

Abstract
The fibroblast growth factor (FGF) 19 subfamily of ligands, FGF19, FGF21, and FGF23, function as hormones that regulate bile acid, fatty acid, glucose, and phosphate metabolism in target organs through activating FGF receptors (FGFR1–4). We demonstrated that Klotho and βKlotho, homologous single-pass transmembrane proteins that bind to FGFRs, are required for metabolic activity of FGF23 and FGF21, respectively. Here we show that, like FGF21, FGF19 also requires βKlotho. Both FGF19 and FGF21 can signal through FGFR1–3 bound by βKlotho and increase glucose uptake in adipocytes expressing FGFR1. Additionally, both FGF19 and FGF21 bind to the βKlotho-FGFR4 complex; however, only FGF19 signals efficiently through FGFR4. Accordingly, FGF19, but not FGF21, activates FGF signaling in hepatocytes that primarily express FGFR4 and reduces transcription of CYP7A1 that encodes the rate-limiting enzyme for bile acid synthesis. We conclude that the expression of βKlotho, in combination with particularFGFR isoforms, determines the tissue-specific metabolic activities of FGF19 and FGF21.
The FGF192 subfamily of ligands, which consists of FGF15 (the mouse ortholog of human FGF19), FGF19, FGF21, and FGF23, has emerged as a novel group of endocrine factors that regulate diverse metabolic processes in adulthood (1). FGF15/19 expression is induced upon feeding in intestinal epithelial cells in response to bile acid released into the intestinal lumen. FGF15/19 then acts on hepatocytes to reduce bile acid synthesis through suppressing transcription of CYP7A1, which encodes the rate-limiting enzyme for bile acid synthesis (2). FGF15/19 also acts on the gall bladder and stimulates its filling with bile. Thus, FGF15/19 functions as an essential component in a postprandial negative feedback loop for bile acid synthesis and release (2, 3). Conversely, FGF21 expression is induced upon fasting in the liver (4, 5). FGF21 secreted from the liver then acts on adipose tissues to induce metabolic adaptation to fasting. Specifically, FGF21 stimulates lipolysis in adipocytes to release fatty acids, which in turn converted to ketones in the liver. FGF21 was originally identified as a hormone that stimulates glucose uptake in adipocytes (6). However, unlike insulin, FGF21 reduces fat storage because it stimulates lipolysis (4, 5). The FGF23 gene was identified as the gene mutated in patients with autosomal dominant hypophosphatemic rickets (7). Autosomal dominant hypophosphatemic rickets patients carry a missense mutation in the FGF23 gene that confers resistance to proteolytic inactivation of FGF23 protein, resulting in increased blood FGF23 levels. Because of its inhibitory activity on phosphate reabsorption and vitamin D biosynthesis in the kidney, high FGF23 blood levels in autosomal dominant hypophosphatemic rickets patients result in phosphate wasting and defects in bone mineralization (8–11).
The recent crystallographic analysis shows that the topology of the heparin-binding regions of FGF19 and FGF23 diverges completely from that of canonical paracrine-acting FGFs, which reduces the affinity of these ligands for heparin/heparan sulfate (12, 13). The weak heparin binding affinity of the FGF19 family members enables them to avoid being captured in extracellular matrices and thus to function as endocrine factors. On the other hand, this weak heparin binding activity reduces the capacity of heparin/heparan sulfate to promotes direct interaction between FGFs and FGFRs (14). Indeed, attempts to demonstrate a direct interaction between FGFRs and the FGF19 family proteins in vitro have failed. These observations imply that FGF19 subfamily members require additional cofactors, besides heparin/heparan sulfates, to stably bind to their cognate FGFRs in their target tissues.
We and others identified the Klotho protein as a cofactor necessary for FGF23 binding to FGFRs and for efficient activation of FGF signaling (15, 16). The klotho gene was originally identified in mice as an aging-suppressor gene that extends life span when overexpressed and accelerates the development of aging-like phenotypes when disrupted (17, 18). The klotho gene encodes a single-pass transmembrane protein and is expressed in limited tissues, most notably in the distal convoluted tubules in the kidney (17). The Klotho protein physically interacts with FGFR1c, 3c, and 4 as well as with FGF23 itself (14) to stabilize FGF23-FGFR interactions. Forced expression of Klotho conferred responsiveness to FGF23 upon various cell types (15). The fact that Klotho is essential for efficient activation of FGF signaling by FGF23 may explain why Klotho-deficient mice and FGF23-deficient mice show many overlapping phenotypes, including hyperphosphatemia, hypervitaminosis D, and multiple aging-like symptoms (19, 20). Furthermore, we showed that βKlotho, a Klotho family member protein, functions as a cofactor necessary for FGF21 binding to FGFRs and effective activation of FGF signaling (21). βKlotho shares 41% amino acid identity with Klotho and is expressed in adipose tissue, liver, and pancreas (22). βKlotho also physically interacts with multiple FGFRs and significantly increases the affinity of FGF21 for the FGFRs in a manner similar to FGF23, Klotho, and FGFRs (21).
Mice deficient in βKlotho expression have overlapping phenotypes with mice deficient in FGF15 or FGFR4 expression, including increased bile acid synthesis and increased hepatic expression of CYP7A1 and CYP8B1, which encode two key enzymes involved in bile acid synthesis (2, 23, 24). Thus, the genetic evidence strongly suggests that FGF15/19, FGFR4, and βKlotho are essential components in the negative regulation of bile acid synthesis. In this report, we provide molecular and cellular evidence indicating that FGF19, like its subfamilymember FGF21, requires βKlotho to stably bind to FGFRs and effectively activate FGF signaling. The salient difference between FGF19 and FGF21 activities lies in their distinct receptor binding specificity and the tissue-specific distribution of their cognate receptors. These findings provide new insights into the mechanism by which the FGF19 subfamily of ligands exert distinct metabolic activities in different target organs.
