

Abstract
Spatial dispersion of repolarization in the form of transmural, trans-septal and apico-basal dispersion of repolarization creates voltage gradients that inscribe the J wave and T wave of the ECG. Amplification of this spatial dispersion of repolarization (SDR) underlies the development of life-threatening ventricular arrhythmias associated with inherited or acquired ion channelopathies giving rise to the long QT, short QT and Brugada syndromes (BrS). This review focuses on the role of spatial dispersion of repolarization in drug-induced arrhythmogenesis associated with the long QT and BrS. In the long QT syndrome, drug-induced amplification of SDR is often secondary to preferential prolongation of the action potential duration (APD) of M cells, whereas in the BrS, it is thought to be due to selective abbreviation of the APD of right ventricular epicardium. Among the challenges ahead is the identification of a means to quantitate SDR non-invasively. This review also discusses the value of the interval between the peak and end of the T wave (Tpeak–Tend, Tp–Te) as an index of SDR and transmural dispersion of repolarization, in particular.
Keywords: LQT syndrome, Brugada syndrome, spatial dispersion of repolarization, channelopathies
Introduction
Heterogeneities of ventricular repolarization have long been implicated in arrhythmogenesis. It is well established that ventricular myocardium is comprised of at least three electrophysiologically and functionally distinct cell types: epicardial, M and endocardial cells [1, 2]. These three principal ventricular myocardial cell types differ with respect to phase 1 and phase 3 repolarization characteristics. Ventricular epicardial and M, but not endocardial, cells generally display a prominent phase 1, due to a large 4-aminopyridine (4-AP) sensitive transient outward current (Ito), giving the action potential a spike and dome or notched configuration. These regional differences in Ito, first suggested on the basis of action potential data [3], have now been directly demonstrated in canine [4], feline [5], rabbit [6], rat [7], and human [8, 9] ventricular myocytes. Differences in the magnitude of the action potential notch and corresponding differences in Ito have also been described between right and left ventricular epicardial and M cells [10, 11]. This distinction is thought to form the basis for why the Brugada syndrome (BrS), a channelopathy-mediated form of sudden death, is a right ventricular disease.
Between surface epicardial and endocardial layers are layers of transitional and M cells. M cells are distinguished by the ability of their action potential to prolong disproportionately relative to the action potential of other ventricular myocardial cells in response to a slowing of rate and/or in response to action potential duration (APD)-prolonging agents [1, 12, 13]. In the dog, the ionic basis for these features of the M cell include the presence of a smaller slowly activating delayed rectifier current (IKs) [14], a larger late sodium current (late INa) [15] and a larger Na–Ca exchange current (INa–Ca) [16]. In the canine heart, the rapidly activating delayed rectifier (IKr) and inward rectifier (IK1) currents are similar in the three transmural cell types. Transmural and apico-basal differences in the density of IKr channels have been described in the ferret heart [17]. IKr message and channel protein are much larger in the ferret epicardium. IKs is larger in M cells isolated from the right vs. left ventricles of the dog [11]. Calcium current (Ica) has been shown to be similar among cells isolated from epicardium, M, and endocardial regions of the left ventricular wall [18, 19]. One study however reported differences in Ca2+ channel properties between epicardial and endocardial canine ventricular cells with endocardial cells displaying a larger current [20].
The distribution of M cells within the ventricular wall has been investigated in greatest detail in the left ventricle of the canine heart. Although transitional cells are found throughout the wall in the canine left ventricle, M cells displaying the longest action potentials [at basic cycle lengths (BCLs) ≥ 2000 ms] are often localized in the deep subendocardium to midmyocardium in the anterior wall [21], deep subepicardium to midmyocardium in the lateral wall [12] and throughout the wall in the region of the right ventricular (RV) outflow tracts [2]. M cells are also present in the deep cell layers of endocardial structures, including papillary muscles, trabecule and the interventricular septum [22]. Unlike Purkinje fibers, M cells are not found in discrete bundles or islets [22, 23], although there is evidence that they may be localized in discrete muscle layers. Cells with the characteristics of M cells have been described in the canine, guinea pig, rabbit, pig and human ventricles [4, 12–14, 21–41].
Transmural, trans-septal and apico-basal heterogeneities of final repolarization of the action potential within ventricular myocardium are thought to be responsible for inscription of the T wave [39, 42]. Studies involving the arterially-perfused wedge have shown that currents flowing down voltage gradients on either side of the M region are in large part responsible for the T wave [39]. The interplay between these opposing forces establishes the height and width of the T wave and the degree to which either the ascending or descending limb of the T wave is interrupted, leading to a bifurcated or notched appearance of the T wave [39]. The voltage gradients result from a more positive plateau potential in the M region than in epicardium or endocardium as well as from differences in the time-course of phase 3 of the action potential of the three predominant ventricular cell types.
Under normal and most long QT conditions, the epicardial response is the earliest to repolarize and the M cell action potential is often the last. Full repolarization of the epicardial action potential is coincident with peak of the T wave and repolarization of the M cells coincides with the end of the T wave. Thus, the repolarization of the M cells in the heart usually determine the QT interval. The interval between the peak and end of the T wave (Tp–Te) has been suggested to provide an index of transmural dispersion of repolarization, which may be of prognostic value [39, 43].
Although apico-basal repolarization gradients have been suggested to play a prominent role in the registration of the T wave, particularly T waves changes associated with cardiac memory [42, 44] studies involving coronary-perfused wedge preparations suggest little or no contribution under baseline conditions and greater contribution of global dispersion of repolarization to T wave changes associated with cardiac memory [39, 45].
The long QT syndrome
Prolongation of the action potential duration of the M cell usually underlies the prolongation of the QT interval on the surface ECG, the time interval between ventricular depolarization and repolarization. Prolongation of the QT can occur as a consequence of congenital defects or in response to drugs that prolong the APD via a reduction in IKs, IKr or the inward rectifier potassium current (IK1), or an increase in calcium current (ICa) or late sodium current (INa). The inherited forms of the long QT syndrome (LQTS) are phenotypically and genotypically diverse, but have in common the appearance of long QT interval in the ECG, an atypical polymorphic ventricular tachycardia known as torsade de pointes (TdP), and, in many but not all cases, a relatively high risk for sudden cardiac death [46–48]. Congenital LQTS is subdivided into ten genotypes distinguished by mutations in at least seven different ion genes and in structural anchoring protein located on chromosomes 3, 4, 6, 7, 11, 17 and 21 (Table 1) [49–56]. Timothy syndrome, also referred to as LQT8, is a rare congenital disorder characterized by multi-organ dysfunction including prolongation of the QT interval, lethal arrhythmias, webbing of fingers and toes, congenital heart disease, immune deficiency, intermittent hypoglycemia, cognitive abnormalities, and autism. Timothy syndrome has been linked to loss of voltage-dependent inactivation due to mutations in Cav1.2, the gene that encodes for an α subunit of the calcium channel [57]. The most recent genes associated with LQTS are CAV3 which encodes caveolin-3 and SCN4B which encodes NaVB4, an auxiliary subunit of the cardiac sodium channel. Mutations in both genes produce a gain of function in late INa, causing an LQT3-like phenotype [55, 56].
Table 1.
Inherited disorders caused by ion channelopathies
| Rhythm | Inheritance | Locus | Ion channel | Gene | ||
|---|---|---|---|---|---|---|
| Long QT syndrome | (RW) | TdP | AD | |||
| LQT1 | 11p15 | IKs | KCNQ1, KvLQT1 | |||
| LQT2 | 7q35 | IKr | KCNH2, HERG | |||
| LQT3 | 3p21 | INa | SCN5A, Nav1.5 | |||
| LQT4 | 4q25 | ANKB, ANK2 | ||||
| LQT5 | 21q22 | IKs | KCNE1, minK | |||
| LQT6 | 21q22 | IKr | KCNE2, MiRP1 | |||
| LQT7 | (Andersen-Tawil syndrome) | 17q23 | IK1 | KCNJ2, Kir 2.1 | ||
| LQT8 | (Timothy syndrome) | 6q8A | ICa | CACNA1C, Cav1.2 | ||
| LQT9 | 3p25 | INa | CAV3, Caveolin-3 | |||
| LQT10 | 11q23.3 | INa | SCN4B. Navb4 | |||
| LQT syndrome (JLN) | TdP | AR | 11p15 | IKs | KCNQ1, KvLQT1 | |
| 21q22 | IKs | KCNE1, minK | ||||
| Brugada syndrome | BrS1 | PVT | AD | 3p21 | INa | SCN5A, Nav1.5 |
| BrS2 | PVT | AD | 3p24 | INa | GPD1L | |
| BrS3 | PVT | AD | 12p13.3 | ICa | CACNA1C, CaV1.2 | |
| BrS4 | PVT | AD | 10p12.33 | ICa | CACNB2β, Cavβ2b | |
AD — autosomal dominant; AR — autosomal recessive; JLN — Jervell and Lange-Nielsen; LQT — long QT; RW — Romano-Ward; TdP — torsade de pointes; VF — ventricular fibrillation; VT — ventricular tachycardia; PVT — polymorphic VT
Two patterns of inheritance have been identified: 1) a rare autosomal recessive disease associated with deafness (Jervell and Lange-Nielsen), caused by 2 genes that encode for the slowly activating delayed rectifier potassium channel (KCNQ1 and KCNE1); and 2) a much more common autosomal dominant form known as the Romano-Ward syndrome, caused by mutations in 10 different genes, including KCNQ1 (KvLQT1; LQT1); KCNH2 (HERG;LQT2); SCN5A (Nav1.5; LQT3); ANKB (LQT4); KCNE1 (minK; LQT5); KCNE2 (MiRP1; LQT6); KCNJ2 (LQT7; Andersen’s syndrome), CACNA1C (Cav1.2; LQT8; Timothy syndrome), CAV3 (Caveolin-3; LQT9) and SCN4B (NaVB4, LQT10). Six of the 10 genes encode for cardiac potassium channels, one for the cardiac sodium channel (SCN5A), one for the β subunit of the sodium channel, one for caveolin-3 and one for a protein called Ankyrin B (ANKB), which is involved in anchoring of ion channels to the cellular membrane.
Acquired LQTS refers to a syndrome similar to the congenital form but caused by exposure to drugs that prolong the duration of the ventricular action potential [58] or QT prolongation secondary to cardiomyopathies such as dilated or hypertrophic cardiomyopathy, as well as to abnormal QT prolongation associated with bradycardia or electrolyte imbalance [59–63].
Table 2 presents a list of drugs known to prolong the QT interval and to induce torsade de pointes. Most of these drugs block IKr, many also block IKs, and some augment late INa, so that in many ways they are similar to congenital forms of LQTS. The acquired form of the disease is far more prevalent than the congenital form, and in some cases may have a genetic predisposition.
Table 2.
