

Abstract
Red blood cells with abnormal hemoglobins (Hb) are frequently associated with increased hemoglobin autoxidation, accumulation of iron in membranes, increased membrane damage and a shorter red cell life span. The mechanisms for many of these changes have not been elucidated. We have shown in our previous studies that hydrogen peroxide formed in association with hemoglobin autoxidation reacts with hemoglobin and initiates a cascade of reactions that results in heme degradation with the formation of two fluorescent emission bands and the release of iron. Heme degradation was assessed by measuring the fluorescent band at ex 321 nm. A 5.6 fold increase in fluorescence was found in red cells from sickle transgenic mice that expressed exclusively human globins when compared to red cells from control mice. When sickle transgenic mice co-express the γM transgene, that expresses HbF and inhibits polymerization, heme degradation is decreased. Mice expressing exclusively hemoglobin C had a 6.9 fold increase in fluorescence compared to control. Heme degradation was also increased 3.5 fold in β-thalassemic mice generated by deletion of murine βmajor. Membrane bound IgG and red cell metHb were highly correlated with the intensity of the fluorescent heme degradation band. These results suggest that degradation of the heme moiety in intact hemoglobin and/or degradation of free heme by peroxides are higher in pathological RBCs. Concomitant release of iron appears to be responsible for the membrane damage that leads to IgG binding and the removal of red cells from circulation.
Keywords: Heme degradation, fluorescence, transgenic mice, sickle cell, thalassemia, hemoglobin
Introduction
Under normal physiological conditions about 3% of the total body hemoglobin undergoes autoxidation every day producing metHb and superoxide [1]. Preservation of red cell integrity and function requires that antioxidant enzymes and metHb reductase are able to cope with the oxidative load. The level of oxidative stress may increase in the presence of destabilizing hemoglobin mutations or chain imbalance leading to thalassemia. Three systems in which oxidative stress has recently been highlighted are sickle cell disease, thalassemia, and hemoglobin C disease [2–7]. Sickle cell disease is caused by a substitution of valine for glutamic acid at the β-6 position in the hemoglobin β-chain. This substitution results in hemoglobin S (HbS) that polymerizes under deoxygenated conditions. Hemoglobin C (HbC) is the result of a mutation at the same β-6 position that leads to intraerythrocytic crystal formation under oxygenated conditions[8]. Both HbS and HbC are less stable than normal adult hemoglobin (HbA). Hence autoxidation is increased and subsequent production of reactive oxygen species (ROS) increases. Normal red cell function is also dependent on balanced expression of hemoglobin α and β chains. Thalassemia is the result of an excess of α or β chains that are less stable than tetrameric hemoglobin leading to the destruction of excess chains, release of heme and heme iron, and oxidative stress. It has been reported that heme and non-heme iron accumulate in the membranes of pathological RBCs [6] and it is believed that redox cycling of this heme iron and non-heme iron is involved in membrane damage and hemolysis.
We have shown in previous studies that heme degradation is a reliable indicator of the formation of ROS in red cells both in vitro and in vivo. This is based on the reaction of hydrogen peroxide with hemoglobin, which initiates a cascade of oxidative reactions, resulting in degradation of heme with the formation of two fluorescent heme degradation products and the release of iron [9–12]. This reaction occurs when either hemoglobin or red cells react with hydrogen peroxide, but also during hemoglobin autoxidation, during the storage of red cells and even in vivo. We have, thus, shown an increase in the fluorescent degradation products for old cells in circulation and for hemoglobin CC patients where the hemoglobin is less stable (communicated). Atamna and Ginsburg reported that degradation of heme by glutathione also takes place in vivo and can account for the accumulation of non-heme iron in the membranes of red cells with unstable hemoglobins [13].
In the present study, we further investigated the effect of unstable hemoglobins on the in vivo formation of fluorescent degradation products using transgenic mouse models. Transgenic mouse models for pathological hemoglobins are useful experimental tools for testing experimental hypothesis and interventions. Transgenic mice expressing HbS [14–16] and HbC [17] were generated by inserting human transgenes that code for human α- and mutant β-chains, and by knocking out murine α- and β-globins. The thalassemia model used here is the result of a homozygous deletion of murine βmajor [18]. We found that heme degradation was elevated in these three models as assessed by measuring the fluorescence band at ex. 321 nm.
Materials and Methods
Preparation of RBCs and Hb
Blood was collected into tubes containing EDTA as an anticoagulant by tail incision and kept on ice. Blood was centrifuged at 3000 RPM for 10 min at 2– 4°C. The RBC pellet was washed twice with 15 volumes of ice cold phosphate buffered saline (PBS), pH 7.4. Cells were diluted to 50% hematocrit with ice cold PBS, pH 7.4, containing 100 µM EDTA. Cells were stored at 4°C and shipped overnight, then stored at 4°C upon receipt until the analysis was completed within 24 hrs.
Measurement of basal RBC fluorescence
20 µl washed RBCs were lysed in 3 ml ice-cold deionized double distilled water. The Hb spectrum of the hemolysate was recorded from 490 nm to 640 nm using a Perkin Elmer lamda 6 spectrophotometer. The concentrations of oxyHb and metHb were determined by a least square-fitting program using spectra of oxyHb and metHb at known concentrations. The Hb concentration of the hemolysate was then adjusted to 50 µM and the fluorescent emission (em) spectrum was measured from 400 nm to 600 nm at an excitation (ex) wavelength of 321 nm using a Perkin Elmer LS50B spectrofluorimeter. The fluorescence intensity of emission maximum at 480 nm was used as a measure of the heme degradation products. The ex and em slit widths were kept at 10 nm.
