

Abstract
In a previous examination using natural all-RNA substrates that contained either a 5′-oxy or 5′-thio leaving group at the cleavage site, we demonstrated that (i) the attack by the 2′-oxygen at C17 on the phosphorus atom is the rate-limiting step only for the substrate that contains a 5′-thio group (R11S) and (ii) the departure of the 5′ leaving group is the rate-limiting step for the natural all-RNA substrate (R11O) in both nonenzymatic and hammerhead ribozyme-catalyzed reactions; the energy diagrams for these reactions were provided in our previous publication. In this report we found that the rate of cleavage of R11O by a hammerhead ribozyme was enhanced 14-fold when Mg2+ ions were replaced by Mn2+ ions, whereas the rate of cleavage of R11S was enhanced only 2.2-fold when Mg2+ ions were replaced by Mn2+ ions. This result appears to be exactly the opposite of that predicted from the direct coordination of the metal ion with the leaving 5′-oxygen, because a switch in metal ion specificity was not observed with the 5′-thio substrate. However, our quantitative analyses based on the previously provided energy diagram indicate that this result is in accord with the double-metal-ion mechanism of catalysis.
Among various catalytic RNAs, the hammerhead-type ribozyme is the smallest and best understood as far as the relationship between structure and function is concerned. Naturally occurring hammerhead ribozymes can be found in some RNA viruses, and they act “in cis” during viral replication by the rolling circle mechanism (1–3). The hammerhead ribozyme has been engineered in such a way that it can act “in trans” against other RNA molecules (4, 5). The trans-acting hammerhead ribozyme developed by Haseloff and Gerlach (5) consists of an antisense section (stems I and III) and a catalytic domain with a flanking stem II and loop section (Fig. 1a). Because of the small size of hammerhead ribozymes, they are well suited for mechanistic studies, being good representatives of catalytic RNAs. Recently, hammerhead ribozymes were crystallized and their structures were analyzed in detail by two independent groups (6–9). The global three-dimensional structures of the two crystallized ribozymes were nearly identical: one domain of the conserved core, which consists of the sequence C3U4G5A6 and is located next to stem I, makes a sharp turn identical to the uridine turn in tRNAs (10). As a result, stem II and stem III are aligned almost colinearly through pseudocontinuous, long A-type helices.
Figure 1.

(a) RNA substrates (R11O and R11S) and trans-acting hammerhead ribozymes (R32 and R31). An expanded view of the cleavage site (between C17 and C1.1) is shown to provide details of the phosphodiester linkage in the unmodified substrate (R11O) and in the modified (R11S) substrate with a phosphorothioate linkage. Ribozyme R31 lacks one nucleotide at the 3′ end as compared with R32 ribozyme. (b) Minimal reaction scheme for hammerhead ribozymes. The reaction catalyzed by the hammerhead ribozyme consists of at least three steps. The substrate (together with metal ions) first binds to the ribozyme (kassoc). The phosphodiester bond of the bound substrate is cleaved by the action of metal ions (kcleav). The cleaved fragments dissociate from the ribozyme, which becomes available for a new series of catalytic events (kdiss). In this study, kcat always reflects kcleav.
It is now well established that ribozymes are metalloenzymes (2, 11–28). Although x-ray analysis of a hammerhead ribozyme identified one potential catalytic metal ion (7–9), the exact number of metal ions required for catalysis remains obscure, because the newly captured conformational intermediate appears to demand further conformational change for the following in-line attack (9). Direct evidence that a Mg2+ ion acts as a Lewis acid by coordinating directly to the leaving 3′-oxygen, thereby stabilizing the developing negative charge on the leaving 3′-oxygen, was reported in the case of ribozyme from Tetrahymena; this evidence was provided by a switch in metal ion specificity with a 3′-thio substrate (14). Recently, evidence for the double-metal-ion mechanism of catalysis was provided for the Tetrahymena ribozyme-catalyzed reaction (29). Base catalysis mediated by Mg2+ hydroxide was proposed on the basis of pH–rate profiles of various metal-ion-catalyzed reactions of the hammerhead ribozyme (13), although this mechanism was recently challenged by the double-metal-ion mechanism of catalysis, wherein metal ions are coordinated directly to the attacking and leaving oxygens (28). The latter double-metal-ion mechanism of catalysis with direct coordination of metal ions with the attacking and the leaving oxygen atoms is nearly identical with the mechanism we proposed on the basis of our previous examination of solvent isotope effects (20) and ab initio calculations (19). Although the number of Mg2+ ions involved in catalysis by hammerhead ribozymes remains obscure, a general double-metal-ion mechanism would be well suited to phosphotransfer catalyzed by ribozymes or by protein enzymes, such as polymerases and alkaline phosphatases (17, 30).
Kuimelis and McLaughlin (26, 27) and our group (23) recently worked on a substrate that contained a single mandatory ribonucleotide with a 5′-thio leaving group at the cleavage site for the hammerhead ribozyme. We found that the 5′-thio substrate (R11S) was two orders of magnitude more susceptible to ribozyme-mediated cleavage than the parental 5′-oxy substrate (R11O). We now report that our observation, that the rate of cleavage of R11O by a hammerhead ribozyme was enhanced 14-fold when Mg2+ ions were replaced by Mn2+ ions, whereas the rate of cleavage of R11S was enhanced only 2.2-fold when Mg2+ ions were replaced by Mn2+ ions, can be best explained by the double-metal-ion mechanism of catalysis.