Drugs with risk of long QT syndrome and torsade de pointes.
| Generic name | Class/clinical use |
|---|---|
| Amiodarone | Anti-arrhythmic/abnormal heart rhythm |
| Arsenic trioxide | Anti-cancer/leukemia |
| Bepridil | Anti-anginal/heart pain |
| Chloroquine | Anti-malarial/malaria infection |
| Chlorpromazine | Anti-psychotic/anti-emetic/schizophrenia/nausea |
| Cisapride | GI stimulant/heartburn |
| Clarithromycin | Antibiotic/bacterial infection |
| Disopyramide | Anti-arrhythmic/abnormal heart rhythm |
| Dofetilide | Anti-arrhythmic/abnormal heart rhythm |
| Domperidone* | Anti-nausea/nausea |
| Droperidol | Sedative; anti-nausea/anesthesia adjunct, nausea |
| Erythromycin | Antibiotic; GI stimulant/bacterial infection; increase GI motility |
| Halofantrine | Anti-malarial/malaria infection |
| Haloperidol | Anti-psychotic/schizophrenia, agitation |
| Ibutilide | Anti-arrhythmic/abnormal heart rhythm |
| Levomethadyl | Opiate agonist/pain control, narcotic dependence |
| Mesoridazine | Anti-psychotic/schizophrenia |
| Methadone | Opiate agonist/pain control, narcotic dependence |
| Pentamidine | Anti-infective/pneumocystis pneumonia |
| Pimozide | Anti-psychotic/Tourette’s tics |
| Procainamide | Anti-arrhythmic/abnormal heart rhythm |
| Quinidine | Anti-arrhythmic/abnormal heart rhythm |
| Sotalol | Anti-arrhythmic/abnormal heart rhythm |
| Sparfloxacin | Antibiotic/bacterial infection |
| Thioridazine | Anti-psychotic/schizophrenia |
The ability of genetic mutations and drugs to amplify spatial dispersion of repolarization within the ventricular myocardium has been identified as the principal arrhythmogenic substrate in both acquired and congenital LQTS. Early- and in some cases delayed-afterdepolarizations have been identified as triggers responsible for the precipitation of torsade de pointes. The accentuation of spatial dispersion, typically secondary to an increase of transmural, trans-septal or apico-basal dispersion of repolarization, and the development of early after-depolarization (EAD)-induced triggered activity underlie the substrate and trigger for the development of torsade de pointes arrhythmias observed under LQTS conditions [64, 65]. Models of the LQT1, LQT2, LQT3, LQT5, LQT6, LQT7 and LQT8 forms of the long QT syndrome have been developed using the canine arterially perfused left ventricular wedge preparation [66–69]. These models suggest that in LQT1–3, 4, and 8 preferential prolongation of the M cell APD can lead to an increase in the QT interval as well as an increase in transmural dispersion of repolarization (TDR), which contributes to the development of spontaneous as well as stimulation-induced TdP [32, 37, 70].
The distinctive characteristics of the M cells are at the heart of the long QT syndrome. The hallmark of the M cell is the ability of its action potential to prolong more than that of epicardium or endocardium in response to a slowing of rate [2, 12, 71]. As previously detailed, this feature of the M cell is due to weaker repolarizing current during phases 2 and 3 secondary to a smaller IKs and a larger late INa and INa–Ca [14–16] compared to epicardial and endocardial cells. These ionic distinctions sensitize the M cells to a variety of pharmacological agents and pathophysiological states. Agents that block IKr, IKs or increase ICa or late INa generally produce a much greater prolongation of the APD of the M cell than of epicardial or endocardial cells.
Experimental models that mimic the clinical congenital syndromes with respect to prolongation of the QT interval, T wave morphology, rate dependence of QT have also been helpful in elucidation of the basis for sympathetic nervous system influences [21, 32, 37–39].
IKs block using chromanol 293B is used to mimic LQT1. IKs block alone produces a homogeneous prolongation of repolarization and refractoriness across the ventricular wall and does not induce arrhythmias. The addition of isoproterenol causes abbreviation of epicardial and endocardial APD but a prolongation or no change in the APD of the M cell, resulting in a marked augmentation of TDR and the development of spontaneous and stimulation-induced TdP [37]. These changes give rise to a broad based T wave and the long QT interval characteristics of LQT1. The development of TdP in the model requires β adrenergic stimulation, consistent with a high sensitivity of congenital LQTS, LQT1 in particular, to sympathetic stimulation [46–48, 72, 73].
IKr block using d-Sotalol has been used to mimic LQT2 and provides a model of the most common form of acquired (drug-induced) LQTS. A greater prolongation of the M cell action potential and slowing of phase 3 of the action potential of all three cell types results in a low amplitude T wave, long QT interval, large transmural dispersion of repolarization and the development of spontaneous as well as stimulation-induced TdP. The addition of hypokalemia gives rise to low-amplitude T waves with a deeply notched or bifurcated appearance, similar to those commonly seen in patients with the LQT2 syndrome [32, 39]. Isoproterenol further exaggerates transmural dispersion of repolarization, thus increasing the incidence of TdP [70].
ATX-II, an agent that increases late INa, is used to mimic LQT3 [32]. ATX-II markedly prolongs the QT interval, delays the onset of the T wave, in some cases also widening it, and produces a sharp rise in transmural dispersion of repolarization as a result of a greater prolongation of the APD of the M cell. The differential effect of ATX-II to prolong the M cell action potential is likely due to the presence of a larger late sodium current in the M cell [15]. ATX-II produces a marked delay in onset of the T wave because of a relatively large effect of the drug on epicardial and endocardial APD. This feature is consistent with the late-appearing T wave (long isoelectric ST segment) observed in patients with the LQT3 syndrome. Also in agreement with the clinical presentation of LQT3, the model displays a steep rate dependence of the QT interval and develops TdP at slow rates. Interestingly, β adrenergic influence in the form of isoproterenol reduces transmural dispersion of repolarization by abbreviating the APD of the M cell more than that of epicardium or endocardium, and thus reducing the incidence of TdP. While the beta-adrenergic blocker propranolol is protective in LQT1 and LQT2 wedge models, it has the opposite effects in LQT3, acting to amplify transmural dispersion and promoting TdP [70].
It is interesting that the response to sympathetic activation displays a very different time-course in the case of LQT1 and LQT2, both in experimental models and in the clinic [65, 74]. In LQT1, β adrenergic stimulation induces an increase in TDR that is most prominent during the first two minutes, but which persists, although to a lesser extent, during steady-state. Torsade de pointes incidence is enhanced during the initial period as well as during steady-state. In LQT2, isoproterenol produces only a transient increase in TDR that persists for less than 2 minutes. Torsade de pointes incidence is therefore enhanced only for a brief period of time. These differences in time-course may explain the important differences in autonomic activity and other gene-specific triggers that contribute to events in patients with different LQTS genotypes [65, 73, 75].
Figure 1 presents a working hypothesis for our understanding of the mechanisms underlying LQTS-related TdP based on available data. The hypothesis presumes the presence of electrical heterogeneity in the form of transmural or trans-septal dispersion of repolarization under baseline conditions and the amplification of TDR by agents that reduce net repolarizing current via a reduction in IKr or IKs or augmentation of ICa or late INa. Conditions that cause a reduction in IKr or augmentation of late INa lead to a preferential prolongation of the M cell action potential. As a consequence, the QT interval prolongs and is accompanied by a dramatic increase in transmural dispersion of repolarization, thus creating a vulnerable window for the development of reentry. The reduction in net repolarizing current also predisposes to the development of EAD-induced triggered activity in M and Purkinje cells, which provide the extrasystole that triggers TdP when it falls within the vulnerable period. Beta-adrenergic agonists further amplify transmural heterogeneity (transiently) in the case of IKr block, but reduce it in the case of INa agonists [30, 70].
Figure 1.
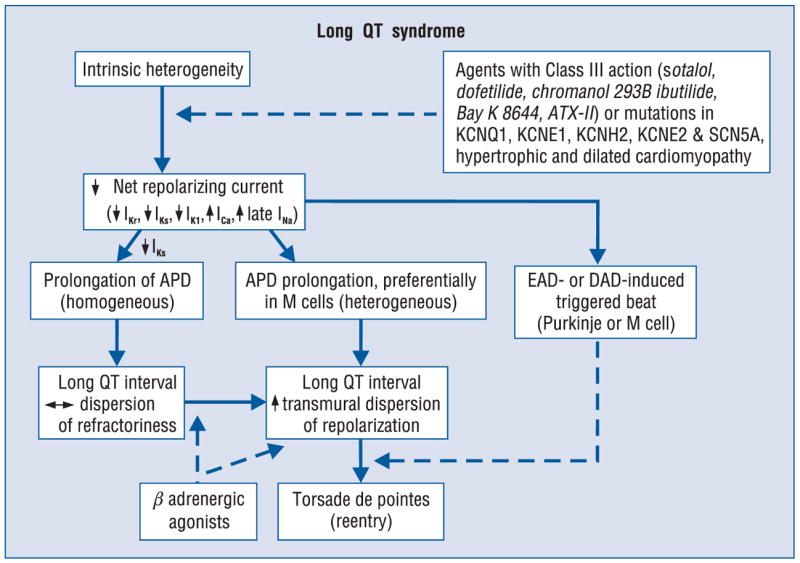
Proposed cellular mechanism for the development of torsade de pointes in the long QT syndromes.
Although conditions that prolong QT are often associated with an increase in TDR, this is not always the case. Amiodarone, a potent antiarrhythmic agent used in the management of both atrial and ventricular arrhythmias, is rarely associated with TdP [76]. Chronic administration of amiodarone produces a greater prolongation of APD in epicardium and endocardium, but less of an increase in APD, or even a decrease at slow rates, in the M region, thereby reducing TDR [77]. In a dog model of chronic complete atrioventricular block and acquired LQTS, 6 weeks of amiodarone was shown to produce a major QT prolongation without producing TdP. In contrast, after 6 weeks of dronedarone, TdP occurred in 4 of 8 dogs displaying the highest spatial dispersion of repolarization (105 ± 20 ms) [78] Sodium pentobarbital is another agent that prolongs the QT interval but reduces TDR. Pentobarbital has been shown to produce a dose-dependent prolongation of the QT interval, accompanied by a reduction in TDR from 51 to 27 ms [36]. TdP is not observed under these conditions, nor can it be induced with programmed stimulation. Amiodarone and pentobarbital have in common the ability to block IKs, IKr, and late INa. This combination produces a preferential prolongation of the APD of epicardium and endocardium so that the QT interval is prolonged, but TDR is actually reduced and TdP does not occur.
Cisapride, another agent that blocks both inward and outward currents, produces a biphasic concentration-dependent prolongation of the QT interval. A parallel biphasic dose-response relationship is seen for TDR, peaking at 0.2 μM, and it is only at this concentration that TdP is observed. Higher concentrations of cisapride further prolong QT, but reduced TDR, thereby preventing TdP induction [79]. This finding suggests that the spatial dispersion of repolarization is more important than the prolongation of the QT interval in determining the substrate for TdP.
The IKs blocker, chromanol 293B, is another agent that increases QT without augmenting TDR. Chromanol 293B prolongs APD of the 3 cell types homogeneously, neither increasing TDR nor widening the T wave. TdP is not observed under these conditions. Although an arrhythmogenic substrate is not present with IKs block alone, it develops very quickly with the introduction of β adrenergic stimulation. Isoproterenol abbreviates the APD of epicardial and endocardial cells but not that of the M cell, resulting in a marked accentuation of TDR [70]. Torsade de pointes readily develops under these conditions.
These observations have advanced our understanding of why long-QT patients, LQT1 in particular, are so sensitive to sympathetic influences, and have provided further evidence in support of the hypothesis that the risks associated with LQTS are not due to the prolongation of the QT interval but rather to an increase in spatial dispersion of repolarization that usually, but not always, accompanies the prolongation of the QT interval.
Thus, QT prolonging agents display very different concentration-dependent behavior. Pure IKr blockers such as sotalol, dofetilide and erythromycin produce a dose-dependent prolongation of the QT interval that is associated with a dose-dependent prolongation of TDR. When TDR reaches the threshold for re-entry, which in the canine wedge preparation is approximately 90 ms, TdP will occur. With more complex agents such as quinidine and cisapride there is a biphasic dose-response relationship. TDR parallels QT, but the two can peak at different concentrations. TdP occurs when and if TDR reaches the threshold value. Still other drugs produce a dose-dependent prolongation of QT, but a smaller increase or even a decrease in TDR; threshold values for TdP are rarely reached. Finally, agents that preferentially block IKs such as chromanol 293B, and agents with multiple ion channel effects including pentobarbital, amiodarone, and the new anti-anginal agent ranolazine, produce a dose-dependent prolongation of the QT interval that is not associated with an increase in TDR. Torsade de pointes rarely, if ever, occurs under these conditions.
Thus, TdP is not produced by drugs that cause a dose-dependent prolongation of QT but a reduction of TDR or little or no increase in TDR. Taken together these findings indicate that arrhythmogenesis in the long QT syndromes is not due to prolongation of QT interval but rather to the increase in TDR that often accompanies prolongation of the QT interval. The ability of sympathetic influences to dramatically increase TDR also explains why LQT1 and LQT2 patients are so sensitive to sympathetic stimulation.