Transgenic mice
Animal studies involving transgenic mice were performed at the Bronx Comprehensive Sickle Cell Center and were approved by the Animal Institute Committee of AECOM. All transgenic mice described below (Table 1) have a single copy of the relevant transgene(s).
Table 1.
Mice used in experiments
| Nick-name | αβ-globin transgene name | Description of transgene | γ construct | Mouse α-knockout | Mouse β-knockout or deletion | % human α | % human β | % human γ | % retics | Hct |
|---|---|---|---|---|---|---|---|---|---|---|
| C57Bl | -- | -- | -- | +//+ | +//+ | 0 | 0 | 0 | 2.2 ± 0.5 | |
| THAL | -- | -- | -- | + // + | Hbbth-1//Hbbth-1¶ | 0 | 0 | 0 | 24.6±3.3 | |
| NY1DD | NY1 | miniLCRα2, miniLCRβS* | -- | + // + | Hbbth-1//Hbbth-1¶ | 45 | 73 | 0 | 4.3± 0.4 | |
| NY1KO γM | NY1 | miniLCRα2, miniLCRβS* | γM £ | Hba0//Hba0 § | Hbb0//Hbb0 † | 100 | 80 | 20 | 30.1±9.6 | |
| NY1KO γH | NY1 | miniLCRα2, miniLCRβS* | γH £ | Hba0//Hba0 § | Hbb0//Hbb0 † | 100 | 60 | 40 | 12.9±2.7 | |
| BERK § | BERK | miniLCR, α1, GγAγδβS | -- | Hba0//Hba0 § | Hbb0//Hbb0 ‡ | 100 | <99 | >1 | 36.5±8.0 | |
| BERK γM § | BERK | miniLCR, α1, GγAγδβS | γM £ | Hba0//Hba0 § | Hbb0//Hbb0 ‡ | 100 | 79 | 21 | 37.2±5.8 | |
| HbC-lo | HbC | miniLCRα2, miniLCRβC** | -- | +//Hba0 § | +//Hbb0 † | 68 | 51 | -- | 3.9 ± 0.6 | |
| HbCKO-high | HbC | miniLCRα2, miniLCRβC** | -- | Hba0//Hba0 § | Hbb0//Hbb0 † | 100 | 100 | -- | 13.9 ± 1.3 |
All mice have a single copy of the relevant transgene(s). A “+” denotes wild type. Hba0 indicates that the mouse α has been knocked out; Hbb0 indicates that mouse β has been knocked out; Hbbth-1 indicates deletion of the mouse βmajor.
[15]
ref. [17]
ref. [35]
mouse βmajor deletion on a C57BL/6J background [18]
[19]
[20]
[36]
Thalassemic mice
Beta-thalassemic mice (Thal or DD) were homozygous for deletion of mouse βmajor [18]. The genotype was determined by IEF and HPLC as previously described [16].
Sickle transgenic mice
Five groups of sickle cell transgenic mice were tested. Group 1 mice (BERK or αHβS αKKβKK) were obtained from Chris Pàszty [14] and backcrossed onto a C57/BL6 background. These mice express 100% of all α-globin as human α and 100% of all β-globin as human βS. Group 2 are BERK mice that also express γM (BERK-γM) as previously described [16]. These mice express 100% human α, 79% human βS, and 21% human γ. Group 3 animals (NY1DD or αHβS αβDD) were generated as described [15]. These mice have a single copy of the NY1 transgene (co-integrated LCRα and LCRβS) [15] and are homozygous for the mouse βmajor deletion [18]. They express 45% of all α-globins as human α and 73% of all β-globins as human βS. Group 4 animals express the NY1 transgene and the γM transgene (NY1KO-γM or αHβS γM αKKβKK) and are homozygous for the mouse α-globin knockout [19] and the mouse β-globin knockout [20] as previously described [16]. These mice express 100% human α, 80% βS, and 20% human γ. Group 5 animals also express NY1 transgene and the γH transgene (NY1KO- γH); they express 100% human α, 60% βS, and 40% human γ as previously described [16].
Hemoglobin C
Two groups of mice expressing hemoglobin C were tested that have a co-integrated LCRα and LCRβC [17]. Group 1 mice (HbC-lo or αHβC αKβK) had a single wild-type copy and a single knocked-out copy of the mouse α-globin and β-globin. They express 68% human α and 51% human βC. Group 2 mice (HbCKO or αHβC αKKβKK) are homozygous for both the mouse α- and β-knockouts and express 100% human α and βC.
Determination of RBC bound IgG
Binding of autologous IgG to RBCs from transgenic mice was detected by using anti-IgG antibody labeled with FITC. Washed RBCs at 2% hematocrit (in PBS buffer without EDTA) were incubated with or without FITC-labeled goat anti mouse IgG in PBS for 60 min on ice. Then unbound IgG was removed by washing 3 times with PBS with 1.2% BSA. Fluorescence of these cells was determined by flow cytometry at an ex of 488 nm and em of 530 nm. Bound IgG fluorescence is calculated by subtraction of control cell fluorescence from the fluorescence of IgG antibody treated cells.