MATERIALS AND METHODS
Synthesis of Ribozymes and Substrates.
Ribozymes (R32, R31, and I-R32) and an unmodified RNA substrate (R11O) were synthesized with a DNA/RNA synthesizer (model 394; PE Applied Biosystems) and purified as described previously (23). A bridging 5′-phosphorothioate linkage in a substrate (R11S) was incorporated at a specific site (indicated within a circle in Fig. 1a) by using 5′-thiol amidite as described previously (23). 31P NMR spectrum (JEOL; 200 MHz for 31P) of R11S confirmed the single bridging thiolinkage at 17.12 ppm (23).
Cleavage of R11O and R11S Substrates by Ribozymes.
For 5′-end-labeling of the R11S substrate, a small aliquot of the synthetic R11S substrate was incubated in buffer [50 mM Tris⋅HCl (pH 6.0)/10 mM MgCl2/0.1 mM EDTA] with polynucleotide kinase (Escherichia coli A 19; Takara Biomedicals, Kyoto) and [γ-32P]ATP (10 μCi/μl, Amersham; 1 μCi = 37 kBq) for 30 min at 37°C. After completion of the incubation, the reaction products were extracted twice with phenol/chloroform (1:1) and precipitated in ethanol in the presence of sodium acetate (23). The labeled R11S substrate was stored dry at −80°C until further use. The R11O substrate was 5′-end-labeled as reported elsewhere (31).
Cleavage reactions were carried out in 50 mM Mes buffer (pH 6.0) in the presence of 0.3 mM metal ions (either Mg2+ or Mn2+) at 28°C under multiple-turnover conditions. Concentrations of ribozyme and substrate were 2.0 μM and from 2 to 50 μM, respectively. The reaction was initiated by the addition of metal ions and aliquots were removed from the reaction mixture at appropriate intervals. These aliquots were then mixed with an equal volume of a solution that contained 100 mM EDTA, 9 M urea, and xylene cyanol and bromophenol blue (0.1% each). Uncleaved substrate and 5′-cleaved products were separated on a 20% polyacrylamide gel that contained 7 M urea. Electrophoresis was carried out with circulating iced water on the back side of the gel to prevent the decomposition of substrates. The extent of cleavage reactions was determined by quantitation of radioactivity in the bands of substrate and product with a Bio-image analyzer (BAS 2000; Fuji). Cleavage rates were obtained from the slopes of the curves for the time-course of reactions at the initial stage (0–3 min), and Km and kcat were calculated from Eadie–Hofstee plots. For measurements of initial rates, for subsequent calculations of kcat and Km, at different concentrations of substrate (four to six different concentrations, spanning the Km), the first 3 min of the reaction were examined. Initial rates were measured in triplicate at four to six different concentrations of substrate and the average values were plotted (Eadie–Hofstee plots in Fig. 2 d–g). Calculated values of kcat and Km are summarized in Table 1. Potential errors in these values were found to be 30% at most from results of duplicate experiments (two sets of Eadie–Hofstee plots).
Figure 2.

(a) Non-ribozyme-mediated hydrolysis of the R11S substrate in the presence of Mg2+ or Mn2+ ions. The autoradiogram shows the background reaction for the first 20 min. Within 20 min, no hydrolysis of R11S occurs in the absence of ribozymes [reaction mediated by Mg2+ ions (lanes 1–6) and Mn2+ ions (lanes 7–12) in 0, 1, 3, 5, 10, and 20 min, respectively, is shown]. The product (P) was produced during the isolation of R11S (S). The R11S substrate (25 μM) was incubated at 28°C in the presence of 50 mM Mes buffer (pH 6.0) and 0.3 mM either Mg2+ or Mn2+ ions (added as MgCl2 or MnCl2) for various times. (b) Time course of the R32 ribozyme-catalyzed reaction with R11O as substrate in the presence of either Mg2+ or Mn2+ ions. In the autoradiogram, lanes 1–8 show the R32 ribozyme-mediated cleavage in the presence of Mg2+ ions and lanes 9–16 show cleavage reactions in the presence of Mn2+ ions (10, 20, 40, 60, 90, 120, 180, and 300 min, respectively). The R32 ribozyme-catalyzed cleavage reaction was carried out by incubating R11O (25 μM) and R32 (2.0 μM) in 50 mM Mes buffer (pH 6.0) with 0.3 mM metal ions (either Mg2+ or Mn2+) at 28°C. Samples of lanes with odd numbers were loaded first and electrophoresis was started. Then samples of lanes with even numbers were added and electrophoresis was restarted. (c) Time course of the R32 ribozyme-catalyzed reaction for the R11S substrate in the presence of either Mg2+ or Mn2+ ions. The ribozyme-catalyzed cleavage reaction was carried out under conditions similar to those described above, except that R11S (25 μM) was used as substrate and the cleavage reaction was allowed to proceed for only 22 min. The lower autoradiogram represents the ribozyme-mediated cleavage in the presence of Mg2+ ions (lanes 1–10 for 0, 0.5, 1.0, 1.5, 2.0, 3.0, 5.0, 8.0, 15.0, and 22.0 min, respectively), and the upper one represents that for Mn2+ ions (lanes 1–10 for the same time intervals). (d) Eadie–Hofstee plot of kinetic data obtained from reactions in 50 mM Mes buffer (pH 6.0), 0.3 mM Mg2+ at 28°C. Initial rates of cleavage were determined with 2.0 μM R32 and 2.0–25 μM R11O. The line yields a Km of 1.2 μM and a Vmax of 0.008 μM⋅min−1. (e) This Eadie–Hofstee plot of the kinetic data was generated under conditions similar to those used in d except Mg2+ ions were replaced by Mn2+ ions. The line yields a Km of 9.5 μM and a Vmax of 0.11 μM⋅min−1. (f) Eadie–Hofstee plot of kinetic data obtained under conditions similar to those described for d except that R11S was used instead of R11O as the substrate. The line yields a Km of 5.5 μM and a Vmax of 0.60 μM⋅min−1. (g) Eadie–Hofstee plot of kinetic data obtained under conditions similar to those described for d except that, in this case, R11S and Mn2+ ions were used. The line yields a Km of 10.2 μM and a Vmax of 1.3 μM⋅min−1.
