truncated structure against this data also revealed a truncated structure; these ‘full-length’ protein crystals were therefore not used further.
The data were integrated and reduced using MOSFLM (Leslie, 1992 ▶) and SCALA and TRUNCATE from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶; Table 1 ▶). Observations were rejected during scaling if they deviated by more than five standard deviations from the mean. The crystals belonged to space group C2221. In both crystals, the asymmetric unit accommodates two molecules of molecular weight 22.6 kDa (including the tag residues and with the C-terminal truncation), corresponding to V M values of 1.98 and 2.16 Å3 Da−1 (Matthews, 1968 ▶) and solvent contents of 38 and 43% for the ‘peak’ and ‘refinement’ crystals, respectively.
Table 1. Data-collection and phasing statistics.
Values in parentheses are for the highest resolution shell.
| Data set | ‘Refinement’ | ‘Peak’ |
|---|---|---|
| X-ray source | Beamline ID23-1, ESRF | |
| Wavelength (Å) | 0.914 | 0.979 |
| No. of images | 160 | 600 |
| Oscillation angle (°) | 0.8 | 0.7 |
| Exposure time per image (s) | 0.4 | 0.7 |
| Beamline attenuators set for transmission level (%) | 100 | 10 |
| Space group | C2221 | C2221 |
| Unit-cell parameters (Å) | a = 81.4, b = 118.2, c = 81.1 | a = 81.6, b = 118.3, c = 74.1 |
| Resolution range (Å) | 23–1.95 (2.00–1.95) | 25–2.10 (2.15–2.10) |
| No. of unique reflections | 28689 | 21346 |
| Completeness (%) | 99.5 (100.0) | 100.0 (100.0) |
| Rmerge† (%) | 9.2 (52.0) | 9.9 (52.0) |
| Multiplicity | 4.9 (5.1) | 16.5 (17.0) |
| I/〈σ(I)〉 | 14.5 (3.2) | 30.4 (5.2) |
| Wilson B factor (Å2) | 19.9 | 22.8 |
| Figure of merit (acentric/centric)‡ | — | 0.43 (0.25)/0.18 (0.17) |
| Phasing power | — | 1.53 (0.55) |
R
merge = 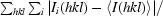
 , where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of I(hkl) for all i measurements.
, where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of I(hkl) for all i measurements.
Figure of merit after phase calculation in SHARP and before density modification.
Using the SAD data, SHELXD (Sheldrick, 2008 ▶) identified ten heavy-atom positions corresponding to nine Se atoms out of an expected ten (excluding the tag) and one cadmium ion, which were used as input for autoSHARP (Vonrhein et al., 2006 ▶). The heavy-atom positions were subsequently refined and phases were calculated using SHARP (de La Fortelle & Bricogne, 1997 ▶; Table 2 ▶), which was followed by solvent flattening using SOLOMON (Abrahams & Leslie, 1996 ▶). The resulting electron-density map was of very good quality. The structure factors and phases were used as input to ARP/wARP (Perrakis et al., 1999 ▶), which successfully constructed 279 residues out of an expected total of 352. This structure was used for refinement against the higher resolution data (starting with five cycles of rigid-body refinement). Subsequent iterative cycles of manual construction using Coot (Emsley & Cowtan, 2004 ▶) and maximum-likelihood refinement using REFMAC5 (Murshudov et al., 1997 ▶) gave a final model of high quality (Fig. 2 ▶) consisting of two monomers of TmrD, 226 water molecules, one cadmium ion and one chloride ion. Data-collection and structural refinement statistics are presented in Tables 1 ▶ and 2 ▶.
Table 2. Structure-refinement statistics.
| Resolution range | 23–1.95 |
| No. of protein atoms | 2572 |
| No. of cadmium/chloride ions | 1/1 |
| No. of water molecules | 226 |
| Rcryst† (%) (No. of reflections) | 21.0 (27186) |
| Rfree† (%) (No. of reflections) | 25.7 (1486) |
| R.m.s.d. bonds (Å) | 0.020 |
| R.m.s.d. angles (°) | 2.16 |
| Average B factor (Å2) | 25.3 |
| Residues not modelled | Monomer A/B, tag residues, 177–183, 1–3; monomer A, 4, 15–18, 126–129 |
| Residues in multiple conformations | B123, B138 |
| Ramachandran plot, nonglycine/proline residues | |
| In most favoured regions | 267 (97.1%) |
| In additionally allowed regions | 8 (2.9%) |
R
cryst = 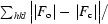
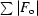 , where F
o is the observed structure-factor amplitude and F
c is the calculated structure-factor amplitude. R
free is calculated based on 5% of reflections not used in refinement.