Results
Heme degradation products in thalassemic transgenic mice
An increase in oxidative stress due to enhanced autoxidation of Hb has been shown in thalassemic RBCs [6]. Heme degradation, associated with oxidative stress, is also expected to increase for these RBCs. To test this hypothesis, heme degradation was measured in RBC of mice that were thalassemic. Figure 1 shows the increase in fluorescence with p<0.001 for the thalassemic mice with homozygous deletion of mouse βmajor relative to control mice (C57/Bl6).
Fig. 1.
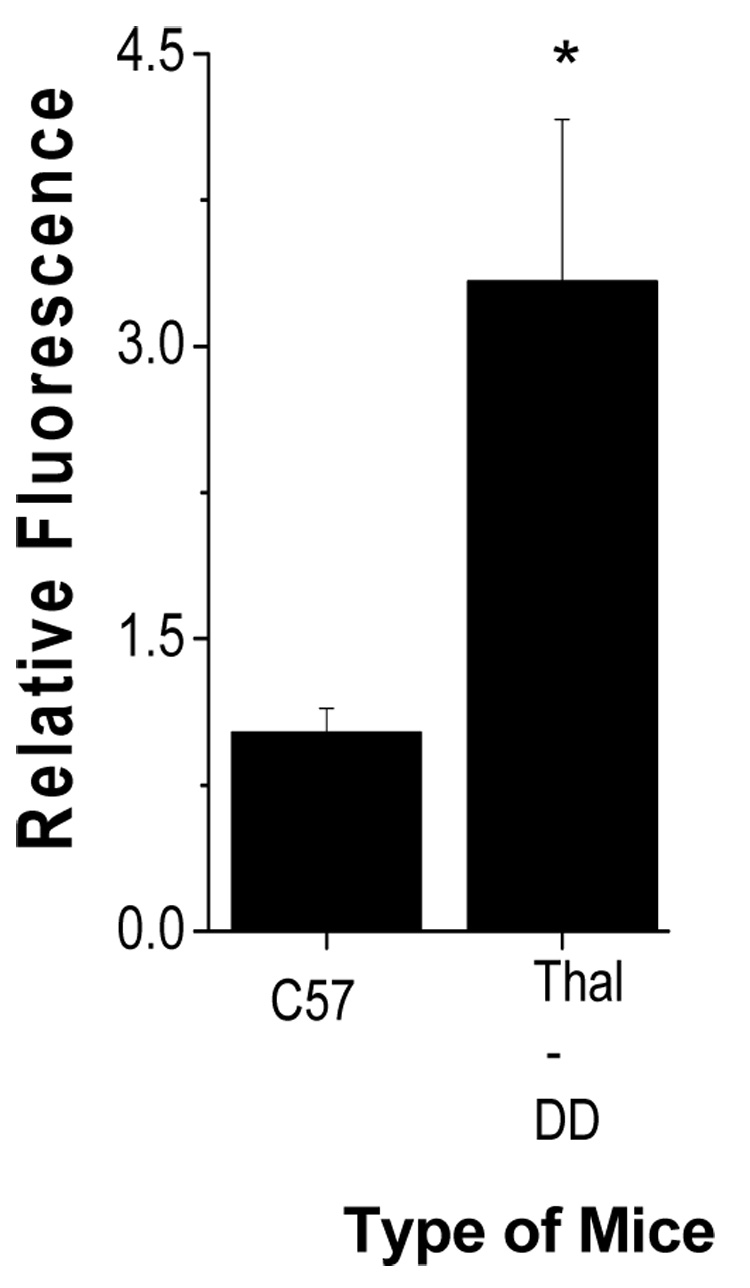
Heme degradation in thalassemic transgenic mice: Hemoglobin concentration of the hemolysate was adjusted to 50 µM and the fluorescence intensity was measured at ex 321 nm and em 480 nm. Details of the procedure used and the characterization of the mouse mutants are given in the Methods section and Table 1. Values are mean ± SD of 4 to 10 animals in each group ( control mice (C57) and thalassemic mice (dd)). *, p <0.001 relative to control.
Heme degradation products in sickle transgenic mice
An increase in oxidative stress is also reported in sickle cells [7]. The sickle transgenic mice are, therefore, expected to have increased heme degradation. Two lines of mice expressing HbS were examined: BERK and NY1 (Fig 2, Table 1). A 5.6 fold increase in fluorescence was found for BERK mice that express exclusively human globins [14] when compared to C57/Bl6 mice (Fig. 2).These mice, in addition to their expression of the human βS gene, also have unbalanced chain synthesis resulting in a β-thalassemia-like condition [16]. Fetal hemoglobin (HbF, α2γ2) inhibits polymer formation and when the γM transgene that resulted in approximately 21% HbF (see Table 1) is co-expressed (BERK-γM), its presence resulted in decreased heme degradation. The presence of γ has two effects: 1) as a beta-like globin it reduces the thalassemia-like behavior in the BERK mouse red cell and 2) it reduces polymer formation. A less severe sickle transgenic model (NY1DD) that has been extensively characterized [15] did not result in significantly increased heme degradation. However, NY1 mice with homozygous knockouts of both mouse α- and β-globin, combined with γM (NY1KO-γM that resulted in expression of about 20% HbF, Table 1) resulted in a significant increase in heme degradation, similar to that of BERK mice. Mice with higher expression of human γ (NY1KO-γH that resulted in expression of about 40% HbF, Table 1) had a partial reversal of the increased heme degradation that is consistent with increased inhibition of polymer formation at higher levels of γ expression [16].