Table 1.
Kinetic parameters for ribozyme-catalyzed cleavage reactions
| M/Sub/Rz | kcat, min−1 | Km, μM | kcat(Mn)/kcat(Mg) | Km(Mn)/ Km(Mg) |
|---|---|---|---|---|
| Mg2+/R11S/R32 | 0.34 | 4.8 | 2.2 | 1.7 |
| Mn2+/R11S/R32 | 0.75 | 8.3 | ||
| Mg2+/R11O/R32 | 0.004 | 1.2 | 14 | 7.9 |
| Mn2+/R11O/R32 | 0.056 | 9.5 | ||
| Mg2+/R11S/R31 | 0.0075 | 7.3 | 4.0 | 2.1 |
| Mn2+/R11S/R31 | 0.03 | 15.6 | ||
| Mg2+/R11S/I-R32 | 0* | |||
| Mn2+/R11S/I-R32 | 0* |
All measurements were made in the presence of 0.3 mM metal ions (M), 50 mM Mes (pH 6.0) at 28°C.
No cleavage of substrates (Sub) was observed within 20 min of the start of the reaction used to determine kinetic parameters for active ribozymes (Rz; R32 and R31) at different concentrations of substrates. The inactive ribozyme (I-R32) differs from R32 by a single mutation of G5 to A.
RESULTS AND DISCUSSION
Stability of R11S and Reaction Conditions.
As described previously (23), because R11S was very labile, we had to identify reaction conditions under which non-ribozyme-mediated hydrolysis could be minimized. Thus, reactions were carried out at pH 6.0 and concentrations of metal ions were kept at 0.3 mM. Moreover, during the isolation of R11S, we could not avoid contamination by a 9-mer product (5′-GCCGUC(S)CC-3′). Although the 9-mer product could potentially serve as a substrate for the ribozymes used in this study (R32 and R31), an examination of a synthetic 9-mer without the phosphorothioate linkage (5′-GCCGUCCC-3′) revealed that its Km was more than 20-fold higher and its kcat was more than 10-fold lower than the corresponding values for the normal 11-mer substrate. Therefore, the 9-mer product did not act as a substrate or as an inhibitor during the measurement of ribozyme-mediated cleavage of R11S (Fig. 2). Note that, even though the 9-mer product (about 20%; indicated by asterisk on the autoradiograms in Fig. 2) was always observed, its concentration remained constant during the kinetic measurements. Therefore, we carried out all our kinetic measurements in the presence of the 9-mer product.
The rates of background reactions at pH 6.0 and with 0.3 mM metal ions were low enough for our purposes: the half-lives of R11S were calculated to be 150 hr and 320 hr, respectively, with Mn2+ and Mg2+ ions (Fig. 2a). Time courses for the ribozyme-catalyzed reactions are shown in Fig. 2b for R11O and in Fig. 2c for R11S. The autoradiogram included in Fig. 2a demonstrates that, within the time frame of the ribozyme-catalyzed reactions (up to 20 min), no background hydrolysis occurs. Nevertheless, for all kinetic measurements, inactive ribozymes [I-R32 (see Table 1); in which G5 was mutated to A (32)] were tested in parallel as controls to confirm that, in a solution that contained an active ribozyme, we were really monitoring ribozyme-catalyzed reactions.
Even at pH 6.0, the ribozyme-catalyzed hydrolysis of R11S was very rapid. Therefore, we were unable to measure reactions under excess ribozyme (single-turnover) conditions. Therefore, all the measurements were carried out under multiple-turnover conditions (Fig. 2). Calculated values of kcat and Km are summarized in Table 1. In addition to our conventional R32 ribozyme, we also used a 31-mer ribozyme (R31) that lacks one nucleotide at the 3′ end, as compared with the parental 32-mer ribozyme (R32). Our previous experiments demonstrated that, in our ribozyme system, when the chemical cleavage step (kcleav in Fig. 1b) was the rate-limiting step, shortening of the binding arm resulted in a decrease in kcat and an increase in Km. By contrast, if the product dissociation step (kdiss) were the rate-limiting step, shortening of the binding arm would result in an increase in kcat (unpublished results). When R11S was treated with R31 in the presence of either Mg2+ or Mn2+ ions, kcat decreased 25- or 40-fold, respectively, and Km doubled in each case, as compared with the values obtained with the parental R32 ribozyme, demonstrating the rate-limiting chemical cleavage (kcleav). As demonstrated previously (23, 25), kcat in our ribozyme-catalyzed reactions represents the rate of the chemical cleavage step (kcleav): note, for example, that, if kcat represented the rate of the product dissociation step (kdiss in Fig. 1b), then values of kcat should have been the same for both R11S and R11O in the same ribozyme-catalyzed reaction because the cleavage products are nearly identical in both cases except at the 3′ end of the 5′ fragment.