MATERIALS AND METHODS
Expression Vectors
Expression vectors for βKlotho and FGFRs designed with a V5 epitope tag at their C terminus were generated by polymerase chain reaction of cDNAs as described previously (15, 21).
Cell Culture and Transfection
Culture of the mouse 3T3-L1 preadipocytes and induction of adipocyte differentiation were described previously (21). Human embryonic kidney cells (HEK293), rat hepatoma cells (H4IIE), and rat myoblastic cells (L6) (American Type Culture Collection, Manassas, VA) were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and penicillin/streptomycin. Transfection of expression vectors was performed 36 h prior to the experiments using Lipofectamine (Invitrogen) and FuGENE HD (Roche Applied Science) for HEK293 cells and L6 cells, respectively, according to the manufacturers protocols.
Preparation of FGF19, FGF21, and FGF23
Human recombinant FGF19, FGF21, and FGF23 (R179Q) were expressed in Escherichia coli, refolded in vitro, and purified by affinity, ion exchange, and size exclusion chromatographies as previously described (25).
Egr-1 Promoter Assay
The human early growth response-1 (Egr-1) gene promoter containing ERK-responsive elements (−580 to +30) (16, 26) was PCR-amplified from human genomic DNA and subcloned into the pEGFP-N1 vector (Clontech, Mountain View, CA). HEK293 cells were transfected with the reporter plasmid using the FuGENE transfection reagent and selected in medium containing 0.8 mg/ml G418 (Sigma- Aldrich). Clones of stable transformants were isolated and plated on 96-well plates, stimulated with FGF2 (Upstate Biotechnology, Lake Placid, NY) for 24 h, and lysed with phosphate- buffered saline containing 1% Triton X-100. Fluorescence of the cell lysates was measured using a microplate reader (FLUOstar OPTIMA, BMG Labtechnologies, Inc., Durham, NC) in the fluorescence mode (excitation, 485 nm; emission, 520 nm). Background signal was determined as fluorescence from the well without cells and subtracted to obtain net fluorescence generated from cells. A clone (E2-7) that showed robust response to FGF2 was cotransfected with human βKlotho and puromycin-resistant vectors and selected in the medium containing 0.5 mg/ml G418 and 0.6 μg/ml puromycin (InvivoGen, San Diego, CA). Clones of the double-transfectants were isolated and stimulated with FGF21 and subjected to the enhanced green fluorescent protein assay as described. A clone (Eβ2) that showed robust response to FGF21 was used for testing FGF19 activity.
Immunoblot Analysis of FGF Signaling
Cells cultured on multi-well plates were serum-starved overnight and then treated for 10 min with human recombinant FGF19, FGF21, FGF23 (R179Q), or FGF2. The cells were snap-frozen in liquid nitrogen, lysed in a buffer containing inhibitors for phosphatases and proteases, and processed for immunoblot analysis using antibodies against phospho-FRS2α(Cell Signaling Technology, Beverly, MA), phospho-44/42 mitogen-activated protein kinase (ERK1/2) (Cell Signaling), and ERK1/2 (Cell Signaling) as previously described (15, 21). The signal intensity was quantified using an image analysis software (ImageQuant, Molecular Dynamics, Sunnyvale, CA). Male 129sv mice at 8 weeks of age were administered either FGF19 (1 μg g−1 body weight), FGF21 (0.3 μg g−1 body weight), or vehicle (10 mM HEPES, pH 7.4, 150 mM NaCl) by injection into the inferior vena cava. Liver, perigonadal fat pads, kidneys, and hind limb muscles were excised 15, 17, 19, and 21 min, respectively, after the injection. The tissues were flash-frozen in liquid nitrogen, homogenized in the lysis buffer, and subjected to immunoblotting. All of the animal experiments were approved by the Institutional Animal Care and Research Advisory Committee of The University of Texas Southwestern Medical Center at Dallas.
FGF19 Pull-down
Cell lysates were prepared from HEK293 cells transfected with FGFR alone or cotransfected with FGFR and βKlotho and incubated with anti-V5-agarose beads (Sigma- Aldrich) at 4°C for 3 h. The beads were washed four times with Tris-buffered saline (TBS) containing 1% Triton X-100 and then incubated with FGF19 (1 μg/ml) at 4°C for 3 h. Thereafter, the beads were washed three times with Krebs-Ringer- HEPES buffer (15) containing 1% Triton X-100 followed by three washes with the same buffer lacking Triton X-100. Beadbound proteins were eluted with Laemmli sample buffer and subjected to immunoblot analysis using antibodies against V5 tag (Invitrogen), βKlotho (R & D Systems, Minneapolis, MN), or FGF19 (R & D Systems).
Knock-down of βKlotho Expression by RNA Interference
3T3-L1 adipocytes were transfected with siRNA duplexes by electroporation as previously described and then used for immunoblot analysis and for glucose uptake assay (21). H4IIE cells were transfected by electroporation as well. Briefly, the cells were harvested using trypsin, washed twice with phosphate- buffered saline, suspended in Opti-MEM (Invitrogen) (1 × 107 cells/ml), mixed with 10 nmol/107 cells of siRNA oligonucleotides (see supplemental Table S1), and electroporated with a gene pulser system (0.24 kV and 960 microfarads capacitance) (Bio-Rad). After electroporation, the cells were incubated at 4°C for 10 min before reseeding onto 6-well plates. Thirty hours after transfection, the cells were serum-starved overnight and used for immunoblot analysis and quantitative RT-PCR.