Fig. 2.
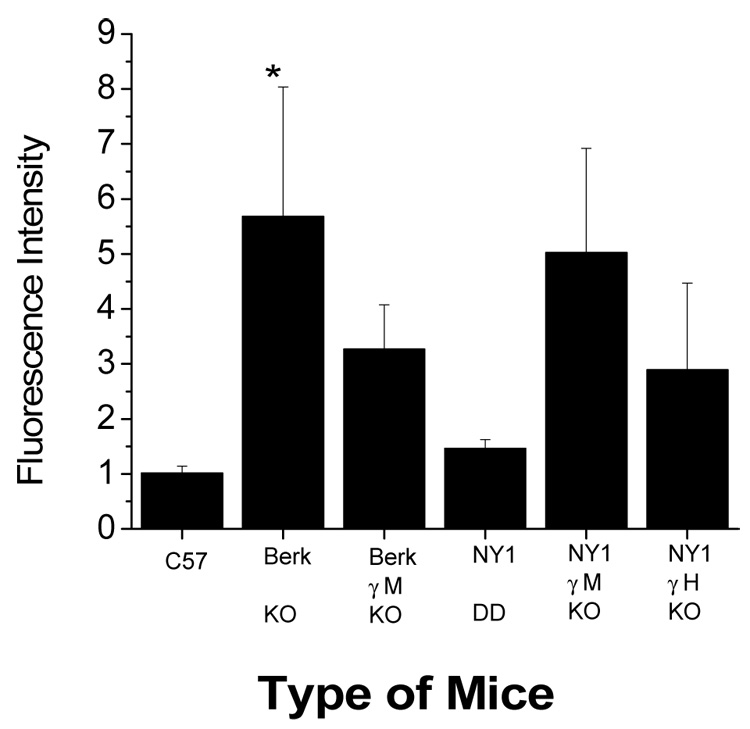
Heme degradation in sickle transgenic mice. Fluorescence was measured as mentioned in Figure 1. The mice corresponding to each column are explained in the methods section and table 1. *, p < 0.001 relative to control.
Heme degradation in Hb CC transgenic mice
Figure 3 shows heme degradation in the red cells from two types of transgenic mice with different levels of human HbC. A partial knockout of both mouse α- and β-globin resulted in mice expressing 68% human α and 51% human βC [17], these mice had a 3 fold increase in fluorescence compared to C57/Bl6 mice (p < 0.001) (Fig. 3). Mice expressing exclusively HbC due to homozygous knockouts of both mouse α- and β-globin resulted in a 6.9 fold increase in fluorescence compared to the control (p < 0.001).
Fig. 3.
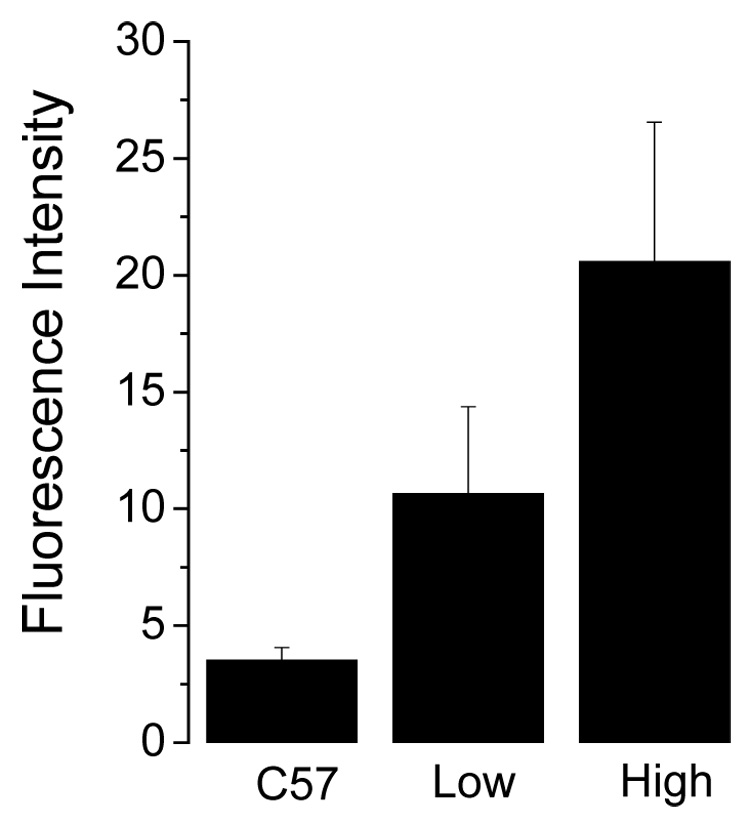
Heme degradation in HbC mice. Fluorescence was measured as mentioned in figure 1. * p <0.001 relative to control.