Tpeak–Tend as a non-invasive marker of spatial dispersion of repolarization
If QT prolongation is not prognostic of risk for the development of life-threatening arrhythmias is there a better index? While a number of promising approaches have been pursued in recent years, our interest has been in the interval between the peak and the end of the T wave (Tpeak–Tend interval). Differences in the time-course of repolarization of these three predominant ventricular myocardial cell types contribute prominently to the inscription of the electrocardiographic T wave recorded in the precordial leads [39]. These differences in action potential morphology lead to the development of opposing voltage gradients on either side of the M region, which contribute to inscription of the T wave, especially those inscribed in the precordial leads (Fig. 1) [39]. Apico-basal gradients are thought to contribute to the T wave as well, especially in the limb lead where the polarity of the T wave may be opposite to that recorded in the precordial leads [42]. The interplay between these opposing transmural forces determines the height and width of the T wave and the extent to which the T wave may be interrupted, resulting in a bifid or notched appearance.
When the T wave is upright, the epicardial response is the earliest to repolarize and the M cell action potential is the last. Full repolarization of the epicardial action potential coincides with the peak of the T wave and repolarization of the M cells is coincident with the end of the T wave. It therefore follows that the duration of the M cell action potential determines the QT interval, whereas the duration of the epicardial action potential determines the QTpeak interval. Another interesting finding stemming from these studies was that the Tpeak–Tend interval may provide an index of transmural dispersion of repolarization [2, 39]. Figure 2 illustrates these relationships under baseline and long QT conditions. ATX-II, a sea anemone toxin, mimics the LQT3 form of the long QT syndrome by augmenting the late sodium current, which helps to sustain the plateau of the action potential. ATX-II, like most agents that prolong the action potential, causes a preferential prolongation of the M cell response, thus producing a dramatic accentuation of the transmural dispersion of repolarization, which is reflected on the ECG as a prolongation of the Tpeak–Tend interval. An increase in transmural dispersion of repolarization is a common feature of all LQTS models and has been shown to provide the substrate for the development of TdP under long QT conditions [2, 32, 70].
Figure 2.
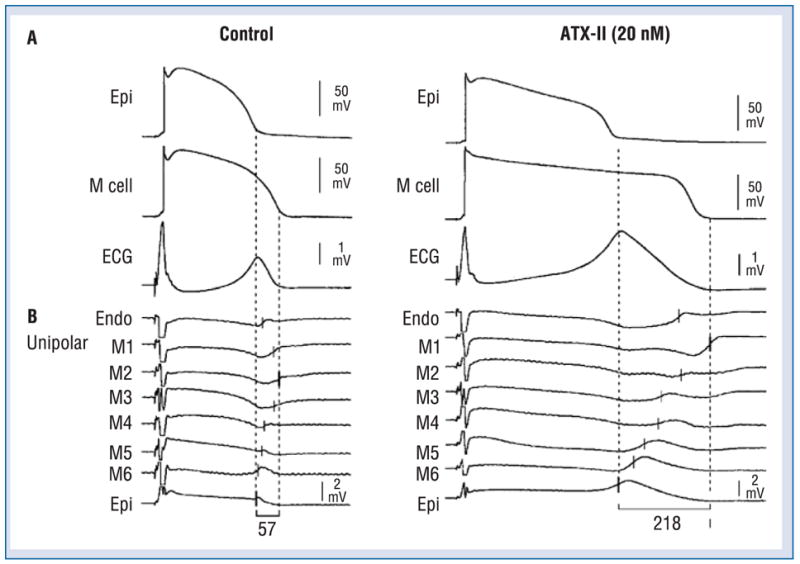
Electrocardiographic Tpeak–Tend interval as a measure of transmural dispersion of repolarization. The figure shows the correspondence among transmembrane, unipolar, and ECG recordings in the absence (left) and the presence (right) of ATX-II (20 nmol/L). Each panel shows: A. Transmembrane action potentials recorded from M (M2) and epicardial sites of a canine left ventricular wedge preparation together with a transmural ECG recorded across the bath (BCL of 2000 ms); B. Eight intramural unipolar electrograms recorded approximately 1.2 mm apart from endocardial (Endo), M (6 sites; M1–M6) and epicardial (Epi) regions (120 μm silver electrodes insulated except at the tip) inserted midway into the wedge preparation. Dashed vertical lines in the unipolar electrograms denote the time maximum of the first derivative (Vmax) of the T wave (ARI, end of activation-recovery interval). Close correspondence between the repolarization time of the cells deep within the wedge and those at the cut surface attest the uniformity of the electrical activity in the respective transmural layers. Modified from [2], with permission.
Based on these and similar studies, the Tpeak–Tend interval in precordial ECG leads was suggested to provide an index of transmural dispersion of repolarization [2]. More recent studies have also provided guidelines for the estimation of transmural dispersion of repolarization in the case of more complex T waves, including negative, biphasic and triphasic T waves [80]. In such cases, the interval from the nadir of the first component of the T wave to the end of the T wave was shown to provide an electrocardiographic approximation of TDR.
While these relationships are relatively straight forward in the coronary-perfused wedge preparation, extrapolation to the surface ECG recorded in vivo must be approached with great caution and will require careful validation. The Tpeak–Tend interval is unlikely to provide an absolute measure of transmural dispersion in vivo, as demonstrated by Xia et al. [81]. However, changes in this parameter are thought to be capable of reflecting changes in spatial dispersion of repolarization, including TDR, and thus may be prognostic of arrhythmic risk under a variety of conditions [82–87]. Takenaka et al. [86] recently demonstrated exercise-induced accentuation of the Tpeak–Tend interval in LQT1 patients. These observations coupled with those of Schwartz et al. [75], demonstrating an association between exercise and risk for TdP in LQT1, patients, pointed to the potential value of Tpeak–Tend in forecasting risk for the development of torsade de pointes. Direct evidence in support of Tpeak–Tend as an index to predict TdP in patients with LQTS has recently been provided by a number of studies. Yamaguchi et al. [88] showed that Tpeak–Tend is more valuable than QTc and QT dispersion as a predictor of TdP in patients with acquired LQTS. Shimizu et al. [85] demonstrated that Tpeak–Tend, but not QTc, predicted sudden cardiac death in patients with hypertrophic cardiomyopathy. In a case-controlled study comparing 30 cases of acquired bradyarrhythmias complicated by TdP and 113 cases with uncomplicated bradyarrhythmias, Topilski et al. [89] found that QT, QTc and Tpeak–Tend intervals were strong predictors of TdP, with the best single discriminator being a prolonged Tpeak–Tend. Watanabe et al. [87] demonstrated that prolonged Tpeak–Tend is associated with inducibility as well as spontaneous development of ventricular tachycardia (VT) in high risk patients with organic heart disease.
These interesting studies demonstrating an association between an increase in Tpeak–Tend and arrhythmic risk notwithstanding, direct validation of Tpeak–Tend measured at the body surface as an index of TDR is still lacking. Guidelines for such validation have been suggested [39, 80, 90]. Because precordial leads view the electrical field across the ventricular wall, Tpeak–Tend would be expected to be most representative of TDR in these leads. The precordial leads are unipolar leads placed on the chest that are referenced to Wilson central terminal. The direction of these leads is radially toward the “center” of the heart, the center of the Einthoven triangle. Unlike the precordial leads, the bipolar limb leads, including leads I, II, and III, do not look across the ventricular wall. While Tpeak–Tend intervals measured in these limb leads may provide an index of TDR, they are more likely to reflect global dispersion, including apico-basal and interventricular dispersion of repolarization [81, 91].
A large increase in TDR is likely to be arrhythmogenic because the dispersion of repolarization and refractoriness occurs over a very short distance (the width of the ventricular wall), creating a steep repolarization gradient [92, 93]. It is the steepness of the repolarization gradient rather than the total magnitude of dispersion that determines its arrhythmogenic potential [92, 93]. Apico-basal or interventricular dispersion of repolarization is less informative because it may or may not be associated with a steep repolarization gradient and thus may or may not be associated with arrhythmic risk.
Another important consideration is that TDR can be highly variable in different regions of the ventricular myocardium, particularly under pathophysiologic conditions. Consequently, it is important to measure Tpeak–Tend independently in each of the precordial leads and it is inadvisable to average Tpeak–Tend among several leads [81]. Yamaguchi et al. [88] in their study of acquired LQTS targeted lead V5. In contrast, because BrS is a right ventricular disorder, Castro et al. [94] targeted lead V2 in their study of Tpeak–Tend in BrS.
The criteria suggested for validation of Tpeak–Tend as an index of TDR therefore include a requirement 1) that individual precordial leads, and not bipolar limb leads, be evaluated; and 2) that TDR be present at baseline and significantly augmented as a result of an intervention.
The quest for direct validation or invalidation of Tpeak–Tend measured at the body surface as an index of TDR based on these fundamental criteria remains unfulfilled [91]. Although most studies to date concur that Tpeak–Tend provides a measure of spatial dispersion of repolarization, the extent to which an augmented Tpeak–Tend interval is prognostic of arrhythmic risk depends on the proximity of the regions displaying disparate repolarization times (i.e., repolarization gradient). Consequently, it would be helpful to know to what extent Tpeak–Tend provides an index of TDR, in which case the differences in refractoriness are likely to be within close proximity. An ideal model in which to test the hypothesis is in the chronic atrioventricular (AV) block dog treated with a Class III antiarrhythmic agent, since changes in Tpeak–Tend could be correlated with TDR in a model that displays prominent TDR, and additionally correlated with the risk for development of TdP.
In addition to pharmacologic agents and pathophysiological states, interventions that reverse the direction of activation of the ventricular wall can augment TDR. In the example illustrated in Figure 3, reversing the direction of activation of the ventricular wall in the arterially-perfused left ventricular wedge preparation is shown to lead to a broader and taller T wave. TDR and Tpeak–Tend are conspicuously augmented. In the presence of cisapride, reversing the direction of activation of the left ventricular (LV) wall, amplifies TDR sufficiently to permit the development of torsade de pointes [79]. Figure 4 shows that similar changes in the T wave are recorded in the human heart in vivo when the direction of transmural activation is reversed [95]. Patients with heart failure were initially paced from an endocardial site facing lead V5–V6 and then from an adjacent site on the epicardial surface. The shift from endocardial to epicardial pacing led to a dramatic increase in TDR and Tp–Te, similar to the changes observed in the wedge preparation. In 4 patients, Tpeak–Tend increased by an average of 24.3%. These data provide evidence in support of important transmural distinctions in repolarization across the ventricular wall, for such a change would not be observed if transmural repolarization was homogeneous, as suggested by some studies [42, 91, 96, 97].
Figure 3.
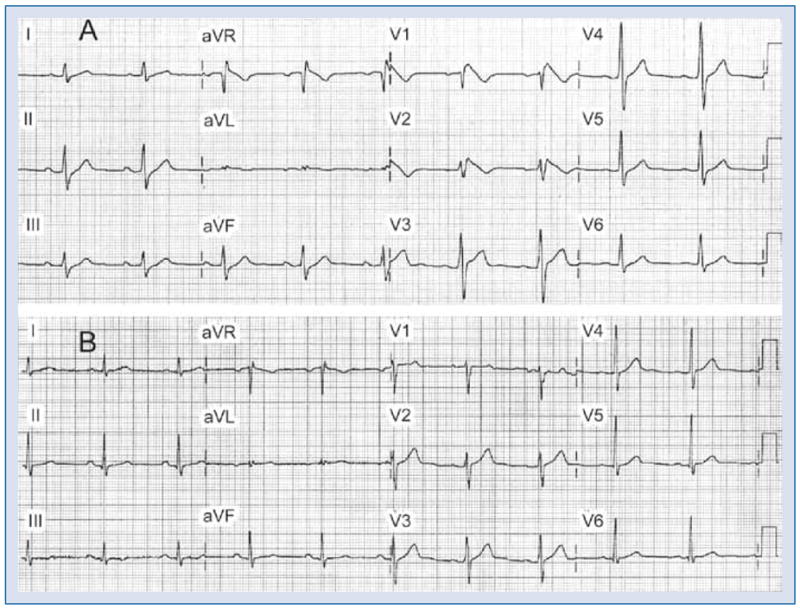
Brugada syndrome phenotype induced by an overdose of desipramine and clonazepam in a 44 year old man previously medicated with desipramine, clonazepam and trazodone. A. ECG shows sinus bradycardia, first degree atrioventricular block (228 ms), prolonged QRS interval (132 ms) and downsloping ST elevation (Type 1) in leads V1–V2, ST elevation in lead V3, upsloping ST depression in leads II, III, and aVF; B. Baseline ECG recorded one year earlier. Modified from [155] with permission.