Energy Diagram.
Examination of nonenzymatic and ribozyme (R32)-catalyzed reactions of R11O and R11S in the presence of 0.3 mM Mg2+ ions at pH 6.0 yielded energy diagrams (23) that are replotted in Fig. 3. Relative energies are, for the natural substrate (R11O; Fig. 3 Left) and a modified substrate with a 5′-bridging thiophosphate linkage (R11S; Fig. 3 Right), of reactions catalyzed by a hammerhead ribozyme (solid lines) as well as of nonenzymatic hydrolysis (broken lines). The chemical cleavage step consists of the attack by the 2′-oxygen at C17 on the phosphorus (TS1) and the departure of the 5′ leaving group (TS2). Among the two separate transition states, TS1 and TS2, according to our molecular orbital calculations (33), TS2 is always a higher energy state than TS1, at least in nonenzymatic hydrolysis (Fig. 3 Top Left). This conclusion was confirmed by the previous observation that 5′-thio substrate was more than four orders of magnitude more susceptible to nonenzymatic hydrolysis than the corresponding 5′-oxy substrate (23, 26, 27, 34, 35). If the formation of the pentacoordinate intermediate [P(V)] were rate-limiting (if TS1 were a higher energy state than TS2), R11S should have been hydrolyzed at a rate slower than the rate of hydrolysis of R11O because the 5′-bridging phosphorothioate linkage would not be expected to enhance the attack by 2′-oxygen (36), and because the transition state for R11S with an apical sulfur atom is expected to be less stable than the corresponding transition state for R11O with an apical oxygen (37). By contrast, in the nonenzymatic hydrolysis of R11S (Top Right), TS1 must be a higher energy state than TS2, because the pKa of a thiol is more than 5 units lower than that of an alcohol. In fact, since a pentacoordinate intermediate for the R11S is not expected to exist (37), the actual, single transition state for R11S should be TS1-like, resulting from complete disappearance of TS2 that had been stabilized by the good sulfur leaving group. The nonenzymatic hydrolysis of R11S via the single transition state, TS1-like (Top Right), is more than four orders of magnitude more rapid than the hydrolysis of R11O via the rate-limiting transition state, TS2 (Top Left) (23).
Figure 3.

Relative energies, for the natural substrate (R11O; Left) and a modified substrate with a 5′-bridging thiophosphate linkage (R11S; Right), of reactions catalyzed by a hammerhead ribozyme (solid lines) as well as of nonenzymatic hydrolysis (broken lines). Among the two separate transition states, TS1 and TS2, according to our molecular orbital calculations (33, 44–49), TS2 is always a higher-energy state than TS1 in nonenzymatic hydrolysis (Top Left). This conclusion was confirmed by analysis of the rates of nonenzymatic hydrolysis of R11O and R11S because R11S was more than four orders of magnitude more susceptible to cleavage than was R11O (23). Because the pKa of a thiol is more than 5 units lower than that of an alcohol, TS1 must be a higher energy state than TS2 in the nonenzymatic hydrolysis of R11S (Top Right). Because of the good thio leaving group, a metal ion catalysis may not be required to cleave phosphorus–sulfur bond in the ribozyme-catalyzed reactions (Bottom Right). The nonenzymatic hydrolysis of R11S via TS1 (Top Right) is more than four orders of magnitude more rapid than that of R11O via TS2 (Top Left) (26, 34, 35). In contrast to the earlier conclusion (26), the departure of the 5′ leaving group is also the rate-limiting step in the hammerhead ribozyme-catalyzed reaction with the natural substrate (Bottom Left). The direct coordination of metal ions with the nucleophilic 2′- and leaving 5′-oxygens stabilizes TS1 and TS2 (Bottom Left). The rate of chemical cleavage of R11O is governed by k1, k2, and k−1 [kcat = k1k2/(k−1 + k2)], whereas that of R11S is governed by k1. Under the conditions of present kinetic measurements, the cleaved fragments dissociate from the ribozyme at a higher rate than the rate of chemical reaction (56).
In contrast to the earlier conclusion (26, 27), the departure of the 5′ leaving group is also the rate-limiting step in the hammerhead ribozyme-catalyzed reaction with the natural substrate (Fig. 3 Bottom Left) because, as can be seen in Table 1, the 5′-thio substrate (R11S) was almost two orders of magnitude more susceptible to the R32-mediated cleavage than was the parental 5′-oxy substrate (R11O): the rate constants were 0.34 and 0.004 min−1, respectively. This conclusion does not depend on the life-time of the pentacoordinate intermediate [P(V)] for R11O. If P(V) for R11O were kinetically insignificant (if the reaction were concerted), the actual, single transition state for R11O would be TS2-like (33).
Our proposed reaction mechanism for the ribozyme-catalyzed reaction is also shown in Fig. 3, wherein metal ions are coordinated directly to the attacking and the leaving oxygen atoms (19, 20). This mechanism is nearly identical with the recently proposed double-metal-ion mechanism (28) except for the fact that, in the latter case, deprotonation of the 2′-OH-Mg was proposed to be the rate-limiting step (see below).