Correlation of heme degradation with Hb oxidation and IgG binding to RBC
Autoxidation of oxyHb produces an equivalent molar ratio of metHb and superoxide. Measurement of metHb is therefore, an indirect indicator of oxidative stress. Under normal physiological conditions this metHb is reduced back to Fe(II)-Hb by metHb reductase. However, the ability to maintain Hb in a reduced state decreases under pathological conditions and metHb, which is non-functional, accumulates. The mean metHb value for each group of sickle, thalassemic and HbC RBCs was compared with the mean fluorescence value for the same group of mice. Figure 4A shows that the metHb content of RBCs was significantly correlated (r=0.9655 p < 0.001) with the fluorescence of the same cells.
Fig. 4.
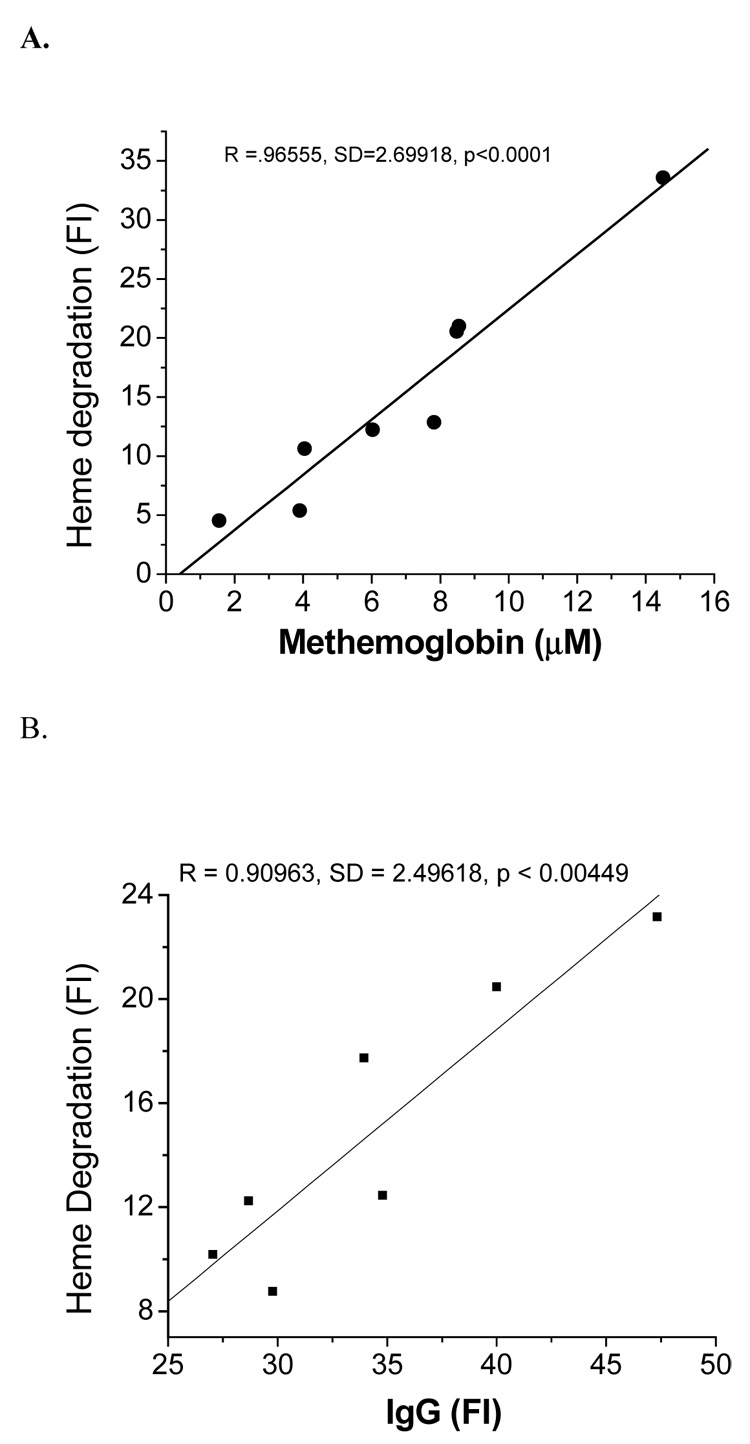
Correlation of heme degradation with metHb and RBC bound IgG: A) Correlation of RBCs metHb with heme degradation of transgenic mice. B) Correlation of RBC bound autologous IgG with heme degradation of transgenic mice. Heme degradation and IgG binding was measured by fluorescence intensity (FI) as described in the methods section.
Binding IgG to senescent cell antigens of the RBC is a trigger for macrophages to recognize senescent cells and remove them from circulation. It has been shown that oxidative stress increases binding of autologous IgG to RBC by inducing the formation of senescent cell antigens [21, 22]. Therefore, the average IgG level of each group of sickle, thalassemia and HbC transgenic mice was compared with the average fluorescence intensity. Binding of IgG antibodies to RBCs was correlated (r = 0.909, p <0.05) to the intensity of the fluorescence (Fig. 4B).
Discussion
Fluorescence in Pathological RBC
Our earlier studies indicate that the reaction of hydrogen peroxide with Hb or autoxidation of oxyHb produces two fluorescent bands in which the dominant band has an em 465 nm at an ex 321 nm [9, 11]. We have also recently reported that fresh red cell lysates have a similar fluorescence band at ex of 321nm with the maximum fluorescence at 480 nm (communicated). The formation of this fluorescence has been attributed to heme degradation by ROS, which are generated from the autoxidation of membrane bound hemoglobin.