Glucose Uptake Assay
3T3-L1 adipocytes transfected with siRNA were stimulated with FGF19 (1 μg/ml), FGF21 (1 μg/ml), or FGF2 (0.1 μg/ml) in Dulbecco's modified Eagle's medium with 0.1% free fatty acid-free bovine serum albumin (Sigma-Aldrich) for 18 h at 37°C and subjected to the glucose uptake assay as described previously (21).
Quantitative RT-PCR
Total RNA was isolated using the RNeasy kit (Qiagen). For quantification of CYP7A1 and SHP mRNA, H4IIE cells transfected with siRNA were stimulated with FGF19, FGF21, or vehicle for 18 h before RNA isolation. 4 μg of total RNA was treated with 0.2 unit of DNase I (Promega, Madison, WI) and then reverse transcribed into first-strand cDNA using the ThemoScript RT-PCR system (Invitrogen) according to the manufacturer's protocol. The specific primers used to quantify gene expression were listed in supplemental Table S2. Quantitative RT-PCR reactions contained 25 ng of cDNA, 150 nM of each primer, and 5 μl of SYBR Green PCR Master mix (Applied Biosystems, Foster City, CA) in a total volume of 10 μl. All of the reactions were performed in triplicate on an Applied Biosystems Prism 7900HT sequence detection system, and relative mRNAlevels were calculated by the comparative threshold cycle method using cyclophilin as the internal control.
Immunoprecipitation
Liver (200 mg) and white adipose tissue (1000 mg) were homogenized in 1 ml of homogenization buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, and 0.5 mM EDTA) containing protease inhibitors. The homogenates were incubated for 30 min at 4°C after the addition of Triton X-100 (final, 1.2% w/v) and then centrifuged twice for 12 min at 18,000 × g to remove debris. The supernatant of liver and white adipose tissue were precleared with 40 μl of protein G-Sepharose or protein A-Sepharose (Amersham Biosciences) conjugated with 20 μg of normal goat or rabbit IgG for 3 h at 4°C, respectively. The precleared lysates of liver and white adipose tissues, respectively, were incubated with 20 μl of protein A-Sepharose conjugated with 20 μg of anti-FGFR4 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) or normal goat IgG and with anti-FGFR1 antibody (Santa Cruz) or normal rabbit IgG for 3 h at 4°C. The beads were washed four times with TBS containing 1% Triton X-100 and three times with TBS. Bead-bound proteins were eluted with Laemmli sample buffer and subjected to immunoblot analysis using anti-βKlotho, anti-FGFR1 (R & D Systems), and anti- FGFR4 (R & D Systems) antibodies.
RESULTS
FGF19 Requires βKlotho to Activate FGF Signaling
Like FGF21 and FGF23, FGF19 failed to robustly activate FGF signaling in HEK293 cells as measured by induction of FRS2α and ERK phosphorylation. Forced expression of Klotho in HEK293 cells caused a selective response to FGF23 but not to FGF19 or FGF21. Conversely, forced expression of βKlotho conferred responsiveness to both FGF21 and FGF19 but not FGF23 (Fig. 1A). Because activation of FGF signaling with FGF23 increases promoter activity and expression of the Egr-1 (early growth response-1) gene in Klotho-expressing cells (16), we tested whether FGF19 and FGF21 also increase Egr-1 promoter activity in βKlotho-expressing cells. Consistent with the results shown in Fig. 1A, FGF19 and FGF21 activated the Egr-1 promoter only in βKlotho-expressing HEK293 cells in a dose-dependent manner, whereas FGF2 activated it independently of βKlotho expression (Fig. 1B). These observations indicate that FGF19 requires βKlotho to activate FGF signaling. We have shown that βKlotho and Klotho enhance binding affinity of FGF21 and FGF23, respectively, to their cognate FGFRs (15, 21). We therefore tested whether βKlotho also enhances binding of FGF19 to its cognate FGFRs. As previously reported, βKlotho binds to FGFR1c and FGFR4 more avidly than does to FGFR2c and FGFR3c (Fig. 1C) and does not interact with “b” isoforms of FGFRs (21). In the absence of βKlotho, FGF19 did not bind to FGFR1c and FGFR4 and bound poorly to FGFR2c and 3c. In the presence of βKlotho, binding of FGF21 to FGFR1c and FGFR4 was significantly increased, whereas binding to FGFR2c and FGFR3 was only slightly increased (Fig. 1C). These observations are reminiscent of our previous findings for FGF21 (21) and indicate that FGF19 also requires βKlotho for binding to FGFR1c and FGFR4 and for robust activation of FGF signaling.
FIGURE 1. FGF19 requires βKlotho for strong binding to FGFR and robust activation of FGF signaling.