The Double-Metal-Ion Mechanism of Catalysis.
We demonstrated previously, from an examination of solvent isotope effects, that a proton-transfer process is not involved in the reactions catalyzed by a hammerhead ribozyme (20, 25). In that study, the rate constant for the ribozyme-mediated cleavage in 1H2O was 4.4 times larger than the corresponding value in 2H2O (D2O). This apparent isotope effect of 4.4 could have been taken as evidence to support an involvement of a proton transfer during the transition state (20, 28). However, because the concentration of the active species (M—O−—R) in D2O is severalfold lower than that in H2O at a fixed pH (38), the reduction in the level of the active species, M—O−—R, in D2O could have been the sole cause of the lower rate of the reaction in D2O. Specifically, the active species represented by M—O−—R is the magnesium-bound 2′-alkoxide. We could estimate the relative concentrations of the active species (M—O−—R) in H2O vs. D2O, assuming that (i) the hydrated metal ions have the same relative ratios of KaH2O/KaD2O as organic acids (38) and (ii) the ratio of the following equilibrium constants (KaM-OH-R/KaM-OD-R) in Eqs. 1 and 2) is the same as that (KaM-OH2/KaM-OD2) in Eqs. 3 and 4, where M and OR represent metal ion and ribose 2′-oxygen, respectively. That is, a change in the equilibrium concentration of metal-bound 2′-alkoxide in the catalytic center (KaM-OH-R/KaM-OD-R) is nearly identical with a change in the equilibrium concentration of metal hydroxide (KaM-OH2/KaM-OD2).
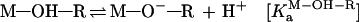 |
1 |
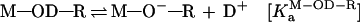 |
2 |
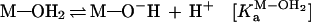 |
3 |
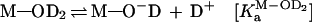 |
4 |
The estimation indicated that the concentration of the active species (M—O−—R) is 4.5 times higher in H2O than that in D2O (20, 25). Therefore, the 4.4-fold lower rate of the reaction in D2O than in H2O was interpreted as the result of the perturbation of the pKa: It was concluded that the reduction in the level of the active species in D2O was the sole cause of the lower rate of ribozyme-catalyzed reactions in D2O (20, 25). Thus, the absence of the actual kinetic isotope effects in the step that leads to cleavage of phosphodiester bonds by ribozymes can be interpreted only in terms of a mechanism in which proton transfer does not take place in the transition state. This observation is consistent with the double-metal-ion mechanism of catalysis (17, 19, 20, 28), in that Mg2+ ions are directly coordinated with the attacking and the leaving oxygens (Fig. 3 Left).
As pointed out by Pontius et al. (28), the direct coordination of the metal ion with the 2′-oxygen of the attacking nucleotide residue, as shown in Fig. 3, polarizes and weakens the 2′-OH bond. As a result, the equilibrium in Eq. 1 shifts to the right, yielding higher concentrations of the active nucleophile, 2′-alkoxide of the ribose (M—O−—R). Therefore, an inverse correlation between the pKa of the metal-bound water molecule (in reality, pKa of the metal-bound ribose 2′-OH) and the ribozyme activity holds (13, 28). Similarly, the direct coordination of the metal ion with the 5′-oxygen of the leaving nucleotide residue, as shown in Fig. 3, weakens the 5′-oxygen-phosphorus bond (19): Metal ions with lower pKa values will weaken the 5′-oxygen-phosphorus bond to a greater extent, and thereby activate the ribozyme-mediated cleavage to a greater extent (19, 28). In this case, the active species should be completely protonated metal-bound water molecules [left species in Eq. 5, where (P)R′O represents the phosphorus-bound 5′-oxygen; see Fig. 3 as well].
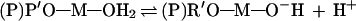 |
5 |
Then, in a pH–rate profile of a ribozyme-catalyzed reaction, one would expect a slope of unity from Eq. 1 and a slope of −1 from Eq. 5, resulting in a bell-shaped pH–rate profile. Such bell-shaped pH–rate profiles are common for imidazole-catalyzed hydrolysis of RNA (39, 40) and RNase A-catalyzed hydrolysis of RNA (41, 42), in that the maximal activity can be found around pH 7, reflecting the pKa of the catalytic molecules (imidazole or histidine). In the case of ribozyme-catalyzed reactions, the activity increases linearly with pH from pH 6 to up to pH 9 (13, 19) in accord with Eq. 1. However, because of the high pKa of the metal-bound water molecules (>10), the decrease in the activity (according to Eq. 5) at an even higher pH is not discernible experimentally.
Experimental Supports for the Double-Metal-Ion Mechanism of Catalysis.
As discussed in the previous section, the absence of the actual kinetic isotope effects indicating that a proton-transfer process is not involved in the reactions catalyzed by a hammerhead ribozyme is consistent with the double-metal-ion mechanism of catalysis (20). The question is: Is there more direct evidence for the double-metal-ion mechanism of catalysis? We believe that quantitative analysis of kinetic parameters listed in Table 1 will provide such evidence. From Eq. 1, we would expect that metal ions with lower pKa will produce higher concentrations of active species (M—O−—R). The pKa (Eq. 3) of Mn2+-bound water molecule is 10.6 and that of Mg2+-bound water molecule is 11.4 (43). This explains the higher catalytic power of Mn2+ in ribozyme reactions (43) because, at a fixed pH, concentrations of active species (Mn2+—O−—R) is roughly 6.3 times higher in Mn2+-containing solution than the corresponding active species (Mg2+—O−—R) in Mg2+-containing solution. However, as can be seen in Table 1, R32-mediated cleavage of R11O is 14 times more efficient in the presence of Mn2+ ions than in the presence of Mg2+ ions, a result that cannot simply be explained by the concentration difference alone of active species (M—O−—R), because the maximum difference should be 6.3 times. This, we believe, suggests an involvement of more than one catalytic metal ion, as depicted in Fig. 3 [The energy diagram for R11O is also consistent with our ab initio molecular orbital calculations (33, 44–49)].