, where F
o is the observed structure-factor amplitude and F
c is the calculated structure-factor amplitude. R
free is calculated based on 5% of reflections not used in refinement.
Figure 2.

Stereo diagram showing a section of the final TmrD structure (corresponding to the central β-sheet) in the final 2F obs − F calc electron-density map contoured at 1.3σ. The diagram was generated using PyMOL (http://www.pymol.org).
3. Results and discussion
3.1. Structure of TmrD
The refined model comprises two monomers in the asymmetric unit: monomer A consists of residues 5–14, 19–125 and 130–176 and monomer B of residues 4–176. None of the purification-tag residues were observed in the electron-density map and they are therefore assumed to be disordered. The two monomers are highly similar, with a root-mean-square deviation (r.m.s.d.) of 0.95 Å (main-chain atoms) between the superposed monomers. The areas of greatest difference occur in helices α7 and α9 (see Fig. 3 ▶ a for helix/strand labelling), although the overall fold is identical. The model also includes a single cadmium ion and chloride ion located at the twofold axis between the two monomers, which are almost certainly from the crystallization buffer, and 226 water molecules.
Figure 3.

(a) Stereo ribbon diagrams of the TmrD monomer showing the overall structure with the β-strands and α-helices labelled. The blue oval shows the LID domain (ATP binding in adenylate kinase and CPT), the yellow oval the Tnbind domain (equivalent to the AMPbind and chloramphenicol-binding domains in adenylate kinase and CPT, respectively) and the purple oval the CORE domain. (b) Topology schematic for the TmrD monomer (circles indicate helices; triangles indicate strands). Purple indicates the central sheet/helix CORE domain of the protein and blue and yellow the two pillar-like extensions forming the sides to the cleft as the LID and Tnbind domains, respectively. The topology diagram was generated with the help of the TOPS server (Westhead et al., 1998 ▶). (c) Stereo ribbon diagram of the TmrD dimer as identified by PISA. The interface helices α2 and α3 are labelled. The ribbon pictures were generated using PyMOL.
The TmrD monomer structure consists of a central five-stranded parallel β-sheet with topology β2↑-β3↑-β1↑-β4↑-β5↑ flanked by seven α-helices on each face of the sheet (Figs. 3 ▶ a and 3 ▶ b). The size-exclusion analysis showed the protein to be a dimer in solution. However, the two monomers of the asymmetric unit do not appear to form the physiological dimer. Instead, PISA (Krissinel & Henrick, 2007 ▶) identified the dimer interface with a complexation significance score of 1.0 between monomer A and the symmetry-related monomer B (symmetry operator: −x + 1/2, y − 1/2, −z + 1/2). This interface gives a total buried surface of 2005 Å2 out of a total of 18 227 Å2, with most of the contacts made between α2 and α3 (Fig. 3 ▶ c) including the formation of a salt bridge between Glu39 (monomer A) and Lys46 (monomer B). The overall shape of the TmrD molecule consists of a core globular region formed by the sheet and most of the helices, but with a large cleft formed by two pillar-like extensions from the core by α2 (one extension) and α5 and α6 (second extension). The cleft is around 15 Å deep by 12 Å wide and contains the conserved Walker A sequence at its base. The 18 C-terminal residues truncated from our TmrD more or less correspond to the predicted amphiphilic helix of TmrB. The C-terminus of our truncated TmrD structure is located at the base of the core globular region away from the pillars and near to the C-terminal end of α1; the truncated residues could form a helix running antiparallel to α1 or act as a membrane anchor (Noda et al., 1992 ▶).