A, forced expression of βKlotho confers responsiveness to FGF19 on HEK293 cells. HEK293 cells were transfected with either mock vector (Mock) or expression vectors for βKlotho and Klotho, respectively, and then stimulated with vehicle, FGF19 (1,000 ng/ml), FGF21 (1,000 ng/ml), FGF23 (300 ng/ml), or FGF2 (100 ng/ml) for 10 min. The cell lysates were subjected to immunoblotting (i.b.) with antibodies against phosphorylated FRS2α (pFRS2α), phosphorylated ERK1/2 (pERK1/2), total ERK1/2 (ERK1/2), βKlotho, or Klotho. A representative result from more than 10 independent experiments is shown. B, dose-dependent activation of Egr-1 promoter with FGF2 (●), FGF19 (▲), and FGF21 (■) in E2-7 cells (HEK293 cells stably transfected with a reporter plasmid containing human Egr-1 promoter and enhanced green fluorescent protein, left panel) and in Eβ2 cells (E2-7 cells stably transfected with a βKlotho expression vector, right panel). The assays were performed in triplicate. C, FGF19 binds to βKlotho-FGFR complexes more efficiently than to FGFR alone. HEK293 cells were transfected with one of the indicated FGFR isoforms (with a V5 tag) alone or with βKlotho. FGFR or FGFR-βKlotho complex were immunoprecipitated from cell lysates on agarose beads carrying an anti-V5 antibody. The beads were then incubated with FGF19, and bead-bound proteins were analyzed by immunoblotting for the presence of βKlotho, FGF19, and FGFR (V5). The difference between b and c isoforms in FGFR13 resides in the C-terminal half of the third immunoglobulin-like domain. Another alternative splicing event occurs within the first immunoglobulin- like domain and acidic box, which generates long (L), middle (M), and short isoforms (S) in FGFR1 and FGFR2. See supplemental Fig. S1 for details.
Adipocytes Respond to Both FGF19 and FGF21
Because differentiated 3T3-L1 adipocytes express βKlotho endogenously and respond to FGF21 (21), we tested whether FGF19 also activates FGF signaling in these cells. Dose-responsiveness of FRS2α and ERK1/2 phosphorylation induced by FGF19 was comparable with that induced by FGF21 (Fig. 2A). We observed significant attenuation of FGF19- and FGF21-induced FRS2α and ERK1/2 phosphorylation by knocking down βKlotho expression using two independent siRNAs (Fig. 2, B and C), indicating that endogenous βKlotho is required for both FGF19 and FGF21 to activate FGF signaling in adipocytes. Quantification of mRNA levels of mouse FGFR1–4 revealed that 3T3-L1 adipocytes predominantly express FGFR1 (Fig. 2D). These observations indicate that activation of FGF signaling by FGF19 and FGF21 in these cells is mediated through the βKlotho-FGFR1 complex. Because FGF21 increases glucose uptake in adipocytes independently of insulin (6, 21), we next tested whether FGF19 exerts similar activity upon adipocytes. Indeed, FGF19 significantly increased glucose uptake within 18 h, which was comparable with that induced by FGF21 (Fig. 2E). These activities of FGF19 and FGF21 are also dependent on βKlotho because they were abolished by knocking down βKlotho expression using two independent siRNAs (Fig. 2E).
FIGURE 2. Both FGF19 and FGF21 activate FGF signaling and increase glucose uptake in adipocytes.

A, dose response of FGF19 and FGF21 signaling in 3T3-L1 adipocytes. The activity of FGF signaling was determined by immunoblot analysis of phosphorylated FRS2α (pFRS2α) and phosphorylated ERK1/2 (pERK1/2) with total ERK1/2 (ERK1/2) used as an internal control. B, knock-down of βKlotho expression by siRNA. 3T3-L1 adipocytes were transfected with a nontargeting random siRNA (Random) or two independent mouse βKlotho siRNAs (βKlotho-1 and βKlotho-2). Expression of βKlotho, with α-actin used as a loading control, was determined by immunoblotting. C, both FGF19 and FGF21 require endogenous βKlotho to activate FGF signaling in adipocytes. 3T3-L1 adipocytes were transfected with a nontargeting random siRNA (Random) or two independent mouse βKlotho siRNAs (βKlotho-1 and βKlotho-2) and then stimulated with FGF19 (1,000 ng/ml), FGF21 (1,000 ng/ml), or FGF2 (100 ng/ml) for 10 min and subjected to immunoblot analysis as in A. D, 3T3-L1 adipocytes predominantly express FGFR1. FGFR1–4 mRNA levels were measured by quantitative RT-PCR in triplicates using the comparative CT method and indicated as the relative fold difference from the lowest expression level of FGFR. The bars indicate means plus S.D. error (n = 3). E, both FGF19 and FGF21 increase glucose uptake in adipocytes in a βKlotho-dependent manner. 3T3-L1 adipocytes were transfected with siRNA as in B and then assayed for glucose uptake after incubation with either vehicle, FGF19 (1,000 ng/ml), or FGF21 (1,000 ng/ml) for 18 h. The results are shown as the means plus S.D. (error bars, n = 3). *, p < 0.05 versus vehicle by Student's t test.
Hepatocytes Respond to FGF19 but Not FGF21
Because the rat hepatoma cell line H4IIE also expresses βKlotho endogenously, we tested whether FGF19 and FGF21 activate FGF signaling in these cells. The dose-response characteristics of FGF19-induced phosphorylation for FRS2α and ERK1/2 in H4IIE cells were similar to that observed in adipocytes (Fig. 3A). In addition, we noted significant attenuation of FGF19-induced FRS2α and ERK1/2 phosphorylation by knocking down βKlotho expression using two independent siRNAs (Fig. 3, B and C), confirming that endogenous βKlotho is required for FGF19 to activate FGF signaling in hepatocytes as well as in adipocytes. However, in contrast to adipocytes, H4IIE hepatocytes are ∼100-fold less sensitive to FGF21 in terms of induction of FRS2α and ERK1/2 phosphorylation (Fig. 3A). Quantification of mRNA levels of rat FGFR1–4 demonstrated that H4IIE hepatocytes predominantly express FGFR4 (Fig. 3D), indicating that FGF19 can activate FGF signaling through the βKlothoFGFR4 complex much more efficiently than FGF21, despite the fact that FGF21 is capable of binding to the βKlotho-FGFR4 complex in vitro (21). Because FGF19 suppresses transcription of CYP7A1 that encodes the rate-limiting enzyme of bile acid synthesis in hepatocytes (27), we next tested whether the ability of FGF19 to suppress CYP7A1 expression also depends on βKlotho. FGF19, but not FGF21, significantly reduced the mRNA level of CYP7A1 and increased that of SHP, an orphan nuclear receptor that negatively regulates CYP7A1 expression (Fig. 3E) (2, 28, 29). Moreover, these activities of FGF19 are dependent on βKlotho because they were attenuated by knock-down of βKlotho expression using two independent siRNAs (Fig. 3E).