Table 1 provides two sets of data that compare the ribozyme-mediated cleavage of a substrate in the presence of either Mg2+ or Mn2+ ions. The first set of data uses the normal substrate R11O and the second set uses the 5′-thio analog R11S. In the first set of data, we observed that the kinetic ratio (Mn2+/Mg2+) for R11O is 14; the magnitude of this ratio can be taken as an indication for the involvement of two required metals, because it is greater than the theoretical maximum of 6.3. The argument here is that this value of 14 is the additive effect of two essential metals.
The second set of data is the same experiment performed on R11S. For this substrate, the corresponding ratio is 2.2, which is significantly lower than the theoretical maximum of 6.3 and it is greatly lower than the experimental value for R11O of 14. This difference can best be interpreted, on the basis of the energy diagram depicted in Fig. 3, that the rate-limiting step of the reaction for R11S has shifted from the cleavage of the P—(5′-O) bond (in R11O) to the formation of the P—(2′-O) bond (in R11S). In R11S the charge build-up at the 5′-leaving group occurs after the rate-limiting step and, therefore, any metal bound at that position is kinetically insignificant. Then, why is this value only 2.2, in the case of R11S, considering the fact that the concentration of the Mn2+-bound nucleophile is 6.3 times higher than that of the Mg2+-bound nucleophile? This value, k1(Mn)/k1(Mg), of 2.2 for the R32-mediated cleavage of R11S means that the nucleophilicity of the Mn2+-bound 2′-O− was lower than that of the Mg2+-bound 2′-O−. This is reasonable because the nucleophile with a higher pKa (Mg2+-bound 2′-O−) is expected to be less stable, more reactive, and a better nucleophile than the nucleophile with a lower pKa (Mn2+-bound 2′-O−).
In short, in the case of the normal substrate R11O, two metals are required because the kinetic ratio [kcat(Mn)/kcat(Mg)] for R11O is “too large” to be accommodated by a single metal model, and the same ratio for R11S is small enough to represent a single metal involvement. The theoretical maximum value of 6.3 is the calculated ratio of catalytically active species based on the difference in pKa values of the two aqueous metal ions, Mg2+ and Mn2+. Anything over this value must then be due to a second metal involvement. If there were no involvement of metal ions in TS2 for the cleavage of R11O, the overall value of k(Mn)/k(Mg) must be lower than 6.3, as in the case of R11S with an extremely weak P—S bond that does not require an acid catalyst for its cleavage. It is also to be noted that the Mn2+ ion with a lower pKa value will weaken the P—(5′-O) bond to a greater extent than does the Mg2+ ion, just as the former weakens a metal-bound O—H bond to a greater extent than does the latter, resulting in the large overall kinetic ratio [kcat(Mn)/kcat(Mg)] for R11O of 14.
The above argument also holds for concerted reactions with a single transition state, TS2-like and TS1-like for R11O and R11S, respectively. The metal in the 2′ position is the one that correlates with the pKa of the aquated metal ion, reflecting the well known slope of unity in the pH–log(rate) profile (13, 19). Unless there is a metal ion coordinating directly with the 5′ oxygen, the kinetic ratio [kcat(Mn)/kcat(Mg)] for R11O should not exceed the theoretical maximum of 6.3, because only at the 5′ position can Mn2+ ion be a better catalyst than the Mg2+ ion owing to its lower pKa value and weakening the P—(5′-O) bond to a greater extent, while at the 2′ position the nucleophilicity of the Mn2+-bound 2′-O− should be lower than that of the Mg2+-bound 2′-O−, as observed in the case of R11S with the kinetic ratio [k1(Mn)/k1(Mg)] smaller than the theoretical maximum of 6.3. Therefore, present kinetic evidence demands that we invoke the double-metal-ion mechanism of catalysis for reactions catalyzed by hammerhead ribozymes (17), regardless of the presence or absence of the pentacoordinate intermediates. Such double-metal-ion catalysis has been proven to be an efficient mechanism for the cleavage of phosphodiester bonds in nonenzymatic reactions (50–55).
Conclusion.
Based on the relative energies (23), for both a natural substrate and a modified substrate with a 5′-bridging thiophosphate linkage, of reactions catalyzed by a hammerhead ribozyme, we could quantitatively analyze the differential metal ion effects. We conclude that (i) the departure of the 5′-leaving group is the rate-limiting step in the hammerhead ribozyme-catalyzed reaction with the natural substrate; (ii) similar to the cofactor that is involved in reactions catalyzed by the Tetrahymena ribozyme (14, 29), a metal cofactor appears to interact with the leaving group at the transition state (TS2) for the natural substrate, although no switch in metal ion specificity was observed for the 5′-thio substrate (because of a change in the rate-limitnig step); and (iii) it seems likely that hammerhead ribozymes exploit the general double-metal-ion mechanism of catalysis, wherein metal ions are coordinating directly to both the attacking and the leaving oxygen atoms (17, 19, 20, 28, 29).