Figure 4.
Schematic representation of right ventricular epicardial action potential changes proposed to underlie the electrocardiographic manifestation of the Brugada syndrome. Modified from [173], with permission.
A clinical consequence of the increase in TDR following reversal of the direction of activation of the ventricular wall was demonstrated by Medina--Ravell et al. [98]. These authors reported the first case in which an upgrade to biventricular pacing in a patient undergoing resynchronization therapy led to the development of torsade de pointes. Pacing via the endocardial electrode in the right ventricular apex was uneventful. In contrast, pacing via the left ventricular epicardial lead in the coronary sinus resulted in an increase in both QT interval and Tpeak–Tend, and the development of torsade de pointes. The available data suggest that epicardial activation of the LV wall during resynchronization therapy is a potential iatrogenic factor in a small subset of patients with prolonged QT intervals and/or in which TDR is prolonged [95].
Brugada syndrome
In that the BrS is believed to be secondary to an exaggeration of the J wave, it seems appropriate to first discuss the basis for the J wave of the ECG. The presence of a prominent action potential notch in epicardium but not endocardium gives rise to a transmural voltage gradient during ventricular activation that manifests as a late delta wave following the QRS or what more commonly is referred to as a J wave [99] or Osborn wave. A distinct J wave is often observed under baseline conditions in the ECG of some animal species, including dogs and baboons. Humans more commonly display a J point elevation rather than a distinct J wave. A prominent J wave in the human ECG is considered pathognomonic of hypothermia [100–102] or hypercalcemia [103, 104].
A transmural gradient in the distribution of Ito is responsible for the transmural gradient in the magnitude of phase 1 and action potential notch, which in turn gives rise to a voltage gradient across the ventricular wall responsible for the inscription of the J wave or J point elevation in the ECG [3, 4, 105]. Direct evidence in support of the hypothesis that the J wave is caused by a transmural gradient in the magnitude of the Ito-mediated action potential notch derives from experiments conducted in the arterially-perfused right ventricular wedge preparation showing a correlation between the amplitude of the epicardial action potential notch and that of the J wave recorded during interventions that alter the appearance of the electrocardiographic J wave, including hypothermia, premature stimulation (restitution), and block of Ito by 4-aminopyridine (4-AP) [99].
The molecular basis for the transmural distribution of Ito has long been a subject of debate. The transmural gradient of Ito in dog has been ascribed to a transmural distribution of KCND3 gene (Kv4.3), which encodes the a subunit of the Ito channel [106] and a transmural gradient of KChIP2, a β subunit that coassembles with and serves as a chaperone for Kv4.3 [107]. The steep transmural gradient of Ito suggests that spatial patterning of Ito is highly regulated in mammalian cardiac myocytes. However, very little is known about transcriptional regulation of Ito and its components. Recently, a transcriptional factor, Iroquois 5, was described and shown to regulate KCND2 expression [108]. The Iroquois homeobox (Irx) genes encode a conserved family of transcription factors that specify the identity of diverse territories in the heart. One of the members of this family, the homeodomain transcription factor 5 (IRX5), has been shown to contribute to the cardiac repolarization gradient. IRX5 causes repression of Kv4.2 expression by recruiting mBop (a cardiac transcriptional repressor), thus forming an inverse Ito gradient that contributes to coordinated contraction of the ventricular wall [18]. In the canine heart, IRX5 is also expressed in an endocardial-to-epicardial gradient, suggesting that it may regulate expression of KChIP2 and/or KCND3 genes.
Myocytes isolated from the epicardial region of the left ventricular wall of the rabbit show a higher density of cAMP-activated chloride current when compared to endocardial myocytes [109]. Ito2, initially ascribed to a K+ current, is now thought to be primarily due to the calcium-activated chloride current (ICl(Ca)) is also thought to contribute to the action potential notch, but it is not known whether this current, differs among the three ventricular myocardial cell types [110].
Transmural activation within the relatively thin wall of RV is relatively rapid causing the J wave to be buried inside the QRS. Thus, although the action potential notch is most prominent in right ventricular epicardium, right ventricular myocardium would be expected to contribute relatively little to the manifestation of the J wave under normal conditions. These observations are consistent with the manifestation of the J wave in ECG leads in which the mean vector axis is transmurally oriented across the left ventricle and septum. Accordingly, the J wave in the dog is most prominent in leads II, III, aVR, aVF, and mid to left precordial leads V3 through V6. A similar picture is seen in the human ECG [104, 111]. In addition, vectorcardiography indicates that the J wave forms an extra loop that occurs at the junction of the QRS and T loops [112]. It is directed leftward and anteriorly, which explains its prominence in leads associated with the left ventricle.
To our knowledge, the first description of the J wave was in the 1920s in animal experiments involving hypercalcemia [103]. The first extensive description and characterization appeared 30 years later by Osborn [113] in a study involving experimental hypothermia in dogs. The appearance of a prominent J wave in the clinic is typically associated with pathophysiological conditions, including hypothermia [100, 111] and hypercalcemia [103, 104]. The prominent J wave induced by hypothermia is the result of a marked accentuation of the spike-and-dome morphology of the action potential of M and epicardial cells (i.e., an increase in both width and magnitude of the notch). In addition to inducing a more prominent notch, hypothermia produces a slowing of conduction which permits the epicardial notch to clear the QRS so as to manifest a distinct J wave. Hypercalcemia-induced accentuation of the J wave [103, 104, 114] may also be explained on the basis of an accentuation of the epicardial action potential notch, possibly as a result of an augmentation of the calcium-activated chloride current and a decrease in ICa [115]. Accentuation of the action potential notch also underlies the electrocardiographic and arrhythmogenic manifestations of the BrS.
The Brugada syndrome is characterized by an accentuated ST segment elevation or J wave appearing principally in the right precordial leads (V1–V3), often followed by a negative T wave. The syndrome, first described in 1992, is generally associated with a high incidence of sudden cardiac death secondary to a rapid polymorphic VT or ventricular fibrillation (VF) [116]. The average age at the time of initial diagnosis or sudden death is 40 ± 22. The youngest patient diagnosed with the syndrome is 2 days of age, and the oldest is 84.
The prevalence of the BrS is estimated at 1–5 per 10,000 inhabitants worldwide. The frequency is lower in western countries and higher (≥ 5 per 10,000) in Southeast Asia, especially Thailand and the Philippines where BrS is considered to be the major cause of sudden death in young individuals. In these countries, the syndrome is often referred to as sudden unexplained nocturnal death syndrome (SUNDS) [117, 118].
Brugada syndrome is inherited via an autosomal dominant mode of transmission. The first gene to be linked to the BrS is SCN5A, the gene encoding for the a subunit of the cardiac sodium channel gene [119]. Mutations in SCN5A are also responsible for the LQT3 form of the long QT syndrome and cardiac conduction disease. A number of mutations have been reported to cause overlapping syndromes; in some cases all three phenotypes are present [120].
Over one hundred mutations in SCN5A have been linked to the syndrome in recent years (see [121] for references; also see www.fsm.it/cardmoc). Only a fraction of these mutations have been studied in expression systems and shown to result in loss of function due either to: 1) failure of the sodium channel to express; 2) a shift in the voltage-and time-dependence of sodium channel current (INa) activation, inactivation or reactivation; 3) entry of the sodium channel into an intermediate state of inactivation from which it recovers more slowly or 4) accelerated inactivation of the sodium channel. In in vitro expression systems, the premature inactivation of the sodium channel is sometimes observed at physiological temperatures, but not at room temperature [122]. Acceleration of INa inactivation was still more accentuated at higher than physiological temperatures, suggesting that the syndrome may be unmasked, and that patients with the BrS may be at an increased risk, during a febrile state [122]. A number of Brugada patients displaying fever-induced polymorphic VT have been identified since the publication of this report [123–132].
Mutation in the SCN5A gene account for approximately 18–30% of BrS cases. A higher incidence of SCN5A mutations has been reported in familial than in sporadic cases [133]. Of note, negative SCN5A results generally do not rule out causal gene mutations, since the promoter region, cryptic splicing mutations or presence of gross rearrangements are generally not part of routine investigation. A recent report by Hong et al. [134] provided the first report of a dysfunctional sodium channel created by an intronic mutation giving rise to cryptic splice site activation in SCN5A in a family with the BrS. The deletion of fragments of segments 2 and 3 of Domain IV of SCN5A caused complete loss of function.
Bezzina et al. [135] recently provided interesting evidence in support of the hypothesis that a SCN5A promoter polymorphism common in Asians modulates variability in cardiac conduction, and may contribute to the high prevalence of BrS in the Asian population. Sequencing of the SCN5A promoter identified a haplotype variant consisting of 6 polymorphisms in near-complete linkage disequilibrium that occurred at an allele frequency of 22% in Asian subjects and was absent in whites and blacks. The results of the study demonstrate that sodium channel transcription in the human heart may vary considerably among individuals and races and be associated with variable conduction velocity and arrhythmia susceptibility.
A second locus on chromosome 3, close to but distinct from SCN5A, has been linked to the syndrome [136] in a large pedigree in which the syndrome is associated with progressive conduction disease, a low sensitivity to procainamide, and a relatively good prognosis. The gene was recently identified as The glycerol-3-phosphate dehydrogenase 1-like Gene (GPD1L). A mutation in GPD1L has been shown to result in a partial reduction of INa [137].
The third and fourth genes associated with the BrS were reported earlier this year and shown to encode the α1 (CACNA1C) and β (CACNB2b) subunits of the L-type cardiac calcium channel. Mutations in the α and β subunits of the calcium channel can also lead to a shorter than normal QT interval, creating a new clinical entity consisting of a combined Brugada/short QT syndrome [138].
We are currently on a very steep learning curve in our understanding of the genotype-phenotype relationship in BrS and the role of genetics in the assignment of risk. It is recommended that all BrS patients, particularly symptomatic patients, undergo genetic screening. The argument for genetic testing is especially compelling in the case of BrS because the clinical manifestations of BrS generally appear later in life, in some cases resulting in sudden death without any warning. Risk stratification of younger family members is best achieved by genetic screening. Identification of mutation carriers among family members of symptomatic probands alerts the physician to closely follow these individuals and to proceed with the implantation of an implantable cardiac defibrillator (ICD) or other therapy if and when appropriate.
Amplification of epicardial and transmural dispersion of repolarization secondary to the presence of genetic defects, pathophysiologic factors and pharmacologic influences, leads to accentuation of the J wave and eventually to loss of the action potential dome, giving rise to extrasystolic activity in the form of phase 2 reentry. Activation of Ito leads to a paradoxical prolongation of APD in canine ventricular tissues [139], but to abbreviation of ventricular APD in species that normally exhibit brief action potentials (e.g., mouse and rat) [140]. Pathophysiologic conditions (e.g., ischemia, metabolic inhibition) and some pharmacologic interventions (e.g., INa or ICa blockers or ATP-sensitive current (IK-ATP), Ito, IKr or IKs activators) can lead to marked abbreviation of APD in canine and feline [141] ventricular cells where Ito is prominent. Under these conditions, canine ventricular epicardium exhibits an all-or-none repolarization as a result of the shift in the balance of currents flowing at the end of phase 1 of the action potential. All-or-none repolarization of the action potential occurs when phase 1 reaches approximately −30 mV. This leads to loss of the action potential dome as the outward currents overwhelm the inward currents. Loss of the dome generally occurs at some epicardial sites but not others, resulting in the development of a marked dispersion of repolarization within the epicardium as well as transmurally, between epicardium and endocardium. Propagation of the action potential dome from the epicardial site at which it is maintained to sites at which it is abolished can cause local re-excitation of the preparation. This mechanism, termed phase 2 reentry, produces extrasystolic beats capable of initiating circus movement reentry [142]. Phase 2 reentry has been shown to occur when right ventricular epicardium is exposed to: 1) K+ channel openers such as pinacidil [143]; 2) sodium channel blockers such as flecainide [144]; 3) increased [Ca2+] [115]; 4) calcium channel blockers such as verapamil; 5) metabolic inhibition [145]; and 6) simulated ischemia [142].