FIGURE 3. FGF19, but not FGF21, activates FGF signaling and suppresses CYP7A1 expression in hepatocytes.

A, dose response of FGF19 and FGF21 signaling in H4IIE hepatocytes. The activity of FGF signaling was determined by immunoblot analysis for phosphorylated FRS2α (pFRS2α) and phosphorylated ERK1/2 (pERK1/2), with total ERK1/2 (ERK1/2) levels serving as an internal control. B, knock-down of βKlotho expression by siRNA. H4IIE cells were transfected with a nontargeting random siRNA (Random) or two independent rat βKlotho siRNAs (βKlotho-1 and βKlotho-3). Expression of βKlotho, and α-actin as a loading control, was determined by immunoblotting. C, FGF19 requires endogenous βKlotho to activate FGF signaling in hepatocytes. H4IIE cells were transfected with a nontargeting random siRNA (Random) or two independent βKlotho siRNAs (βKlotho-1 and βKlotho-3) and stimulated with FGF19, FGF21, or FGF2 at the indicated concentrations for 10 min, and the cell lysates were subjected to immunoblot analysis as in A. D, H4IIE cells predominantly express FGFR4. FGFR1–4 mRNA levels were measured by quantitative RT-PCR in triplicate using the comparative CT method and indicated as the relative fold difference from the lowest expression level of FGFR. The bars indicate the means plus S.D. error (n = 3). E, FGF19 suppresses CYP7A1 expression and increases SHP expression in hepatocytes in a βKlotho-dependent manner. H4IIE cells were transfected with siRNA as in B and then assayed for CYP7A1 and SHP mRNA levels after incubation with either vehicle, FGF19 (50 ng/ml or 100 ng/ml), or FGF21 (100 ng/ml) for 10 h. The results are presented as the relative fold difference from vehicle-treated samples. The bars indicate the means plus S.D. error (n = 3).
FGF19 and FGF21 Signal through Distinct FGFR Isoforms
To determine which FGFR isoforms are responsible for activation of FGF signaling by FGF19 and FGF21, we reconstituted expression of βKlotho and individual FGFR isoforms in the rat myoblastic cell line L6 and asked which FGFR isoforms could confer responsiveness to FGF19 or FGF21. L6 cells are useful for this purpose because they do not respond to FGF19 and FGF21 even when transfected with the βKlotho expression vector. We transfected L6 cells with expression vectors for βKlotho and each of the FGFR isoforms and then stimulated the transfectants with FGF19 or FGF21. Both FGF19 and FGF21 activated FGF signaling in L6 cells coexpressing βKlotho and either FGFR1c, 2c, or 3c (Fig. 4A), but not in control L6 cells expressing FGFR1c, FGFR2c, or FGFR3c alone (Fig. 4B). In contrast, L6 cells coexpressing βKlotho and FGFR4 responded to FGF19 but very poorly to FGF21 (Fig. 4C), which is evocative of the cellular response to FGF19 and FGF21 in hepatocytes (Fig. 3A). We conclude that FGF21 can strongly bind to but cannot signal effectively through the βKlotho-FGFR4 complex. These observations raise the possibility that FGF21 functions as an endogenous surrogate ligand for the βKlotho-FGFR4 complex. Moreover, the physiological role of FGFR2c and FGFR3c in FGF19 signaling remains to be determined.
FIGURE 4. FGF19, but not FGF21, signals through the βKlotho-FGFR4 complex.

A, L6 cells were cotransfected with expression vectors for βKlotho and one of the indicated FGFR isoforms, then stimulated with FGF19 (1,000 ng/ml), FGF21 (1,000 ng/ml), or FGF2 (100 ng/ml) for 10 min. FGF signaling activity was determined by immunoblot (i.b.) analysis for phosphorylated FRS2α (pFRS2α) and phosphorylated ERK1/2 (pERK1/2) and total ERK1/2 (ERK1/2) levels performed as an internal control (upper panel). The signal intensity of phosphorylated ERK1/2 was quantified, corrected with that of corresponding total ERK1/2, and indicated as fold increase from vehicle-treated samples in each FGFR group (lower panel). A representative result from three independent experiments is shown. B, as in A, except that βKlotho was not transfected. C, dose response of FGF19 and FGF21 signaling in L6 cells cotransfected with expression vectors for βKlotho and FGFR4. The cell lysates were subjected to immunoblot analysis as in A.