Acknowledgments
We greatly appreciate the very helpful comments of Prof. Peter H. von Hippel and Dr. William B. Lott. We also thank Drs. Nassim Usman, Francine E. Wincott, Anthony DiRenzo, and Victor Mokler at Ribozyme Pharmaceuticals for the synthesis of R11S (23).
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: R32, 32-mer hammerhead ribozyme; I-R32, 32-mer inactive hammerhead ribozyme; R31, 31-mer hammerhead ribozyme; R11O, 11-mer RNA substrate; R11S, 11-mer RNA substrate that contains a 5′-thio leaving group at the cleavage site.
References
- 1.Symons R H. Trends Biochem Sci. 1989;14:445–450. doi: 10.1016/0968-0004(89)90103-5. [DOI] [PubMed] [Google Scholar]
- 2.Bratty J, Chartrand P, Ferbeyre G, Cedergren R. Biochim Biophys Acta. 1993;1216:345–359. doi: 10.1016/0167-4781(93)90001-t. [DOI] [PubMed] [Google Scholar]
- 3.Symons R H. Annu Rev Biochem. 1992;61:641–671. doi: 10.1146/annurev.bi.61.070192.003233. [DOI] [PubMed] [Google Scholar]
- 4.Uhlenbeck O C. Nature (London) 1987;328:596–600. doi: 10.1038/328596a0. [DOI] [PubMed] [Google Scholar]
- 5.Haseloff J, Gerlach W L. Nature (London) 1988;334:585–591. doi: 10.1038/334585a0. [DOI] [PubMed] [Google Scholar]
- 6.Pley H W, Flaherty K M, McKay D B. Nature (London) 1994;372:68–74. doi: 10.1038/372068a0. [DOI] [PubMed] [Google Scholar]
- 7.Scott W G, Flinch J T, Klug A. Cell. 1995;81:991–1002. doi: 10.1016/s0092-8674(05)80004-2. [DOI] [PubMed] [Google Scholar]
- 8.Scott W G, Klug A. Trends Biochem Sci. 1996;21:220–224. [PubMed] [Google Scholar]
- 9.Scott W G, Murray J B, Arnold J R P, Stoddard B L, Klug A. Science. 1996;274:2065–2069. doi: 10.1126/science.274.5295.2065. [DOI] [PubMed] [Google Scholar]
- 10.Quigley G J, Rich A. Science. 1976;194:796–806. doi: 10.1126/science.790568. [DOI] [PubMed] [Google Scholar]
- 11.Kazakov S, Altman S. Proc Natl Acad Sci USA. 1991;88:9193–9197. doi: 10.1073/pnas.88.20.9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kazakov S, Altman S. Proc Natl Acad Sci USA. 1992;89:7939–7943. doi: 10.1073/pnas.89.17.7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dahm S C, Derrick W B, Uhlenbeck O C. Biochemistry. 1993;32:13040–13045. doi: 10.1021/bi00211a013. [DOI] [PubMed] [Google Scholar]
- 14.Piccirilli J A, Vyle J S, Caruthers M H, Cech T R. Nature (London) 1993;361:85–88. doi: 10.1038/361085a0. [DOI] [PubMed] [Google Scholar]
- 15.Yarus M. FASEB J. 1993;7:31–39. doi: 10.1096/fasebj.7.1.8422972. [DOI] [PubMed] [Google Scholar]
- 16.Uchimaru T, Uebayasi M, Tanabe K, Taira K. FASEB J. 1993;7:137–142. doi: 10.1096/fasebj.7.1.8422960. [DOI] [PubMed] [Google Scholar]
- 17.Steitz T A, Steitz J A. Proc Natl Acad Sci USA. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pyle A M. Science. 1993;261:709–714. doi: 10.1126/science.7688142. [DOI] [PubMed] [Google Scholar]
- 19.Uebayasi M, Uchimaru T, Koguma T, Sawata S, Shimayama T, Taira K. J Org Chem. 1994;59:7414–7420. [Google Scholar]
- 20.Sawata S, Komiyama M, Taira K. J Am Chem Soc. 1995;117:2357–2358. [Google Scholar]
- 21.Bassi G S, Mollegaard N E, Murchie A I H, von Kitzing E, Lilley D M J. Nat Struct Biol. 1995;2:45–55. doi: 10.1038/nsb0195-45. [DOI] [PubMed] [Google Scholar]
- 22.Orita M, Vinayak R, Andrus A, Warashina M, Chiba A, Kaniwa H, Nishikawa F, Nishikawa S, Taira K. J Biol Chem. 1996;271:9447–9454. doi: 10.1074/jbc.271.16.9447. [DOI] [PubMed] [Google Scholar]
- 23.Zhou D-M, Usman N, Wincott F E, Matulic-Adamic J, Orita M, Zhang L-H, Komiyama M, Kumar P K R, Taira K. J Am Chem Soc. 1996;118:5862–5866. [Google Scholar]
- 24.Zhou D-M, Kumar P K R, Zhang L-H, Taira K. J Am Chem Soc. 1996;118:8969–8970. [Google Scholar]
- 25.Kumar P K R, Zhou D-M, Yoshinari K, Taira K. In: Catalytic RNA: Nucleic Acids and Molecular Biology. Eckstein F, Lilley D M J, editors. Vol. 10. Berlin: Springer; 1996. pp. 217–230. [Google Scholar]