Table 3 provides a list of the drugs thus far identified as capable of inducing a Brugada ECG phenotype. Antiarrhythmic agents exhibiting potent use-dependent INa block have been shown to induce or unmask the Brugada ECG. These agents include ajmaline, flecainide, procainamide, disopyramide, propafenone and pilsicainide [146–148]. Antidepressants and antipsychotic agents have also been reported to induce the Brugada pattern. A study of 98 patients experiencing an overdose of tricyclic antidepressants reported that 15 of them displayed an ECG consistent with BrS [149]. The overall mortality was 3% among all patients but 6.7% among patients that displayed a Brugada phenotype. Rouleau et al. [150] described 3 cases of psychotropic drug-induced BrS ECG, occurring during concomitant administration of amitryptiline and a phenotiazine (case 1), overdose of fluoxetine (case 2), and co-administration of trifluoperazine and loxapine (case 3). Babaliaros and Hurst [151] described a Brugada pattern in patients receiving increasing doses of imipramine. Akhtar and Goldschlager [152] recently reported a case of BrS following overdose of desipramine and clonazepam (Fig. 3). Chow et al. [153] reported a similar case following desipramine. Bolognesi et al. [154] described a Brugada ECG pattern following overdose of amitryptyline and with maprotiline. Two additional cases of BrS were reported following overdose with nortryptiline [155, 156] or lithium [157]. The available data suggest most cases of antidepressant and antipsychotic-induced BrS phenotype occurs as a consequence of drug overdose or drug combination.
Table 3.
|
Exaggerated or otherwise abnormal J waves have long been linked to idiopathic ventricular fibrillation as well as to the BrS [116, 158–162]. The Brugada syndrome is characterized by exaggerated J wave that manifests as an ST segment elevation in the right precordial leads [116]. A number of studies have highlighted the similarities between the conditions that predispose to phase 2 reentry and those that attend the appearance of the BrS. Loss of the action potential dome in epicardium, but not endocardium generates a transmural current that manifests on the ECG as an ST segment elevation, similar to that encountered in patients with the BrS [99, 145, 163]. Evidence in support of a phase 2 reentrant mechanism in humans was recently provided by Thomsen et al. [164, 165].
Parasympathetic agonists like acetylcholine facilitate loss of the action potential dome [166] by suppressing ICa and/or augmenting potassium current. Beta-adrenergic agonists restore the dome by augmenting ICa. Agents that inhibit the sodium channel current facilitate loss of the canine right ventricular action potential dome via a negative shift in the voltage at which phase 1 begins [144, 167]. These findings are consistent with accentuation of ST segment elevation in patients with the BrS following vagal maneuvers or Class I antiarrhythmic agents as well as normalization of the ST segment elevation following β adrenergic agents and phosphodiesterase III inhibitors [99, 168, 169]. Loss of the action potential dome is more readily induced in right vs. left canine ventricular epicardium [10, 145, 163] because of the more prominent Ito-mediated phase 1 in action potentials in this region of the heart. As previously noted, this distinction is believed to be the basis for why the BrS is a right ventricular disease.
In the past, much of the focus was on the ability of a reduction in sodium channel current to unmask the BrS and create and arrhythmogenic substrate. A recent report shows that a combination of INa and ICa block is more effective than INa inhibition alone in precipitating the BrS in the arterially-perfused wedge preparation [170].
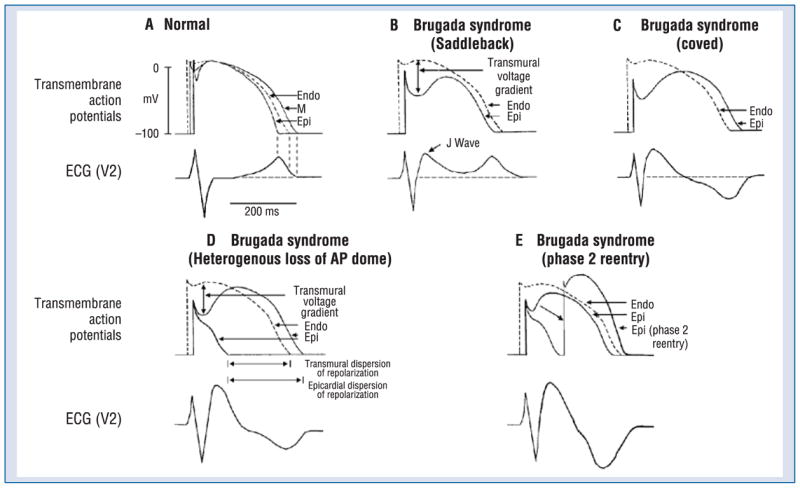
The ST segment elevation associated with the BrS has be attributed to: 1) conduction delay in the right ventricular epicardial free wall in the region of the outflow tract (RVOT) [171] and/or 2) accentuation of the right ventricular epicardial action potential that may lead to loss of the action potential dome [172]. The cellular mechanisms thought to be responsible for the development of the Brugada phenotype via hypothesis 2 is schematically illustrated in Figure 4 [173, 174].
The ST segment is usually isoelectric because of the absence of transmural voltage gradients at the level of the action potential plateau (Fig. 4A). Accentuation of the right ventricular notch under pathophysiologic conditions leads to exaggeration of transmural voltage gradients and thus to accentuation of the J wave or to J point elevation. When epicardial repolarization precedes repolarization of the cells in the M and endocardial regions the T wave remains positive. This results in a saddleback configuration of the repolarization waves (Fig. 4B). Further accentuation of the notch may be accompanied by a prolongation of the epicardial action potential such that the direction of repolarization across the right ventricular wall and transmural voltage gradients are reversed, leading to the development of a coved-type ST segment elevation and inversion of the T wave (Fig. 4C), typically observed in the ECG of Brugada patients. A delay in epicardial activation may also contribute to inversion of the T wave. The downsloping ST segment elevation observed in the experimental wedge models often appears as an R′, suggesting that the appearance of a right bundle branch block (RBBB) morphology in Brugada patients may be due at least in part to early repolarization of right ventricular (RV) epicardium, rather than major impulse conduction block in the right bundle.
Gussak et al. [175] pointed out that a majority of RBBB-like morphologies encountered in cases of BrS do not fit the criteria for RBBB. Moreover, attempts by Miyazaki et al. [168] to record delayed activation of the RV in Brugada patients met with failure.
It is important to point out that although the typical Brugada morphology is present in Figure 4B and C, an arrhythmogenic substrate is absent. The arrhythmogenic substrate is thought to develop when a further shift in the balance of current leads to loss of the action potential dome at some epicardial sites but not others (Fig. 4D). Loss of the action potential dome in epicardium but not endocardium results in the development of a marked transmural dispersion of repolarization and refractoriness, responsible for the development of a vulnerable window during which a premature impulse or extrasystole can induce a reentrant arrhythmia. Conduction of the action potential dome from sites at which it is maintained to sites at which it is lost causes local re-excitation via a phase 2 reentry mechanism, leading to the development of a very closely-coupled extrasystole, which captures the vulnerable window across the wall, thus triggering a circus movement reentry in the form of VT/VF (Fig. 4E) [142, 176]. The phase 2 reentrant beat fuses with the T wave of the basic response, thus accentuating the negative T wave. This morphology is often observed in the clinic preceding the onset of polymorphic VT.
Studies involving the arterially-perfused right ventricular wedge preparation provide evidence in support of these hypotheses [176]. Aiba et al. [177] used a high resolution optical mapping system that allows simultaneous recording of transmembrane action potentials from 256 sites along the transmural surface of the arterially-perfused canine right-ventricular wedge preparation to demonstrate that a steep repolarization gradient between the region at which the dome is lost and the region at which it is maintained is essential for the development of a closely coupled phase 2 reentrant extrasystole. This study also showed that reentry initially rotates in the epicardium and gradually shifts to a transmural orientation, responsible for nonsustained polymorphic VT or VF.
Kurita et al. placed monophasic action potential (MAP) electrodes on the epicardial and endocardial surfaces of the RVOT in patients with the BrS and demonstrated an accentuated notch in the epicardial response, thus providing support for this mechanism in humans [178, 179].
Thus, accentuation of the right ventricular epicardial action potential notch underlies the ST segment elevation. Eventual loss of the dome of the right ventricular epicardial action potential further exaggerates ST segment elevation. A vulnerable window is created both within epicardium, as well as transmurally, which serves as the substrate for the development of reentry (Fig. 5).
Figure 5.
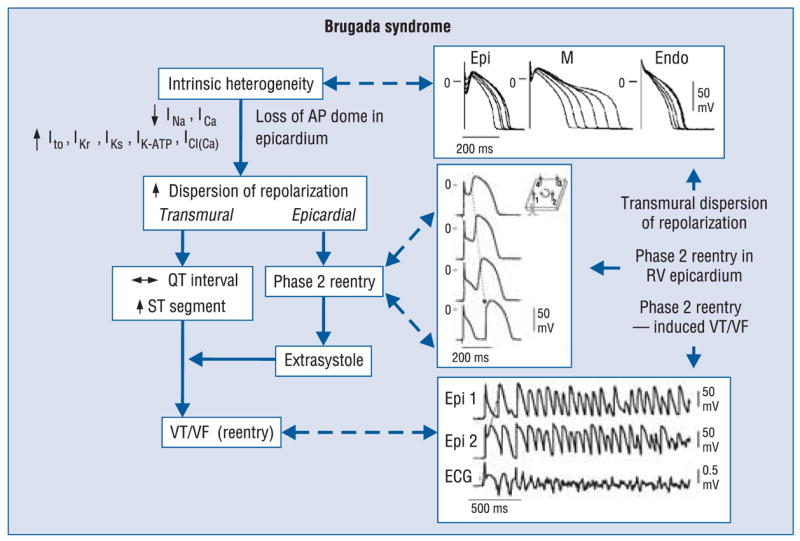
Proposed mechanism for the Brugada syndrome. A shift in the balance of currents serves to amplify existing heterogeneities by causing loss of the action potential dome at some epicardial, but not endocardial sites. A vulnerable window develops as a result of the dispersion of repolarization and refractoriness within epicardium as well as across the wall. Epicardial dispersion leads to the development of phase 2 reentry, which provides the extrasystole that captures the vulnerable window and initiates ventricular tachycardia/ventricular fibrillation (VT/VF) via a circus movement reentry mechanism. Modified from [174], with permission.
Phase 2 reentry provides the extrasystole that serves as the trigger that precipitates episodes of ventricular tachycardia and fibrillation in the BrS. Evidence in support of this hypothesis was recently provided in an arterially-perfused canine right ventricular experimental model of the BrS [176]. The VT and VF generated in these preparations is usually polymorphic, resembling a rapid form of torsade de pointes. This activity is likely related to the migrating spiral wave shown to generate a pattern resembling a polymorphic VT [180, 181].
Role of TDR in drug-induced arrhythmogenesis
In the acquired long QT syndrome, QT interval increases as a function of drug concentration, whereas in BrS it remains largely unchanged. What these two drug-induced syndromes have in common is an amplification of spatial dispersion of repolarization, TDR in particular, which results in the development of TdP when dispersion reaches the threshold for reentry.