Several studies have shown that the autoxidation of oxyHb as well as binding of Hb to cell membranes increases in human hemoglobinapathies. The heme degradation and resultant fluorescence formation is, thus, expected to increase with such pathological RBCs. This expected relationship between RBC oxidative stress and the intensity of the fluorescence signal is supported in transgenic mice models by a comparison of blood from normal mice with transgenic mice expressing human sickle Hb (HbS), human hemoglobin C (HbC) and mouse models of β-thalassemia (Fig. 1–3). In all of these cases, heme degradation increases from 1.5 to 6.9 fold depending on the severity of the generated disease state for thalassemia [6], sickle cell [7] and hemoglobin C [23]. The correlation of the fluorescence with an increase in the level of metHb supports the contention that heme degradation as a measure of oxidative stress is coupled to Hb autoxidation (Fig. 4A).
For HbC and HbS the increased levels of metHb are attributed to the increased rates of autoxidation for these Hbs [7, 23]. For thalassemia increased rates of autoxidation can be attributed to degradation of excess α-chains. This effect is comparable to the 17 fold increase in rates of autoxidation found when tetrameric Hb dissociates into dimers [3].
Two lines of sickle transgenic mice were studied, BERK mice were compared to NY1KO-γM mice, because we have previously shown that the hematological indices of these two types of mice are similar [16]. The greater heme degradation for the BERK mice than the NY1KO-γM mice can be attributed to the presence of thalassemia in addition to the presence of the unstable Hb, HbS. Increasing the amount of HbF decreases the amount of degradation product present, which may be due either to reduction of polymer formation by γ, or to stabilization of βS in the heterotetramer (αH2βS γ). Both HbS [24] and HbC [25] have an excess positive charge relative to HbA and have been shown to preferentially bind to Band 3. The presence of γ or αmouse in heterotetramers may reduce the amount of membrane-associated, unstable β-chains. The low level of degradation products in NY1DD mice may, thus, be due to reduced polymer formation (the residual mouse α-chains inhibit polymer formation as efficiently as human γ) [26] or the more efficient interaction of murine α-globin with murine alpha Hb sparing protein (AHSP) [27]. Since NY1DD and NY1KO-γM mice have about the same percent βS, but very different levels of heme degradation products, we speculate that inefficient interaction of human α with murine AHSP may also contribute to the oxidative environment of murine RBCs that express exclusively human Hbs. Similarly, mouse α and its interaction with murine AHSP may stabilize βC in the HbC-lo mouse or reduce the amount of membrane-associated, unstable β-chains.
These results support the contention that heme degradation occurs in vivo and that the level of the heme degradation products reflect the extent of oxidative stress.
Membrane damage associated with in vivo heme degradation
Hb is known to bind to membrane Band 3 protein especially under hypoxic conditions [28]. HbS and HbC have a higher affinity for band 3 than HbA [24, 25]. Hydrogen peroxide generated by autoxidation of membrane associated Hb may be relatively inaccessible to catalase and generate more heme degradation products [29]. Higher metHb levels are found in these pathological RBCs (Fig 4A). Degradation of heme releases iron, which can accumulate in the membrane. MetHb binds the heme less tightly than the Fe(II) forms of hemoglobin. The higher levels of metHb in the transgenic mice can, therefore, result in the release of hemin, which would be expected to deposit in the hydrophobic membrane [30]. Redox cycling of hemin and iron initiates lipid peroxidation to form lipid hydroperoxide. These lipid hydroperoxides also degrade heme to form fluorescent products.
A relationship between heme degradation and the removal of senescent cells from circulation is implied by our finding that the level of autologous IgG binding to the membrane correlates with the level of cellular heme degradation products (Fig.4B).The relationship between heme degradation and IgG binding can be attributed to a direct effect of the degradation products. It can, however, also be attributed to the release of iron associated with heme degradation and enhanced denaturation of hemoglobin that is expected when one of the hemes of hemoglobin are degraded. Both of theses factors (heme free membrane iron and hemoglobin denaturation) have been linked to the aggregation or clustering of Band 3 protein, which is has been implicated as a signal for macrophages to clear senescent RBCs from circulation [31, 32].
In transgenic mice, the increase in heme degradation and binding of IgG may reflect the level of oxidative stress. During cellular aging, heme degradation has been shown to reflect the accumulated oxidative stress that cells have been exposed to in the circulation. It is, thus, the resultant membrane damage from this accumulated oxidative stress that results in IgG binding and the removal of senescent cells.
Fluorescence reflects the oxidative stress of RBC
Oxidative stress is widely assessed by lipid peroxidation, which is frequently evaluated by measuring malondialdehyde (MDA), a low-molecular weight end product of lipid peroxidation, using the thiobarbituric acid reaction. This method is not specific for MDA because thiobarbituric acid also reacts with other aldehydes to give the same absorption maximum [33]. Measurements of fluorescence in lipid extracts are also widely used to determine oxidative stress. These fluorescent products are attributed to formation of conjugated Schiff base compounds through the interaction of aldehydes with the amino group of phospholipids [34]. Determination of MDA, lipid hydroperoxides and 4-hydroxynonenol by HPLC or mass spectroscopy is a more reliable method to assess lipid peroxidation.