Interestingly, monomer A has a significantly higher average B factor (28.1 Å2 for the protein atoms) compared with monomer B (23.8 Å2) and this is also reflected in the identification of fewer water molecules around monomer A as well as by the two regions of missing residues in the chain. In the crystal, monomer B makes an interlocking contact with the symmetry-related monomer B′ mostly through interactions between residues 120–131 and their symmetry equivalents. For monomer A no such equivalent interaction takes place. These contacts act to stabilize amino acids 15–18 and 126–129, which were not located in monomer A.
3.2. Comparison to structural homologues and functional prediction
All structural comparisons were carried out using monomer B since this represents the most complete model of the TmrD monomer.
A search of the PDB using SSM (Krissinel & Henrick, 2004 ▶, 2005 ▶) found that TmrD was most similar to PDB entry 1nks, adenylate kinase from Sulfolobus acidocaldarius (Vonrhein et al., 1998 ▶), with 2.43 Å r.m.s.d. (main-chain atoms) over 148 matched residues, and the series of entries, 1qhn/1qhx/1qhy and 1grq/1grr, for chloramphenicol phosphotransferase (CPT) from Streptomyces venezuelae (Izard & Ellis, 2000 ▶; Izard, 2001 ▶), with an average 2.69 Å r.m.s.d. (main-chain atoms) over 146 matched residues (Fig. 4 ▶). The folds of the three proteins are very similar (Fig. 5 ▶), although both TmrD and CPT lack the extended β-sheet loop between β3 and α4 that is present in adenylate kinase. TmrD also lacks a similar β-sheet loop extending over the active site in CPT from its equivalent of the ‘AMPbind’ domain (see below). In the apo TmrD structure determined here, these differences result in a more open and exposed cleft between the two pillars described above. Sequence comparisons of TmrD with the adenylate kinase and CPT using ClustalW (with default settings) give scores of just 8 (corresponding to 18% identity over 177 matched residues) and 5 (corresponding to 19% identity over 178 matched residues), respectively. However, the strong structural similarities together with sequence conservation of the Walker A nucleotide-binding sequence suggest a role for TmrD as a phosphotransferase. CPT provides an alternative pathway for S. venezuelae to avoid inhibition by one of its own products, chloramphenicol, which in most other bacteria is achieved by chloramphenicol acetyltransferases. CPT inactivates chloramphenicol by forming the 3′-phospho ester (Fig. 1 ▶) using ATP as the phosphoryl donor (Mosher et al., 1995 ▶).
Figure 4.

Sequence alignment of TmrD, TmrB and CPT. The alignment of TmrD and TmrB was made using ClustalW, while the alignment of TmrD and CPT was made using SSM with the TmrD structure and the apo CPT structure (PDB code 1qhn); the figure was produced using ESPript (Gouet et al., 1999 ▶). The residues marked with asterisks are the conserved Walker A sequence. Boxed residues show identity. For the CPT sequence, residues with an orange background make hydrogen-bond contacts via their side chain to the ATP and those with a green background make hydrogen-bond contacts via their main chain to the ATP; the residue with a red background is the catalytic residue. For the TmrD sequence, the same colour scheme indicates the residues that are hypothesized to make similar contacts for the phosphorylation of tunicamycin. Also for TmrD, triangles mark the Tnbind domain and circles the LID domain.
Figure 5.

Stereo diagrams (in the same orientation as in Fig. 3a ▶) showing the structural superposition of TmrD in blue, apo CPT in magenta (PDB code 1qhn) and adenylate kinase in green (PDB code 1nks; nucleotide-free monomer E). The adenylate kinase extended β-sheet loop can be clearly seen in green in the centre of the upper images, as can the CPT extension in magenta over the active site. The structural superposition was made using SSM and the diagrams were generated with PyMOL.
TmrD contains one cis-peptide link between Gln148 and Thr149, allowing a sharp turn to be made into β5. Neither CPT nor adenylate kinase possess the equivalent cis-peptide: CPT has a two-residue insertion at this point and adenylate kinase has a much larger amino-acid insertion that extends both the β-sheet and helix.