Genetic predisposition
The degree to which a genetic predisposition contributes to the clinical manifestation of drug-induced arrhythmogenesis is not well defined. Available data suggest that up to 10–15% percent of individuals who develop TdP following exposure to QT-prolonging drugs possess mutations associated with the LQTS and may be considered to have a subclinical form of the congenital syndrome [182–186].
Abbot et al. [187] were among the first to show that a polymorphism (i.e., a genetic variation that is present in greater than 1% of the population) in an ion channel gene is associated with a predisposition to drug-induced TdP. They identified a polymorphism (T8A) of the KCNE2 gene encoding for MiRP, a beta subunit of the IKr channel, that is present in 1.6% of the population and is associated with TdP related to quinidine and to sulfamethoxazole//trimethoprim administration. This finding suggests that common genetic variations may increase the risk for development of drug related arrhythmias. Yang et al. [185] showed that DNA variants in the coding regions of congenital long-QT disease genes predisposing to acquired LQTS can be identified in approximately 10% to 15% of affected subjects, predominantly in genes encoding ancillary subunits, providing further support for the hypothesis that subclinical mutations and polymorphisms may predispose to drug-induced TdP. Splawski et al. [188] further advanced this concept by identifying a heterozygous polymorphism involving substitution of serine with tyrosine in codon 1103 (S1103Y) in the sodium channel gene SCN5A (S1102Y in the shorter splice variant of SCN5A) among Africans and African Americans that increases the risk for acquired TdP. The polymorphism was present in 57% of 23 patients with pro-arrhythmic episodes, but in only 13% of controls. These findings suggest that carriers of such polymorphisms can be identified and excluded from treatment with drugs that are associated with a risk of proarrhythmia. It is conceivable that such testing could be applied widely since accurate results could be made available rapidly at relatively low cost.
Another common polymorphism that has been associated with acquired forms of LQTS and TdP is K897T in KCNH2 [189]. Most functional expression studies have reported that K897T reduces IKr, [190–192] although one study has reported an increase in HERG current [193].
The action of antidepressants to precipitate the BrS may also have a genetic disposition. For example, the SCN5A promoter haplotype (so-called Hap B) has been shown to be associated with longer PR and QRS intervals as well as a more exaggerated response to sodium channel blockers [135].
Genetic defects can also contribute to drug-induced channelopathies by influencing the metabolism of drugs. In the case of relatively pure IKr blockers, there is a clear relationship between plasma levels of drug and the incidence of TdP. Genetic variants of the genes encoding for enzymes responsible for drug metabolism could alter pharmacokinetics so as to cause wide fluctuations in plasma levels, thus exerting a significant proarrhythmic influence [194, 195]. For example, in the case of cytochrome CYP2D6, which is involved in the metabolism of some QT prolonging drugs (terodiline, thioridazine), multiple polymorphisms have been reported that reduce or eliminate its function; 5–10% of Caucasians and African-Americans lack a functional CYP2D6. Numerous proteins, including drug transport molecules and other drug metabolizing enzymes, are involved in drug absorption, distribution, and elimination and genetic variants of each of these has the potential to modulate drug concentrations and effects. Multiple substrates and inhibitors of the cytochrome P450 enzymes have been identified. A comprehensive database can be found at http://medicine.iupui.edu/flockhart/.
Acknowledgments
The authors do not report any conflict of interest regarding this work.
Supported by grant HL47678 from NHLBI and grants from the American Heart Association and NYS and Florida Grand Lodges F. & A.M.
References
- 1.Antzelevitch C, Sicouri S, Litovsky SH, et al. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res. 1991;69:1427–1449. doi: 10.1161/01.res.69.6.1427. [DOI] [PubMed] [Google Scholar]
- 2.Antzelevitch C, Shimizu W, Yan GX, et al. The M cell: Its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. 1999;10:1124–1152. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 3.Litovsky SH, Antzelevitch C. Transient outward current prominent in canine ventricular epicardium but not endocardium. Circ Res. 1988;62:116–126. doi: 10.1161/01.res.62.1.116. [DOI] [PubMed] [Google Scholar]
- 4.Liu DW, Gintant GA, Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res. 1993;72:671–687. doi: 10.1161/01.res.72.3.671. [DOI] [PubMed] [Google Scholar]
- 5.Furukawa T, Myerburg RJ, Furukawa N, Bassett AL, Kimura S. Differences in transient outward currents of feline endocardial and epicardial myocytes. Circ Res. 1990;67:1287–1291. doi: 10.1161/01.res.67.5.1287. [DOI] [PubMed] [Google Scholar]
- 6.Fedida D, Giles WR. Regional variations in action potentials and transient outward current in myocytes isolated from rabbit left ventricle. J Physiol (Lond) 1991;442:191–209. doi: 10.1113/jphysiol.1991.sp018789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark RB, Bouchard RA, Salinas-Stefanon E, Sanchez-Chapula J, Giles WR. Heterogeneity of action potential waveforms and potassium currents in rat ventricle. Cardiovasc Res. 1993;27:1795–1799. doi: 10.1093/cvr/27.10.1795. [DOI] [PubMed] [Google Scholar]
- 8.Wettwer E, Amos GJ, Posival H, Ravens U. Transient outward current in human ventricular myocytes of subepicardial and sub-endocardial origin. Circ Res. 1994;75:473–482. doi: 10.1161/01.res.75.3.473. [DOI] [PubMed] [Google Scholar]
- 9.Nabauer M, Beuckelmann DJ, Uberfuhr P, Steinbeck G. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation. 1996;93:168–177. doi: 10.1161/01.cir.93.1.168. [DOI] [PubMed] [Google Scholar]
- 10.Di Diego JM, Sun ZQ, Antzelevitch C. Ito and action potential notch are smaller in left vs. right canine ventricular epicardium. Am J Physiol. 1996;271:H548–H561. doi: 10.1152/ajpheart.1996.271.2.H548. [DOI] [PubMed] [Google Scholar]
- 11.Volders PG, Sipido KR, Carmeliet E, Spatjens RL, Wellens HJ, Vos MA. Repolarizing K+ currents ITO1 and IKs are larger in right than left canine ventricular midmyocardium. Circulation. 1999;99:206–210. doi: 10.1161/01.cir.99.2.206. [DOI] [PubMed] [Google Scholar]
- 12.Sicouri S, Antzelevitch C. A subpopulation of cells with unique electrophysiological properties in the deep subepicardium of the canine ventricle. The M cell Circ Res. 1991;68:1729–1741. doi: 10.1161/01.res.68.6.1729. [DOI] [PubMed] [Google Scholar]
- 13.Anyukhovsky EP, Sosunov EA, Rosen MR. Regional differences in electrophysiologic properties of epicardium, midmyocardium and endocardium: In vitro and in vivo correlations. Circulation. 1996;94:1981–1988. doi: 10.1161/01.cir.94.8.1981. [DOI] [PubMed] [Google Scholar]
- 14.Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. Circ Res. 1995;76:351–365. doi: 10.1161/01.res.76.3.351. [DOI] [PubMed] [Google Scholar]
- 15.Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol. 2001;281:H689–H697. doi: 10.1152/ajpheart.2001.281.2.H689. [DOI] [PubMed] [Google Scholar]
- 16.Zygmunt AC, Goodrow RJ, Antzelevitch C. I(NaCa) contributes to electrical heterogeneity within the canine ventricle. Am J Physiol Heart Circ Physiol. 2000;278:H1671–H1678. doi: 10.1152/ajpheart.2000.278.5.H1671. [DOI] [PubMed] [Google Scholar]
- 17.Brahmajothi MV, Morales MJ, Reimer KA, Strauss HC. Regional localization of ERG, the channel protein responsible for the rapid component of the delayed rectifier, K+ current in the ferret heart. Circ Res. 1997;81:128–135. doi: 10.1161/01.res.81.1.128. [DOI] [PubMed] [Google Scholar]
- 18.Cordeiro JM, Greene L, Heilmann C, Antzelevitch D, Antzelevitch C. Transmural heterogeneity of calcium activity and mechanical function in the canine left ventricle. Am J Physiol Heart Circ Physiol. 2004;286:H1471–H1479. doi: 10.1152/ajpheart.00748.2003. [DOI] [PubMed] [Google Scholar]
- 19.Banyasz T, Fulop L, Magyar J, Szentandrassy N, Varro A, Nanasi PP. Endocardial versus epicardial differences in L-type calcium current in canine ventricular myocytes studied by action potential voltage clamp. Cardiovasc Res. 2003;58:66–75. doi: 10.1016/s0008-6363(02)00853-2. [DOI] [PubMed] [Google Scholar]
- 20.Wang HS, Cohen IS. Calcium channel heterogeneity in canine left ventricular myocytes. J Physiol. 2003;547:825–833. doi: 10.1113/jphysiol.2002.035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan GX, Shimizu W, Antzelevitch C. Characteristics and distribution of M cells in arterially-perfused canine left ventricular wedge preparations. Circulation. 1998;98:1921–1927. doi: 10.1161/01.cir.98.18.1921. [DOI] [PubMed] [Google Scholar]
- 22.Sicouri S, Antzelevitch C. Electrophysiologic characteristics of M cells in the canine left ventricular free wall. J Cardiovasc Electrophysiol. 1995;6:591–603. doi: 10.1111/j.1540-8167.1995.tb00435.x. [DOI] [PubMed] [Google Scholar]
- 23.Sicouri S, Fish J, Antzelevitch C. Distribution of M cells in the canine ventricle. J Cardiovasc Electrophysiol. 1994;5:824–837. doi: 10.1111/j.1540-8167.1994.tb01121.x. [DOI] [PubMed] [Google Scholar]
- 24.Antzelevitch C, Sicouri S. Clinical relevance of cardiac arrhythmias generated by afterdepolarizations. Role of M cells in the generation of U waves, triggered activity and torsade de pointes. J Am Coll Cardiol. 1994;23:259–277. doi: 10.1016/0735-1097(94)90529-0. [DOI] [PubMed] [Google Scholar]
- 25.Stankovicova T, Szilard M, De Scheerder I, Sipido KR. M cells and transmural heterogeneity of action potential configuration in myocytes from the left ventricular wall of the pig heart. Cardiovasc Res. 2000;45:952–960. doi: 10.1016/s0008-6363(99)00418-6. [DOI] [PubMed] [Google Scholar]
- 26.Sicouri S, Antzelevitch C. Drug-induced afterdepolarizations and triggered activity occur in a discrete subpopulation of ventricular muscle cell (M cells) in the canine heart: Quinidine and Digitalis. J Cardiovasc Electrophysiol. 1993;4:48–58. doi: 10.1111/j.1540-8167.1993.tb01211.x. [DOI] [PubMed] [Google Scholar]
- 27.Drouin E, Charpentier F, Gauthier C, Laurent K, Le Marec H. Electrophysiological characteristics of cells spanning the left ventricular wall of human heart: Evidence for the presence of M cells. J Am Coll Cardiol. 1995;26:185–192. doi: 10.1016/0735-1097(95)00167-x. [DOI] [PubMed] [Google Scholar]
- 28.Weissenburger J, Nesterenko VV, Antzelevitch C. Transmural heterogeneity of ventricular repolarization under baseline and long QT conditions in the canine heart in vivo: Torsades de pointes develops with halothane but not pentobarbital anesthesia. J Cardiovasc Electrophysiol. 2000;11:290–304. doi: 10.1111/j.1540-8167.2000.tb01798.x. [DOI] [PubMed] [Google Scholar]