While theses methods measure oxidative stress they do not generally distinguish between different sources of oxidative stress present in the circulatory system. Heme degradation has, however, been shown (Nagababu et al communicated) to specifically reflect oxidative stress that originates from the red cell.
The measurement of fluorescence in cell lysates by spectrofluorimeter is an easy and highly sensitive method to assess RBC oxidative stress especially in pathological conditions and avoids the highly laborious and time consuming HPLC and mass spectrometry methods required for reliable measures of lipid peroxidation. In addition, the determination of fluorescent degradation products makes it possible to delineate the contribution of red cells to oxidative stress. This approach is particularly valuable in studying red cell mutants where we want to evaluate the contribution of the altered hemoglobin to oxidative stress.
In summary, fluorescence arising from heme degradation increases in RBCs with unstable Hbs. The increased fluorescence intensity reflects increased oxidative stress. Associated with heme degradation is an increase in free iron and hemoglobin instability, which together result in membrane damage and the exposure of senescent antigens, one of the major pathways for removal of aged cells from circulation.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute on Aging and by the Bronx Comprehensive Sickle Cell Center, NHLBI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jaffe ER, Neurmann G. A Comparision of the Effect of Menadione, Methylene Blue and Ascorbic Acid on the Reduction of Methemoglobin in Vivo. Nature. 1964;202:607–608. doi: 10.1038/202607a0. [DOI] [PubMed] [Google Scholar]
- 2.Shikama K. The Molecular Mechanism of Autoxidation for Myoglobin and Hemoglobin: A Venerable Puzzle. Chem. Rev. 1998;98:1357–1374. doi: 10.1021/cr970042e. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Levy A, Rifkind JM. Autoxidation of hemoglobin enhanced by dissociation into dimers. J. Biol. Chem. 1991;266:24698–24701. [PubMed] [Google Scholar]
- 4.Collman JP, Boulatov R, Sunderland CJ, Fu L. Functional analogues of cytochrome c oxidase, myoglobin, and hemoglobin. Chem. Rev. 2004;104:561–588. doi: 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]
- 5.M.H.F. Steinberg BG, Higgs DR, Nagel RL. Disorders of hemoglobin: genetics, Pathophysiology and Clinical Management. UK: cambridge University press; 2001. [Google Scholar]
- 6.Shinar E, Rachmilewitz EA. Oxidative denaturation of red blood cells in thalassemia. Semin. Hematol. 1990;27:70–82. [PubMed] [Google Scholar]
- 7.Hebbel RP. The sickle erythrocyte in double jeopardy: autoxidation and iron decompartmentalization. Semin. Hematol. 1990;27:51–69. [PubMed] [Google Scholar]
- 8.Hirsch RE, Raventos-Suarez C, Olson JA, Nagel RL. Ligand state of intraerythrocytic circulating HbC crystals in homozygote CC patients. Blood. 1985;66:775–777. [PubMed] [Google Scholar]
- 9.Nagababu E, Rifkind JM. Formation of fluorescent heme degradation products during the oxidation of hemoglobin by hydrogen peroxide. Biochem. Biophys. Res. Commun. 1998;247:592–596. doi: 10.1006/bbrc.1998.8846. [DOI] [PubMed] [Google Scholar]
- 10.Nagababu E, Rifkind JM. Heme degradation by reactive oxygen species. Antioxid. Redox Signal. 2004;6:967–978. doi: 10.1089/ars.2004.6.967. [DOI] [PubMed] [Google Scholar]
- 11.Nagababu E, Rifkind JM. Heme degradation during autoxidation of oxyhemoglobin. Biochem. Biophys. Res. Commun. 2000;273:839–845. doi: 10.1006/bbrc.2000.3025. [DOI] [PubMed] [Google Scholar]
- 12.Nagababu E, Rifkind JM. Reaction of hydrogen peroxide with ferrylhemoglobin: superoxide production and heme degradation. Biochemistry. 2000;39:12503–12511. doi: 10.1021/bi992170y. [DOI] [PubMed] [Google Scholar]
- 13.Atamna H, Ginsburg H. Heme degradation in the presence of glutathione. A proposed mechanism to account for the high levels of non-heme iron found in the membranes of hemoglobinopathic red blood cells. J. Biol. Chem. 1995;270:24876–24883. doi: 10.1074/jbc.270.42.24876. [DOI] [PubMed] [Google Scholar]
- 14.Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, et al. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science. 1997;278:876–878. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 15.Fabry ME, Nagel RL, Pachnis A, Suzuka SM, Costantini F. High expression of human beta S- and alpha-globins in transgenic mice: hemoglobin composition and hematological consequences. Proc. Natl. Acad. Sci. U S A. 1992;89:12150–12154. doi: 10.1073/pnas.89.24.12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fabry ME, Suzuka SM, Weinberg RS, Lawrence C, Factor SM, et al. Second generation knockout sickle mice: the effect of HbF. Blood. 2001;97:410–418. doi: 10.1182/blood.v97.2.410. [DOI] [PubMed] [Google Scholar]