In light of the structural similarity, the characteristic nucleoside monophosphate kinase (NMK) domains (Schulz et al., 1990 ▶) CORE, LID (binding ATP in the case of both NMK and TmrD) and AMPbind (binding AMP in the case of NMK and binding tunicamycin in our case; Tnbind) can easily be identified in the TmrD structure (Fig. 3 ▶ a). In NMKs, the LID and AMPbind domains are known to undergo considerable induced-fit structural movement during the catalytic cycle from the apoenzyme to bound substrates and product release (see, for example, Schulz et al., 1990 ▶; Vonrhein et al., 1995 ▶). In the structure presented here no substrates are bound, probably leaving the TmrD structure with ‘open’ conformations of these two domains. Indeed, compared with the various states of the subunits in the adenylate kinase (1nks) crystal structure, the TmrD structure shows an open LID domain (similar to the nucleotide-free subunit E of 1nks) and a Tnbind domain that is between the open and closed conformations of the adenylate kinase structure (between the open subunit E and the closed subunit F with both AMP and ATP bound). The CPT structures determined by Izard & Ellis (2000 ▶) do not show significant changes in conformation between the apo form and the structures with substrates bound; this was suggested to be a consequence of the binding of a sulfate ion to the ATP site in the apo form which may have resulted in some of the induced-fit movements being replicated in that crystal form. Izard and Ellis reported circular-dichroism experiments showing that large changes in secondary structure occurred on binding sulfate or ATP but not chloramphenicol (Izard & Ellis, 2000 ▶). The TmrD structure reported here may thus represent a closer analogue of the true apo form of CPT, showing a more open form, particularly of the LID domain, compared with the CPT structures.
Experiments to cocrystallize tunicamycin with TmrD failed and soaking crystals with tunicamycin resulted in very rapid crystal cracking and deterioration. Trials to find alternative crystallization conditions in the presence of both tunicamycin and nonhydrolysable ATP (ATPγS) were also not successful. Given the relatively large size of tunicamycin (molecular weight of ∼850 Da), these results are perhaps not surprising. In the crystal form used in this work, the likely binding location of tunicamycin is blocked in monomer B by the symmetry-related Arg124 and Asp125. This is not the case in monomer A, where the crystal lattice could just allow sufficient space for the antibiotic to bind, although significant movement of the protein would be required to bind tunicamycin, which might explain the crystal cracking observed in the soaking experiments.
Therefore, despite the expected induced-fit movements on binding ATP and/or tunicamycin, the CPT structure with chloramphenicol bound (PDB code 1qhy) and the structure of uridine (from the single-crystal structure of uridine determined by Green et al., 1975 ▶) were used to model the uracil-ribose component of tunicamycin in our TmrD structure (Fig. 6 ▶). The structural alignment and modelling show that a tunicamycin molecule could be bound and oriented in the active site such that Glu38 could be the essential catalytic residue equivalent to Asp37 of CPT, which functions as a general base to extract a proton from the chloramphenicol hydroxyl group to be phosphorylated prior to nucleophilic attack on the ATP. Under these conditions, a phosphate could be transferred from ATP to hydroxyl1 or hydroxyl2 of the tunicamycin ribose ring (Fig. 1 ▶). Either of the hydroxyl moieties could be the target of phosphorylation, depending upon the exact orientation adopted by tunicamycin in the active site. Indeed, hydroxyl3 may also be susceptible if tunicamycin adopts a conformation other than that of the single-crystal structure; rotation around the uracil-ribose bond could place hydroxyl3 closer to Glu38. Tunicamycin is a far larger molecule than chloramphenicol but the modelling indicates that the lengthy tunicamine sugar, N-acetylglucosamine and fatty-acid moieties would naturally be directed out of the active site to the enzyme surface, where they may form positive interactions around the area of β2 and α2.
Figure 6.

Stereo diagram of the hypothetical TmrD active site with the uracil-ribose component of tunicamycin modelled using the crystal structures of CPT and uridine. The TmrD Cα trace is shown in red and the CPT Cα trace is shown in blue. Several of the residues pertinent to the CPT reaction are shown (indicated with a C after the residue name and green C atoms) together with their TmrD structural equivalents (indicated with a T after the residue name and grey C atoms). ATP is shown as bound in CPT (from PDB entry 1qhx aligned with TmrD using SSM). Since TmrD is in the ‘open’ conformation, it is difficult to model ATP as it might be bound in TmrD. The movements of the LID domain and of the Tnbind domain required to go from the open to closed conformation can be easily observed, with Glu38 T moving in towards the uracil and Arg124 T and Gln127 T moving in towards the ATP. The figure was generated using PyMOL.