- 29.Sicouri S, Quist M, Antzelevitch C. Evidence for the presence of M cells in the guinea pig ventricle. J Cardiovasc Electrophysiol. 1996;7:503–511. doi: 10.1111/j.1540-8167.1996.tb00557.x. [DOI] [PubMed] [Google Scholar]
- 30.Li GR, Feng J, Yue L, Carrier M. Transmural heterogeneity of action potentials and Ito1 in myocytes isolated from the human right ventricle. Am J Physiol. 1998;275:H369–H377. doi: 10.1152/ajpheart.1998.275.2.H369. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Sinovas A, Cinca J, Tapias A, Armadans L, Tresanchez M, Soler-Soler J. Lack of evidence of M-cells in porcine left ventricular myocardium. Cardiovasc Res. 1997;33:307–313. doi: 10.1016/s0008-6363(96)00205-2. [DOI] [PubMed] [Google Scholar]
- 32.Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade de pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038–2047. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- 33.El-Sherif N, Caref EB, Yin H, Restivo M. The electrophysiological mechanism of ventricular arrhythmias in the long QT syndrome: Tridimensional mapping of activation and recovery patterns. Circ Res. 1996;79:474–492. doi: 10.1161/01.res.79.3.474. [DOI] [PubMed] [Google Scholar]
- 34.Weirich J, Bernhardt R, Loewen N, Wenzel W, Antoni H. Regional- and species-dependent effects of K+-channel blocking agents on subendocardium and mid-wall slices of human, rabbit, and guinea pig myocardium. Pflugers Arch. 1996;431:R130. (abstract) [Google Scholar]
- 35.Burashnikov A, Antzelevitch C. Acceleration-induced action potential prolongation and early afterdepolarizations. J Cardiovasc Electrophysiol. 1998;9:934–948. doi: 10.1111/j.1540-8167.1998.tb00134.x. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu W, McMahon B, Antzelevitch C. Sodium pentobarbital reduces transmural dispersion of repolarization and prevents torsade de pointes in models of acquired and congenital long QT syndrome. J Cardiovasc Electrophysiol. 1999;10:156–164. doi: 10.1111/j.1540-8167.1999.tb00656.x. [DOI] [PubMed] [Google Scholar]
- 37.Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long QT syndrome: Effects of b-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarization and torsade de pointes. Circulation. 1998;98:2314–2322. doi: 10.1161/01.cir.98.21.2314. [DOI] [PubMed] [Google Scholar]
- 38.Shimizu W, Antzelevitch C. Cellular and ionic basis for T-wave alternans under Long QT conditions. Circulation. 1999;99:1499–1507. doi: 10.1161/01.cir.99.11.1499. [DOI] [PubMed] [Google Scholar]
- 39.Yan GX, Antzelevitch C. Cellular basis for the normal T wave and the electrocardiographic manifestations of the long QT syndrome. Circulation. 1998;98:1928–1936. doi: 10.1161/01.cir.98.18.1928. [DOI] [PubMed] [Google Scholar]
- 40.Balati B, Varro A, Papp JG. Comparison of the cellular electrophysiological characteristics of canine left ventricular epicardium, M cells, endocardium and Purkinje fibres. Acta Physiol Scand. 1998;164:181–190. doi: 10.1046/j.1365-201X.1998.00416.x. [DOI] [PubMed] [Google Scholar]
- 41.McIntosh MA, Cobbe SM, Smith GL. Heterogeneous changes in action potential and intracellular Ca2+ in left ventricular myocyte sub-types from rabbits with heart failure. Cardiovasc Res. 2000;45:397–409. doi: 10.1016/s0008-6363(99)00360-0. [DOI] [PubMed] [Google Scholar]
- 42.Janse MJ, Sosunov EA, Coronel R, et al. Repolarization gradients in the canine left ventricle before and after induction of short-term cardiac memory. Circulation. 2005;112:1711–1718. doi: 10.1161/CIRCULATIONAHA.104.516583. [DOI] [PubMed] [Google Scholar]
- 43.Antzelevitch C. The M cell. Invited Editorial Comment. Journal of Cardiovascular Pharmacology and Therapeutics. 1997;2:73–76. doi: 10.1177/107424849700200109. [DOI] [PubMed] [Google Scholar]
- 44.Cohen IS, Giles WR, Noble D. Cellular basis for the T wave of the electrocardiogram. Nature. 1976;262:657–661. doi: 10.1038/262657a0. [DOI] [PubMed] [Google Scholar]
- 45.Jeyaraj D, Wilson LD, Zhong J, et al. Mechanoelectrical feedback as novel mechanism of cardiac electrical remodeling. Circulation. 2007;115:3145–3155. doi: 10.1161/CIRCULATIONAHA.107.688317. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz PJ. The idiopathic long QT syndrome: Progress and questions. Am Heart J. 1985;109:399–411. doi: 10.1016/0002-8703(85)90626-x. [DOI] [PubMed] [Google Scholar]
- 47.Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome: Prospective longitudinal study of 328 families. Circulation. 1991;84:1136–1144. doi: 10.1161/01.cir.84.3.1136. [DOI] [PubMed] [Google Scholar]
- 48.Zipes DP. The long QT interval syndrome: A Rosetta stone for sympathetic related ventricular tachyarrhythmias. Circulation. 1991;84:1414–1419. doi: 10.1161/01.cir.84.3.1414. [DOI] [PubMed] [Google Scholar]
- 49.Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 50.Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 51.Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001;105:511–519. doi: 10.1016/s0092-8674(01)00342-7. [DOI] [PubMed] [Google Scholar]
- 52.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 53.Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 54.Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet. 1997;17:338–340. doi: 10.1038/ng1197-338. [DOI] [PubMed] [Google Scholar]
- 55.Cronk LB, Ye B, Kaku T, et al. Novel mechanism for sudden infant death syndrome: Persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007;4:161–166. doi: 10.1016/j.hrthm.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Medeiros-Domingo A, Kaku T, Tester DJ, et al. SCN4B-encoded sodium channel {beta}4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–142. doi: 10.1161/CIRCULATIONAHA.106.659086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Splawski I, Timothy KW, Sharpe LM, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 58.Bednar MM, Harrigan EP, Anziano RJ, Camm AJ, Ruskin JN. The QT interval. Prog Cardiovasc Dis. 2001;43:1–45. doi: 10.1053/pcad.2001.21469. [DOI] [PubMed] [Google Scholar]
- 59.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 60.Sipido KR, Volders PG, De Groot SH, et al. Enhanced Ca(2+) release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: Potential link between contractile adaptation and arrhythmogenesis. Circulation. 2000;102:2137–2144. doi: 10.1161/01.cir.102.17.2137. [DOI] [PubMed] [Google Scholar]
- 61.Volders PG, Sipido KR, Vos MA, et al. Downregulation of delayed rectifier K(+) currents in dogs with chronic complete atrioventricular block and acquired torsades de pointes. Circulation. 1999;100:2455–2461. doi: 10.1161/01.cir.100.24.2455. [DOI] [PubMed] [Google Scholar]
- 62.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: Role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–2552. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 64.Belardinelli L, Antzelevitch C, Vos MA. Assessing predictors of drug-induced torsade de pointes. Trends Pharmacol Sci. 2003;24:619–625. doi: 10.1016/j.tips.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 65.Antzelevitch C, Shimizu W. Cellular mechanisms underlying the Long QT syndrome. Curr Opin Cardiol. 2002;17:43–51. doi: 10.1097/00001573-200201000-00007. [DOI] [PubMed] [Google Scholar]
- 66.Shimizu W, Antzelevitch C. Effects of a K(+) channel opener to reduce transmural dispersion of repolarization and prevent torsade de pointes in LQT1, LQT2, and LQT3 models of the long--QT syndrome. Circulation. 2000;102:706–712. doi: 10.1161/01.cir.102.6.706. [DOI] [PubMed] [Google Scholar]
- 67.Antzelevitch C. Heterogeneity of cellular repolarization in LQTS: The role of M cells. Eur Heart J. 2001;(suppl 3):K2–K16. [Google Scholar]
- 68.Tsuboi M, Antzelevitch C. Cellular basis for electrocardiographic and arrhythmic manifestations of Andersen-Tawil syndrome (LQT7) Heart Rhythm. 2006;3:328–335. doi: 10.1016/j.hrthm.2005.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sicouri S, Timothy KW, Zygmunt AC, et al. Cellular basis for the electrocardiographic and arrhythmic manifestations of Timothy syndrome: Effects of ranolazine. Heart Rhythm. 2007;4:638–647. doi: 10.1016/j.hrthm.2006.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shimizu W, Antzelevitch C. Differential effects of beta-adrenergic agonists and antagonists in LQT1, LQT2 and LQT3 models of the long QT syndrome. J Am Coll Cardiol. 2000;35:778–786. doi: 10.1016/s0735-1097(99)00582-3. [DOI] [PubMed] [Google Scholar]
- 71.Anyukhovsky EP, Sosunov EA, Gainullin RZ, Rosen MR. The controversial M cell. J Cardiovasc Electrophysiol. 1999;10:244–260. doi: 10.1111/j.1540-8167.1999.tb00667.x. [DOI] [PubMed] [Google Scholar]
- 72.Crampton RS. Preeminence of the left stellate ganglion in the long Q-T syndrome. Circulation. 1979;59:769–778. doi: 10.1161/01.cir.59.4.769. [DOI] [PubMed] [Google Scholar]
- 73.Ali RH, Zareba W, Moss A, et al. Clinical and genetic variables associated with acute arousal and nonarousal-related cardiac events among subjects with long QT syndrome. Am J Cardiol. 2000;85:457–461. doi: 10.1016/s0002-9149(99)90772-5. [DOI] [PubMed] [Google Scholar]
- 74.Noda T, Takaki H, Kurita T, et al. Gene-specific response of dynamic ventricular repolarization to sympathetic stimulation in LQT1, LQT2 and LQT3 forms of congenital long QT syndrome. Eur Heart J. 2002;23:975–983. doi: 10.1053/euhj.2001.3079. [DOI] [PubMed] [Google Scholar]
- 75.Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 76.Antzelevitch C. Role of transmural dispersion of repolarization in the genesis of drug-induced torsades de pointes. Heart Rhythm. 2005;2:S9–S15. doi: 10.1016/j.hrthm.2004.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sicouri S, Moro S, Litovsky SH, Elizari MV, Antzelevitch C. Chronic amiodarone reduces transmural dispersion of repolarization in the canine heart. J Cardiovasc Electrophysiol. 1997;8:1269–1279. doi: 10.1111/j.1540-8167.1997.tb01018.x. [DOI] [PubMed] [Google Scholar]
- 78.van Opstal JM, Schoenmakers M, Verduyn SC, et al. Chronic amiodarone evokes no torsade de pointes arrhythmias despite QT lengthening in an animal model of acquired long-QT syndrome. Circulation. 2001;104:2722–2727. doi: 10.1161/hc4701.099579. [DOI] [PubMed] [Google Scholar]
- 79.Di Diego JM, Belardinelli L, Antzelevitch C. Cisapride-induced transmural dispersion of repolarization and torsade de pointes in the canine left ventricular wedge preparation during epicardial stimulation. Circulation. 2003;108:1027–1033. doi: 10.1161/01.CIR.0000085066.05180.40. [DOI] [PubMed] [Google Scholar]
- 80.Emori T, Antzelevitch C. Cellular basis for complex T waves and arrhythmic activity following combined I(Kr) and I(Ks) block. J Cardiovasc Electrophysiol. 2001;12:1369–1378. doi: 10.1046/j.1540-8167.2001.01369.x. [DOI] [PubMed] [Google Scholar]
- 81.Xia Y, Liang Y, Kongstad O, et al. In vivo validation of the coincidence of the peak and end of the T wave with full repolarization of the epicardium and endocardium in swine. Heart Rhythm. 2005;2:162–169. doi: 10.1016/j.hrthm.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 82.Wolk R, Stec S, Kulakowski P. Extrasystolic beats affect transmural electrical dispersion during programmed electrical stimulation. Eur J Clinical Invest. 2001;31:293–301. doi: 10.1046/j.1365-2362.2001.00817.x. [DOI] [PubMed] [Google Scholar]