Tissue-specific Activity of the FGF19 Subfamily Members
The experiments using cultured cells demonstrate that the expression of βKlotho in combination with particular FGFR isoforms determines the cellular responses to FGF19 and FGF21. To confirm this in vivo, we first determined which FGFR iso-forms are expressed in different target organs for FGF19 and FGF21. Quantification of mRNA levels revealed that white adipose tissue and liver predominantly express FGFR1 and FGFR4, respectively (Fig. 5A). Next we addressed whether βKlotho constitutively bound to FGFR1 and FGFR4 in these tissues. βKlotho was coimmunoprecipitated with FGFR1 and FGFR4 from lysates of white adipose tissue and liver, respectively (Fig. 5B). Last, we administered recombinant FGF19 or FGF21 intravenously into mice and determined the level of activation of FGF signaling in skeletal muscle, white adipose tissue, kidney, and liver. FGF19 activated FGF signaling robustly in liver and weakly in white adipose tissue as evidenced by induction of ERK phosphorylation; FGF21, however, activated FGF signaling in white adipose tissue but not in liver (Fig. 5C). These data mirror the data obtained using cultured hepatocyte and adipocyte cell lines (Figs. 2 and 3).
FIGURE 5. FGF19- and FGF21-dependent signaling in white adipose tissue and liver in vivo.
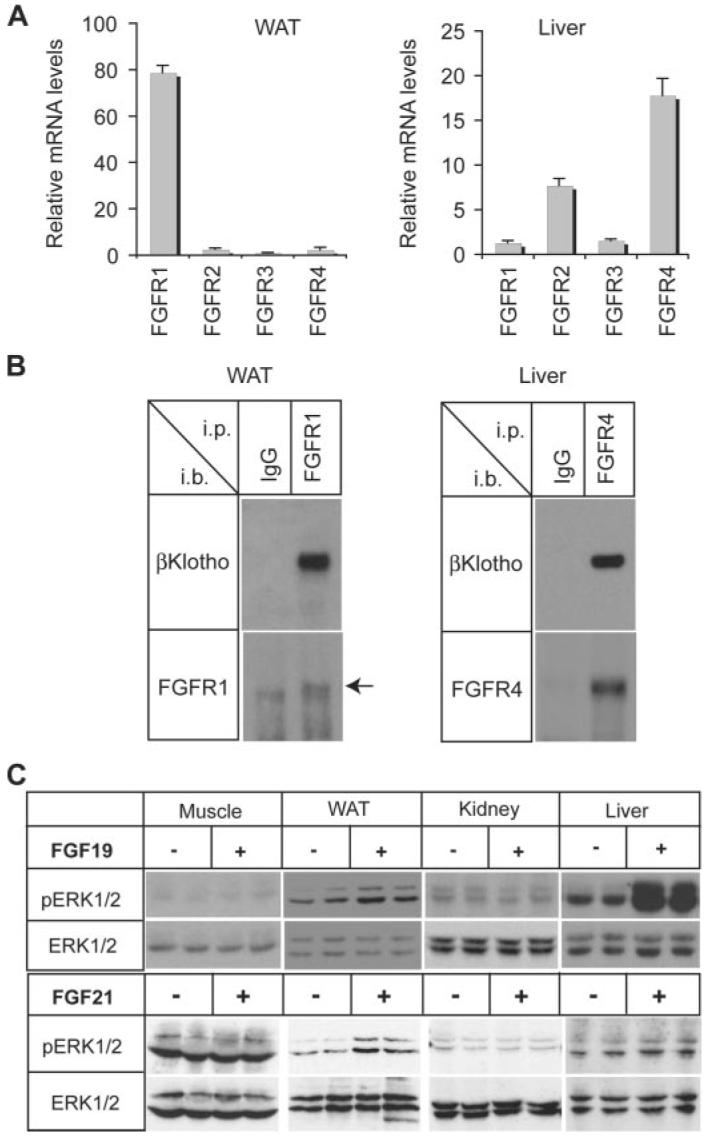
A, tissue distribution of FGFRs. FGFR1–4 mRNA levels in white adipose tissue and liver from wild-type mice were measured by quantitative RT-PCR in triplicates using the comparative CT method and indicated as the relative fold difference from the lowest expression level of FGFR. The bars indicate the means plus S.D. error (n = 3). B, βKlotho constitutively binds to FGFR1 and FGFR4 in mouse tissues. Tissue lysates from white adipose tissue (WAT) and liver were immunoprecipitated (i.p.) with anti-FGFR1 antibody and anti-FGFR4 antibody, respectively, and immunoblotted (i.b.) with the same anti-FGFR antibody or anti-βKlotho antibody. Normal IgG was used instead of anti-FGFR antibody as a negative control. The arrow indicates the FGFR1 specific band. C, tissue-specific activation of FGF signaling by FGF19 and FGF21. Hind limb muscles (Muscle), white adipose tissue (WAT), kidney, and liver were excised from mice treated either with vehicle (n = 4), FGF19 (n = 2), or FGF21 (n = 2). Tissue lysates were prepared for immunoblot analysis using the antibodies indicated.
DISCUSSION
In this report, we have identified three factors that dictate the tissue-specific activity of FGF19 and FGF21: (i) FGF19, like FGF21, requires βKlotho for strong binding to FGFRs and robust activation of FGF signaling; (ii) only FGF19 signals efficiently through FGFR4; and (iii) adipocytes express βKlotho and FGFR1, whereas hepatocytes express βKlotho and FGFR4. These findings explain why both FGF19 and FGF21 increase glucose uptake in adipocytes (Fig. 2), why FGF19 but not FGF21 suppresses expression of CYP7A1 in hepatocytes (Fig. 3), and why endogenous FGF21 cannot compensate for lack of FGF15 (the mouse ortholog of FGF19) and fails to suppress hepatic expression of CYP7A1 in FGF15-deficient mice (2).