- 26.Kuimelis R G, McLaughlin L W. J Am Chem Soc. 1995;117:11019–11020. [Google Scholar]
- 27.Kuimelis R G, McLaughlin L W. Biochemistry. 1996;35:5308–5317. doi: 10.1021/bi952994p. [DOI] [PubMed] [Google Scholar]
- 28.Pontius B W, Lott W B, von Hippel P H. Proc Natl Acad Sci USA. 1997;94:2290–2294. doi: 10.1073/pnas.94.6.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinstein L B, Jones B C N M, Cosstick R, Cech T R. Nature (London) 1997;388:648–653. doi: 10.1038/42076. [DOI] [PubMed] [Google Scholar]
- 30.Strater N, Lipscomb W N, Klabunde T, Krebs B. Angew Chem Int Ed Engl. 1996;35:2024–2055. [Google Scholar]
- 31.Shimayama T, Nishikawa F, Nishikawa S, Taira K. Nucleic Acids Res. 1993;21:2605–2611. doi: 10.1093/nar/21.11.2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inokuchi Y, Yuyama N, Hirashima A, Nishikawa S, Ohkawa J, Taira K. J Biol Chem. 1994;269:11361–11366. [PubMed] [Google Scholar]
- 33.Taira K, Uchimaru T, Storer J W, Yelimiena A, Uebayasi M, Tanabe K. J Org Chem. 1993;58:3009–3017. [Google Scholar]
- 34.Liu X, Reese C B. Tetrahedron Lett. 1995;36:3413–3416. [Google Scholar]
- 35.Thomson J B, Patel B K, Jimenez V, Eckstein F. J Org Chem. 1996;61:6273–6281. doi: 10.1021/jo960795l. [DOI] [PubMed] [Google Scholar]
- 36.Komiyama M, Bender M L. Bull Chem Soc Jpn. 1980;53:1073–1076. [Google Scholar]
- 37.Uchimaru T, Stec W J, Taira K. J Org Chem. 1997;62:5793–5800. [Google Scholar]
- 38.Jencks W P. Catalysis in Chemistry and Enzymology. New York: McGraw-Hill; 1969. pp. 250–253. [Google Scholar]
- 39.Breslow R, Labelle M. J Am Chem Soc. 1986;108:2655–2659. [Google Scholar]
- 40.Breslow R, Xu R. Proc Natl Acad Sci USA. 1993;90:1201–1207. doi: 10.1073/pnas.90.4.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.del Rosario E J, Hammes G G. Biochemistry. 1969;8:1884–1889. doi: 10.1021/bi00833a017. [DOI] [PubMed] [Google Scholar]
- 42.Richard F M, Wyckoff H W. In: The Enzymes. 3rd Ed. Boyer P D, editor. Vol. 4. New York: Academic; 1971. pp. 647–806. [Google Scholar]
- 43.Pan T, Long D V, Uhlenbeck O C. In: The RNA World. Gesteland R F, Atkins J F, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. pp. 271–302. [Google Scholar]
- 44.Taira K, Uchimaru T, Tanabe K, Uebayasi M, Nishikawa S. Nucleic Acids Res. 1991;19:2747–2753. doi: 10.1093/nar/19.10.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uchimaru T, Uebayasi M, Hirose T, Tsuzuki S, Yelimiena A, Tanabe K, Taira K. J Org Chem. 1996;61:1599–1608. doi: 10.1021/jo951517+. [DOI] [PubMed] [Google Scholar]
- 46.Taira K, Uebayashi M, Maeda H, Furukawa K. Protein Eng. 1990;3:691–701. doi: 10.1093/protein/3.8.691. [DOI] [PubMed] [Google Scholar]
- 47.Uchimaru T, Tanabe K, Nishikawa S, Taira K. J Am Chem Soc. 1991;113:4351–4353. [Google Scholar]
- 48.Storer J W, Uchimaru T, Tanabe K, Uebayasi M, Nishikawa S, Taira K. J Am Chem Soc. 1991;113:5216–5217. [Google Scholar]
- 49.Uchimaru T, Tsuzuki S, Storer J W, Tanabe K, Taira K. J Org Chem. 1994;59:1835–1843. [Google Scholar]
- 50.Wall M, Hynes R C, Chin J. Angew Chem Int Ed Engl. 1993;32:1633–1635. [Google Scholar]
- 51.Tsubouchi A, Bruice T C. J Am Chem Soc. 1994;116:11614–11615. [Google Scholar]
- 52.Ishikubo A, Yashiro M, Komiyama M. Nucleic Acids Res Symp Ser. 1995;34:85–86. [PubMed] [Google Scholar]
- 53.Yashiro, M., Ishikubo, A. & Komiyama, M. (1995) J. Chem. Soc. Chem. Commun. 1793–1794.
- 54.Young M J, Chin J. J Am Chem Soc. 1995;117:10577–10578. [Google Scholar]
- 55.Chapman W H, Jr, Breslow R J. J Am Chem Soc. 1995;117:5462–5469. [Google Scholar]
- 56.Warashina, M., Takagi, Y., Sawata, S., Zhou, D.-M., Kuwabara, T. & Taira, K. (1997) J. Org. Chem. 62, in press.