Other active-site residues important to phosphotransferase activity are structurally conserved when compared with CPT: the Walker A signature components Gly12 (Gly10 in CPT), Gly17 (Gly15) and Lys18 (Lys15), which would coordinate the phosphoryl group to be transferred, and Thr19 (Ser17), which would coordinate a magnesium ion, and also Arg124 (Arg133), which would coordinate an ATP phosphoryl group. Gln127 could be the equivalent of Arg136 of CPT in stabilizing the ATP phosphoryl groups, although Arg121 could achieve the same result with a small shift of α5.
3.3. Tunicamycin-resistance protein-family sequences
The first ten results from a sequence-similarity search with BLAST (Altschul et al., 1997 ▶) using the D. radiodurans TmrD sequence as the search target are all annotated as either tunicamycin-resistance proteins or antibiotic resistance proteins. The next five hits are described as ATP- or GTP-binding proteins. To see where the greatest sequence conservation occurs in the tunicamycin-resistance family and to which structural elements the sequences correspond, a multiple alignment of the first ten sequences and TmrD was made using ClustalW (Supplementary Fig. 1 ▶ 1). The first 50 residues form the most conserved region, which contains the first β-strand (β1) of the CORE domain, the GXXXXGKT sequence (starting from residue 12 in TmrD) of the characteristic Walker A ATP-binding motif and the TmrDbind domain (equivalent to the AMPbind domain in NMKs). This first section also contains Glu38, which is proposed to be the catalytic residue that functions as a general base in TmrD and is conserved in all 11 sequences apart from that of B. subtilis, which contains a glutamine. This glutamine could not act as an effective general base to abstract a proton from the tunicamycin hydroxyl group prior to phosphorylation. However, with the likely structural rearrangement of TmrD on binding substrates, it is also possible that Glu36 (an aspartate residue in all of the other ten sequences) could be the general base. The other regions with highest conservation correspond to the central β-strands (β3 and β4) of the CORE domain. Arg121, part of the LID domain and suggested above as being able to stabilize the ATP phosphoryl groups, is conserved in all of the aligned sequences.
The alignments show the Streptomyces avermitilis (a free-living soil bacterium used commercially to produce compounds for human and veterinary medicine) sequence to be unusual in the tunicamycin-resistance protein family in having an insertion of 15 or so residues between α5 and α6, which form the ATP-binding LID domain. The LID domain is likely to undergo conformational changes upon binding ATP and these additional residues may therefore have a strong bearing over some of the reaction steps. The sequence from Oceanobacillus iheyensis (an alkaliphilic and extremely halotolerant organism) is also unusual in that it is shorter than all the others, terminating just after the LID-domain and therefore lacking β5 and α7 (which wrap around one side of the monomer, but are not involved in the active site, substrate binding or dimer formation) and the putative C-terminal membrane anchor.
4. Conclusion
The sequence similarity of TmrD to the B. subtilis TmrB sequence and the clear structural similarity between the TmrD, CPT and adenylate kinase structures suggest that the tunicamycin-resistance proteins are themselves phosphotransferases, probably inactivating tunicamycin by phosphate transfer to one of the hydroxyls on the ribose ring of the uracil nucleotide of tunicamycin. Further studies need to be performed in order to confirm this hypothesis.
Supplementary Material
PDB reference: TmrD, 2vli, r2vlisf
Acknowledgments
The authors gratefully acknowledge Marjolaine Noirclerc-Savoye and Benoit Gallet (RoBioMol) and the protein-analysis team at the Institut de Biologie Structurale for the construction and testing of the protein-expression system and mass-spectrometry services and the High Throughput Crystallization Laboratory of the EMBL Grenoble Outstation for initial screening of crystallization conditions. The Norwegian Structural Biology Centre (NorStruct) is supported by a grant from the National Program in Functional Genomics (FUGE) of the Research Council of Norway. Work for this project was carried out in the laboratories of the Partnership for Structural Biology.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: WD5095).