The capacity of FGF19 to activate FGF signaling through FGFR4 and other FGFRs may be of physiological significance in the regulation of gall bladder filling by FGF19. βKlotho and all four FGFRs are expressed in the gall bladder, with FGFR3 expression being the most abundant (3). Administration of FGF19 causes an increase in gall bladder volume within 15 min not only in wild-type mice but also in mice defective in FGFR4 expression (3), indicating that gall bladder filling induced by FGF19 can be mediated through other FGFRs.
Although FGF19 has been known to act on liver and gall bladder, the fact that FGF19 can signal through the βKlotho-FGFR1 complex has raised the possibility that FGF19 may also act on adipocytes. Indeed, FGF19-overexpressing (30) and FGF21-overexpressing (6) transgenic mice display overlapping phenotypes involving adipose tissues, which include reduced fat mass and resistance to obesity induced by a high fat diet. This potential extrahepatic activity of FGF19 may be explained by assuming that high levels of circulating FGF19 act on adipocytes through the βKlotho-FGFR1 complex in FGF19-overexpressing transgenic mice and evoke responses similar to those induced by FGF21.
Whether or not FGF15/19 contributes to the regulation of adipocyte function under physiological settings remains to be determined. Activation of FGF signaling in response to injected FGF19 in white adipose tissue was less prominent than that observed in liver (Fig. 5C), whereas 3T3-L1 adipocytes and H4IIE hepatocytes in culture responded to FGF19 similarly (Figs. 2 and 3). One possible explanation for this discrepancy is that most FGF19 injected via the inferior vena cava bound to hepatocytes and/or extracellular matrices during the first pass through the liver and did not reach peripheral white adipose tissues at a high concentration. Although the affinity of the FGF19 subfamily members to heparin/heparan sulfate is much lower than the other FGF family members, FGF19 exhibits a higher affinity to heparin than FGF21 and FGF23 (12, 31) and therefore could be trapped by extracellular matrices in the liver to some extent. If this is the case, contribution of FGF15/19 to the regulation of adipocyte function could be minimal under physiological conditions, because FGF19 is secreted from the intestines and first reaches the liver via the portal vein before being delivered to white adipose tissue through systemic circulation. Thus, the ability of FGF19 to weakly bind to heparin/heparan sulfate may contribute to the restriction of metabolic activity of FGF19 to the liver and gall bladder.
Several factors explain why FRS2α and ERK phosphorylation induced by FGF19 or FGF21 is often less robust than that induced by FGF2. First, it is unlikely that all FGFRs always exist as βKlotho-bound forms, because (i) an equilibrium should exist in the interaction between FGFR, βKlotho, and the βKlotho-FGFR complex and (ii) βKlotho may be less abundant than FGFRs. In fact, βKlotho mRNA measured by quantitative RT-PCR is only ∼1% of FGFR1 mRNA in 3T3-L1 adipocytes (data not shown), suggesting that the majority of FGFRs do not bind to βKlotho and therefore cannot be activated by FGF19/21. Second, the fact that each FGFR isoform has a different affinity to βKlotho (21) implies that not only the relative amounts of βKlotho and FGFRs but also the composition of FGFR isoforms affect the number of βKlotho-FGFR complexes formed on the cell surface and thus responsiveness to FGF19/21. Lastly, our data suggest that FGF2 signals through FGFR1 more efficiently than the other FGFR isoforms: (i) The difference between FGF2- and FGF19/21-induced phosphorylation of FRS2α and ERK is more prominent in FGFR1-dominant cells (HEK293 and 3T3-L1; Figs. 1 and 2) than in FGFR4-dominant cells (H4IIE; Fig. 3) and (ii) L6 cells transfected with FGFR1c showed a stronger response to FGF2 than those transfected with the other FGFRs (Fig. 4). In fact, FGF2 is known to have a higher affinity to FGFR1c than to the other FGFR isoforms (32). These factors may contribute to the potential of FGF2 and FGF19/21 in activation of FGF signaling in different cell types.
The FGF19 subfamily members function as hormones that act on specific target organs to control metabolism. On the other hand, many tissues express one or more FGFR iso-forms that potentially function as receptors for these FGFs. Our studies have established that tissue-specific expression of Klotho and βKlotho, in combination with restricted FGFR isoform expression, determine the target organs of the FGF19 subfamily members. FGF19, FGF21, and FGF23 participate in the regulation of multiple metabolic processes involving bile acid, glucose, fatty acids, ketones, phosphate, calcium, and vitamin D and thus may play important roles in the pathophysiology underlying diverse human metabolic disorders, including diabetes, obesity, hypercholesterolemia, chronic kidney diseases, and bone disorders. These metabolically active FGFs and their interacting Klotho gene family members will be potential targets for therapeutic interventions in multiple metabolic disorders.
Supplementary Material
Acknowledgments
We thank O. Sineshchekova and L. Wang for assistance with experiments.
Footnotes
This work was supported in part by grants from the Eisai Research Fund (to M. K.), the Ellison Medical Foundation (to M. K.), the Ted Nash Long Life Foundation (to M. K.), the Irma T. Hirschl Fund (to M. M.), and the Robert A. Welch Foundation (to S. A. K.) and by National Institutes of Health Grants R01AG19712 (to M. K.), DK067158 (to S. A. K.), R01AG25326 (to M. K. and K. P. R.), and R01DE13686 (to M. M.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and supplemental Fig. S1.