- 17.Fabry ME, Romero JR, Suzuka SM, Gilman JG, Feeling-Taylor A, et al. Hemoglobin C in transgenic mice: effect of HbC expression from founders to full mouse globin knockouts. Blood Cells Mol. Dis. 2000;26:331–347. doi: 10.1006/bcmd.2000.0313. [DOI] [PubMed] [Google Scholar]
- 18.Skow LC, Burkhart BA, Johnson FM, Popp RA, Popp DM, et al. A mouse model for beta-thalassemia. Cell. 1983;34:1043–1052. doi: 10.1016/0092-8674(83)90562-7. [DOI] [PubMed] [Google Scholar]
- 19.Paszty C, Mohandas N, Stevens ME, Loring JF, Liebhaber SA, et al. Lethal alpha-thalassaemia created by gene targeting in mice and its genetic rescue. Nat. Genet. 1995;11:33–39. doi: 10.1038/ng0995-33. [DOI] [PubMed] [Google Scholar]
- 20.Shehee WR, Oliver P, Smithies O. Lethal thalassemia after insertional disruption of the mouse major adult beta-globin gene. Proc. Natl. Acad. Sci. U S A. 1993;90:3177–3181. doi: 10.1073/pnas.90.8.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lutz HU, Bussolino F, Flepp R, Fasler S, Stammler P, et al. Naturally occurring anti-band-3 antibodies and complement together mediate phagocytosis of oxidatively stressed human erythrocytes. Proc. Natl. Acad. Sci. U S A. 1987;84:7368–7372. doi: 10.1073/pnas.84.21.7368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lutz HU, Fasler S, Stammler P, Bussolino F, Arese P. Naturally occurring anti-band 3 antibodies and complement in phagocytosis of oxidatively-stressed and in clearance of senescent red cells. Blood Cells. 1988;14:175–203. [PubMed] [Google Scholar]
- 23.Saad S, Salles SI, Velho PE. Decreased reduced glutathione and glutathione reductase activity in subjects with hemoglobin C. Nouv Rev Fr Hematol. 1991;33:11–14. [PubMed] [Google Scholar]
- 24.Shaklai N, Sharma VS, Ranney HM. Interaction of sickle cell hemoglobin with erythrocyte membranes. Proc. Natl. Acad. Sci. U S A. 1981;78:65–68. doi: 10.1073/pnas.78.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiss GH, Ranney HM, Shaklai N. Association of hemoglobin C with erythrocyte ghosts. J. Clin. Invest. 1982;70:946–952. doi: 10.1172/JCI110706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhoda MD, Domenget C, Vidaud M, Bardakdjian-Michau J, Rouyer-Fessard P, et al. Mouse alpha chains inhibit polymerization of hemoglobin induced by human beta S or beta S Antilles chains. Biochim. Biophys. Acta. 1988;952:208–212. doi: 10.1016/0167-4838(88)90117-3. [DOI] [PubMed] [Google Scholar]
- 27.Feng L, Gell DA, Zhou S, Gu L, Kong Y, et al. Molecular mechanism of AHSP-mediated stabilization of alpha-hemoglobin. Cell. 2004;119:629–640. doi: 10.1016/j.cell.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 28.Shaklai N, Yguerabide J, Ranney HM. Interaction of hemoglobin with red blood cell membranes as shown by a fluorescent chromophore. Biochemistry. 1977;16:5585–5592. doi: 10.1021/bi00644a031. [DOI] [PubMed] [Google Scholar]
- 29.Nagababu E, Chrest FJ, Rifkind JM. Hydrogen-peroxide-induced heme degradation in red blood cells: the protective roles of catalase and glutathione peroxidase. Biochim. Biophys. Acta. 2003;1620:211–217. doi: 10.1016/s0304-4165(02)00537-8. [DOI] [PubMed] [Google Scholar]
- 30.Winterbourn CC. Oxidative denaturation in congenital hemolytic anemias: the unstable hemoglobins. Semin. Hematol. 1990;27:41–50. [PubMed] [Google Scholar]
- 31.Low PS. Role of hemoglobin denaturation and band 3 clustering in initiating red cell removal. Adv. Exp. Med. Biol. 1991;307:173–183. doi: 10.1007/978-1-4684-5985-2_16. [DOI] [PubMed] [Google Scholar]
- 32.Turrini F, Arese P, Yuan J, Low PS. Clustering of integral membrane proteins of the human erythrocyte membrane stimulates autologous IgG binding, complement deposition, and phagocytosis. J. Biol. Chem. 1991;266:23611–23617. [PubMed] [Google Scholar]
- 33.Janero DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radic. Biol. Med. 1990;9:515–540. doi: 10.1016/0891-5849(90)90131-2. [DOI] [PubMed] [Google Scholar]
- 34.Malshet VG, Tappel AL, Burns VM. Fluorescent products of lipid peroxidation. II. Methods for analysis and characterization. Lipids. 1974;9:328–332. doi: 10.1007/BF02533109. [DOI] [PubMed] [Google Scholar]
- 35.Gilman JG. Developmental changes of human Gg and Ag and mouse embryonic ey1, ey2, and bh1 in transgenic mice with HS4-Gg-Ag. Blood. 1995;86:648a. [Google Scholar]
- 36.Ciavatta DJ, Ryan TM, Farmer SC, Townes TM. Mouse model of human beta zero thalassemia: targeted deletion of the mouse beta maj- and beta min-globin genes in embryonic stem cells. Proc. Natl. Acad. Sci. U S A. 1995;92:9259–9263. doi: 10.1073/pnas.92.20.9259. [DOI] [PMC free article] [PubMed] [Google Scholar]