References
- Abrahams, J. P. & Leslie, A. G. W. (1996). Acta Cryst. D52, 30–42. [DOI] [PubMed]
- Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997). Nucleic Acids Res.25, 3389–3402. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. (1999). Bioinformatics, 15 305–308. [DOI] [PubMed]
- Green, E. A., Rosenstein, R. D., Shiono, R., Abraham, D. J., Trus, B. L. & Marsh, R. E. (1975). Acta Cryst. B31, 102–107.
- Harada, S., Yoda, K., Mori, M., Tanimoto, A., Furusato, T., Yamene, K., Takatsuki, A., Yamasaki, M. & Tamura, G. (1988). Agric. Biol. Chem.52, 1785–1789.
- Izard, T. (2001). Protein Sci.10, 1508–1513. [DOI] [PMC free article] [PubMed]
- Izard, T. & Ellis, J. (2000). EMBO J.19, 2690–2700. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2005). CompLife 2005, edited by M. R. Berthold, R. Glen, K. Diederichs, O. Kohlbacher & I. Fischer, pp. 67–78. Berlin, Heidelberg: Springer-Verlag.
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol.372, 774–797. [DOI] [PubMed]
- La Fortelle, E. de & Bricogne, G. (1997). Methods Enzymol.276, 472–494. [DOI] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Leslie, A. G. W. (1992). Jnt CCP4/ESF–EACBM Newsl. Protein Crystallogr.26
- Makarova, K. S., Aravind, L., Wolf, Y. I., Tatusov, R. L., Minton, K. W., Koonin, E. V. & Daly, M. J. (2001). Microbiol. Mol. Biol. Rev.65, 44–79. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Mosher, R. H., Camp, D. J., Yang, K., Brown, M. P., Shaw, W. V. & Vining, L. C. (1995). J. Biol. Chem.270, 27000–27006. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Noda, Y., Takatsuki, A., Yoda, K. & Yamasaki, M. (1995). Biosci. Biotechnol. Biochem.59, 321–322. [DOI] [PubMed]
- Noda, Y., Urakawa, I., Yoda, K. & Yamasaki, M. (1996). J. Gen. Appl. Microbiol.42, 343–347.
- Noda, Y., Yoda, K., Takatsuki, A. & Yamasaki, M. (1992). J. Bacteriol.174, 4302–4307. [DOI] [PMC free article] [PubMed]
- Nomura, S., Yamane, K., Sasaki, T., Yamasaki, M., Tamura, G. & Maruo, B. (1978). J. Bacteriol.136, 818–821. [DOI] [PMC free article] [PubMed]
- Nurizzo, D., Mairs, T., Guijarro, M., Rey, V., Meyer, J., Fajardo, P., Chavanne, J., Biasci, J.-C., McSweeney, S. & Mitchell, E. (2006). J. Synchrotron Rad.13, 227–238. [DOI] [PubMed]
- Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Nature Struct. Biol.6, 458–463. [DOI] [PubMed]
- Schulz, G. E., Muller, C. W. & Diederichs, K. (1990). J. Mol. Biol.231, 627–630. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Takatsuki, A., Arima, K. & Tamura, G. (1971). J. Antibiot.24, 215–223. [DOI] [PubMed]
- Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1993). J. Mol. Biol.229, 105–124. [DOI] [PubMed]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2006). Methods Mol. Biol.364, 215–230. [DOI] [PubMed]
- Vonrhein, C., Bonisch, H., Schafer, G. & Schulz, G. E. (1998). J. Mol. Biol.282, 167–179. [DOI] [PubMed]
- Vonrhein, C., Schlauderer, G. J. & Schulz, G. E. (1995). Structure, 3, 483–490. [DOI] [PubMed]
- Walker, J. E., Saraste, M., Runswick, M. J. & Gay, N. J. (1982). EMBO J.1, 945–951. [DOI] [PMC free article] [PubMed]
- Westhead, D. R., Hatton, D. C. & Thornton, J. M. (1998). Trends Biochem. Sci.23, 35–36. [DOI] [PubMed]
- White, O. et al. (1999). Science, 286, 1571–1577. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: TmrD, 2vli, r2vlisf