The abbreviations used are: FGF, fibroblast growth factor; FGFR, FGF receptor; ERK, extracellular signal-regulated kinase; TBS, Tris-buffered saline; siRNA, small interfering RNA; RT, reverse transcription.
REFERENCES
- 1.Itoh N, Ornitz DM. Trends Genet. 2004;20:563–569. doi: 10.1016/j.tig.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 2.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Choi M, Moschetta A, Bookout AL, Peng L, Umetani M, Holmstrom SR, Suino-Powell K, Xu HE, Richardson JA, Gerard RD, Mangelsdorf DJ, Kliewer SA. Nat. Med. 2006;12:1253–1255. doi: 10.1038/nm1501. [DOI] [PubMed] [Google Scholar]
- 4.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, Elmquist JK, Gerard RD, Burgess SC, Hammer RE, Mangelsdorf DJ, Kliewer SA. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 6.Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, Gromada J, Brozinick JT, Hawkins ED, Wroblewski VJ, Li DS, Mehrbod F, Jaskunas SR, Shanafelt AB. J. Clin. Investig. 2005;115:1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White KE, Evans WE, O'Rlordan JLH, Speer MC, Econs MJ, Lorenz-Deplereux B, Grabowski M, Meitinger T, Storm TM. Nat. Genet. 2000;26:345–348. [Google Scholar]
- 8.Quarles LD. Am. J. Physiol. 2003;285:E1–E9. doi: 10.1152/ajpendo.00016.2003. [DOI] [PubMed] [Google Scholar]
- 9.Schiavi SC, Kumar R. Kidney Int. 2004;65:1–14. doi: 10.1111/j.1523-1755.2004.00355.x. [DOI] [PubMed] [Google Scholar]
- 10.White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Kidney Int. 2001;60:2079–2086. doi: 10.1046/j.1523-1755.2001.00064.x. [DOI] [PubMed] [Google Scholar]
- 11.Jonsson KB, Zahradnik R, Larsson T, White KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H, Ljunggren O, Oba K, Yang IM, Miyauchi A, Econs MJ, Lavigne J, Juppner H. N. Engl. J. Med. 2003;348:1656–1663. doi: 10.1056/NEJMoa020881. [DOI] [PubMed] [Google Scholar]
- 12.Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, Xu C, Neubert T, Zhang F, Linhardt RJ, Yu X, White KE, Inagaki T, Kliewer SA, Yamamoto M, Kurosu H, Ogawa Y, Kuro-o M, Lanske B, Razzaque MS, Mohammadi M. Mol. Cell. Biol. 2007;27:3417–3428. doi: 10.1128/MCB.02249-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harmer NJ, Pellegrini L, Chirgadze D, Fernandez-Recio J, Blundell TL. Biochemistry. 2004;43:629–640. doi: 10.1021/bi035320k. [DOI] [PubMed] [Google Scholar]
- 14.Mohammadi M, Olsen SK, Ibrahimi OA. Cytokine Growth Factor Rev. 2005;16:107–137. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 15.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M. J. Biol. Chem. 2006;281:6120–6123. doi: 10.1074/jbc.C500457200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Nature. 2006;444:770–774. doi: 10.1681/01.asn.0000926868.48235.3d. [DOI] [PubMed] [Google Scholar]
- 17.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima Y. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 18.Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, Kahn CR, Rosenblatt KP, Kuro-o M. Science. 2005;309:1829, 1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. J. Clin. Investig. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Razzaque MS, Sitara D, Taguchi T, St-Arnaud R, Lanske B. FASEB J. 2006;20:720–722. doi: 10.1096/fj.05-5432fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogawa Y, Kurosu H, Yamamoto M, Nandi A, Rosenblatt KP, Goetz R, Eliseenkova AV, Mohammadi M, Kuro-o M. Proc. Natl. Acad. Sci. U. S. A. 2007;104:7432–7437. doi: 10.1073/pnas.0701600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito S, Kinoshita S, Shiraishi N, Nakagawa S, Sekine S, Fujimori T, Nabeshima Y. Mech. Dev. 2000;98:115–119. doi: 10.1016/s0925-4773(00)00439-1. [DOI] [PubMed] [Google Scholar]
- 23.Ito S, Fujimori T, Furuya A, Satoh J, Nabeshima Y, Nabeshima Y. J. Clin. Investig. 2005;115:2202–2208. doi: 10.1172/JCI23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu C, Wang F, Kan M, Jin C, Jones RB, Weinstein M, Deng CX, McKeehan WL. J. Biol. Chem. 2000;275:15482–15489. doi: 10.1074/jbc.275.20.15482. [DOI] [PubMed] [Google Scholar]
- 25.Plotnikov AN, Hubbard SR, Schlessinger J, Mohammadi M. Cell. 2000;101:413–424. doi: 10.1016/s0092-8674(00)80851-x. [DOI] [PubMed] [Google Scholar]
- 26.Bauer I, Hohl M, Al-Sarraj A, Vinson C, Thiel G. Arch Biochem. Biophys. 2005;438:36–52. doi: 10.1016/j.abb.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 27.Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, Goodwin B, Jones SA. Genes Dev. 2003;17:1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, Shan B, Russell DW, Schwarz M. Dev. Cell. 2002;2:713–720. doi: 10.1016/s1534-5807(02)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, Chua SS, Wei P, Heyman RA, Karin M, Moore DD. Dev. Cell. 2002;2:721–731. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 30.Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, Stephan JP, Tsai SP, Powell-Braxton L, French D, Stewart TA. Endocrinology. 2002;143:1741–1747. doi: 10.1210/endo.143.5.8850. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. J. Biol. Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. J. Biol. Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.