- 83.Tanabe Y, Inagaki M, Kurita T, et al. Sympathetic stimulation produces a greater increase in both transmural and spatial dispersion of repolarization in LQT1 than LQT2 forms of congenital long QT syndrome. J Am Coll Cardiol. 2001;37:911–919. doi: 10.1016/s0735-1097(00)01200-6. [DOI] [PubMed] [Google Scholar]
- 84.Frederiks J, Swenne CA, Kors JA, et al. Within-subject electrocardiographic differences at equal heart rates: Role of the autonomic nervous system. Pflugers Arch. 2001;441:717–724. doi: 10.1007/s004240000487. [DOI] [PubMed] [Google Scholar]
- 85.Shimizu M, Ino H, Okeie K, et al. T-peak to T-end interval may be a better predictor of high-risk patients with hypertrophic cardiomyopathy associated with a cardiac troponin I mutation than QT dispersion. Clin Cardiol. 2002;25:335–339. doi: 10.1002/clc.4950250706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Takenaka K, Ai T, Shimizu W, et al. Exercise stress test amplifies genotype-phenotype correlation in the LQT1 and LQT2 forms of the long-QT syndrome. Circulation. 2003;107:838–844. doi: 10.1161/01.cir.0000048142.85076.a2. [DOI] [PubMed] [Google Scholar]
- 87.Watanabe N, Kobayashi Y, Tanno K, et al. Transmural dispersion of repolarization and ventricular tachyarrhythmias. J Electrocardiol. 2004;37:191–200. doi: 10.1016/j.jelectrocard.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 88.Yamaguchi M, Shimizu M, Ino H, et al. T wave peak-to-end interval and QT dispersion in acquired long QT syndrome: A new index for arrhythmogenicity. Clin Sci (Lond) 2003;105:671–676. doi: 10.1042/CS20030010. [DOI] [PubMed] [Google Scholar]
- 89.Topilski I, Rogowski O, Rosso R, et al. The morphology of the QT interval predicts torsade de pointes during acquired bradyarrhythmias. J Am Coll Cardiol. 2007;49:320–328. doi: 10.1016/j.jacc.2006.08.058. [DOI] [PubMed] [Google Scholar]
- 90.Antzelevitch C. Tpeak-Tend interval as an index of transmural dispersion of repolarization. Eur J Clin Invest. 2001;31:555–557. doi: 10.1046/j.1365-2362.2001.00849.x. [DOI] [PubMed] [Google Scholar]
- 91.Opthof T, Coronel R, Wilms-Schopman FJ, et al. Dispersion of repolarization in canine ventricle and the electrocardiographic T wave: T(p-e) interval does not reflect transmural dispersion. Heart Rhythm. 2007;4:341–348. doi: 10.1016/j.hrthm.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 92.Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome. Circulation. 2002;105:1247–1253. doi: 10.1161/hc1002.105231. [DOI] [PubMed] [Google Scholar]
- 93.Aiba T, Shimizu W, Hidaka I, et al. Cellular basis for trigger and maintenance of ventricular fibrillation in the Brugada syndrome model: High-resolution optical mapping study. J Am Coll Cardiol. 2006;47:2074–2085. doi: 10.1016/j.jacc.2005.12.064. [DOI] [PubMed] [Google Scholar]
- 94.Castro HJ, Antzelevitch C, Tornes BF, et al. Tpeak-Tend and Tpeak-Tend dispersion as risk factors for ventricular tachycardia/ventricular fibrillation in patients with the Brugada syndrome. J Am Coll Cardiol. 2006;47:1828–1834. doi: 10.1016/j.jacc.2005.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fish JM, Brugada J, Antzelevitch C. Potential proarrhythmic effects of biventricular pacing. J Am Coll Cardiol. 2005;46:2340–2347. doi: 10.1016/j.jacc.2005.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Taggart P, Sutton P, Opthof T, Coronel R, Kallis P. Electrotonic cancellation of transmural electrical gradients in the left ventricle in man. Prog Biophys Mol Biol. 2003;82:243–254. doi: 10.1016/s0079-6107(03)00025-7. [DOI] [PubMed] [Google Scholar]
- 97.Opthof T. In vivo dispersion in repolarization and arrhythmias in the human heart. Am J Physiol Heart Circ Physiol. 2006;290:H77–H78. doi: 10.1152/ajpheart.00954.2005. [DOI] [PubMed] [Google Scholar]
- 98.Medina-Ravell VA, Lankipalli RS, Yan GX, et al. Effect of epicardial or biventricular pacing to prolong QT interval and increase transmural dispersion of repolarization. Does resynchronization therapy pose a risk for patients predisposed to long QT or torsade de pointes? Circulation. 2003;107:740–746. doi: 10.1161/01.cir.0000048126.07819.37. [DOI] [PubMed] [Google Scholar]
- 99.Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93:372–379. doi: 10.1161/01.cir.93.2.372. [DOI] [PubMed] [Google Scholar]
- 100.Clements SD, Hurst JW. Diagnostic value of ECG abnormalities observed in subjects accidentally exposed to cold. Am J Cardiol. 1972;29:729–734. doi: 10.1016/0002-9149(72)90178-6. [DOI] [PubMed] [Google Scholar]
- 101.Thompson R, Rich J, Chmelik F, Nelson WL. Evolutionary changes in the electrocardiogram of severe progressive hypothermia. J Electrocardiol. 1977;10:67–70. doi: 10.1016/s0022-0736(77)80034-4. [DOI] [PubMed] [Google Scholar]
- 102.RuDusky BM. The electrocardiogram in hypothermiathe J wave and the Brugada syndrome. Am J Cardiol. 2004;93:671–672. doi: 10.1016/j.amjcard.2003.10.065. [DOI] [PubMed] [Google Scholar]
- 103.Kraus F. Ueber die wirkung des kalziums auf den kreislauf. Dtsch Med Wochenschr. 1920;46:201–203. [Google Scholar]
- 104.Sridharan MR, Horan LG. Electrocardiographic J wave of hypercalcemia. Am J Cardiol. 1984;54:672–673. doi: 10.1016/0002-9149(84)90273-x. [DOI] [PubMed] [Google Scholar]
- 105.Antzelevitch C, Sicouri S, Lukas A, Nesterenko VV, Liu DW, Di Diego JM. Regional differences in the electrophysiology of ventricular cells: Physiological and clinical implications. In: Zipes DP, Jalife J, editors. Cardiac electrophysiology: From cell to bedside. 2. W.B. Saunders Co; Philadelphia: 1995. pp. 228–245. [Google Scholar]
- 106.Zicha S, Xiao L, Stafford S, et al. Transmural expression of transient outward potassium current subunits in normal and failing canine and human hearts. J Physiol. 2004;561:735–748. doi: 10.1113/jphysiol.2004.075861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rosati B, Pan Z, Lypen S, et al. Regulation of KChIP2 potassium channel beta subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol. 2001;533:119–125. doi: 10.1111/j.1469-7793.2001.0119b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Costantini DL, Arruda EP, Agarwal P, et al. The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell. 2005;123:347–358. doi: 10.1016/j.cell.2005.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Takano M, Noma A. Distribution of the isoprenaline-induced chloride current in rabbit heart. Pflugers Arch. 1992;420:223–226. doi: 10.1007/BF00374995. [DOI] [PubMed] [Google Scholar]
- 110.Zygmunt AC. Intracellular calcium activates chloride current in canine ventricular myocytes. Am J Physiol. 1994;267:H1984–H1995. doi: 10.1152/ajpheart.1994.267.5.H1984. [DOI] [PubMed] [Google Scholar]
- 111.Eagle K. Images in clinical medicine. Osborn waves of hypothermia. N Engl J Med. 1994;10:680. doi: 10.1056/NEJM199403103301005. [DOI] [PubMed] [Google Scholar]
- 112.Emslie-Smith D, Sladden GE, Stirling GR. The significance of changes in the electrocardiogram in hypothermia. Br Heart J. 1959;21:343–351. doi: 10.1136/hrt.21.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Osborn JJ. Experimental hypothermia: Respiratory and blood pH changes in relation to cardiac function. Am J Physiol. 1953;175:389–398. doi: 10.1152/ajplegacy.1953.175.3.389. [DOI] [PubMed] [Google Scholar]
- 114.Sridharan MR, Johnson JC, Horan LG, Sohl GS, Flowers NC. Monophasic action potentials in hypercalcemic and hypothermic „J” waves-a comparative study. Am Fed Clin Res. 1983;31:219. [Google Scholar]
- 115.Di Diego JM, Antzelevitch C. High [Ca2+]-induced electrical heterogeneity and extrasystolic activity in isolated canine ventricular epicardium: Phase 2 reentry. Circulation. 1994;89:1839–1850. doi: 10.1161/01.cir.89.4.1839. [DOI] [PubMed] [Google Scholar]
- 116.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome: A multicenter report. J Am Coll Cardiol. 1992;20:1391–1396. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 117.Nademanee K. Sudden unexplained death syndrome in southeast Asia. Am J Cardiol. 1997;79 (6A):10–11. doi: 10.1016/s0002-9149(97)00113-6. [DOI] [PubMed] [Google Scholar]
- 118.Vatta M, Dumaine R, Varghese G, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11:337–345. doi: 10.1093/hmg/11.3.337. [DOI] [PubMed] [Google Scholar]
- 119.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanisms for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 120.Grant AO, Carboni MP, Neplioueva V, et al. Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J Clin Invest. 2002;110:1201–1209. doi: 10.1172/JCI15570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Antzelevitch C, Brugada P, Brugada J, Brugada R. The Brugada syndrome: From bench to bedside. Blackwell Futura; Oxford: 2005. [Google Scholar]
- 122.Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–809. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- 123.Saura D, Garcia-Alberola A, Carrillo P, Pascual D, Martinez--Sanchez J, Valdes M. Brugada-like electrocardiographic pattern induced by fever. PACE. 2002;25:856–859. doi: 10.1046/j.1460-9592.2002.t01-1-00856.x. [DOI] [PubMed] [Google Scholar]
- 124.Porres JM, Brugada J, Urbistondo V, Garcia F, Reviejo K, Marco P. Fever unmasking the Brugada syndrome. PACE. 2002;25:1646–1648. doi: 10.1046/j.1460-9592.2002.01646.x. [DOI] [PubMed] [Google Scholar]
- 125.Antzelevitch C, Brugada R. Fever and the Brugada syndrome. PACE. 2002;25:1537–1539. doi: 10.1046/j.1460-9592.2002.01537.x. [DOI] [PubMed] [Google Scholar]
- 126.Mok NS, Priori SG, Napolitano C, Chan NY, Chahine M, Baroudi G. A newly characterized SCN5A mutation underlying Brugada syndrome unmasked by hyperthermia. J Cardiovasc Electrophysiol. 2003;14:407–411. doi: 10.1046/j.1540-8167.2003.02379.x. [DOI] [PubMed] [Google Scholar]
- 127.Ortega-Carnicer J, Benezet J, Ceres F. Fever-induced ST-segment elevation and T-wave alternans in a patient with Brugada syndrome. Resuscitation. 2003;57:315–317. doi: 10.1016/s0300-9572(03)00057-1. [DOI] [PubMed] [Google Scholar]
- 128.Dinckal MH, Davutoglu V, Akdemir I, Soydinc S, Kirilmaz A, Aksoy M. Incessant monomorphic ventricular tachycardia during febrile illness in a patient with Brugada syndrome: Fatal electrical storm. Europace. 2003;5:257–261. doi: 10.1016/s1099-5129(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 129.Patruno N, Pontillo D, Achilli A, Ruggeri G, Critelli G. Electrocardiographic pattern of Brugada syndrome disclosed by a febrile illness: Clinical and therapeutic implications. Europace. 2003;5:251–255. doi: 10.1016/s1099-5129(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 130.Peng J, Cui YK, Yuan FH, Yi SD, Chen ZM, Meng SR. Fever and Brugada syndrome: report of 21 cases. Di Yi Jun Yi Da Xue Xue Bao. 2005;25:432–434. [PubMed] [Google Scholar]
- 131.Dulu A, Pastores SM, McAleer E, Voigt L, Halpern NA. Brugada electrocardiographic pattern in a postoperative patient. Crit Care Med. 2005;33:1634–1637. doi: 10.1097/01.ccm.0000169881.09304.a4. [DOI] [PubMed] [Google Scholar]
- 132.Aramaki K, Okumura H, Shimizu M. Chest pain and ST elevation associated with fever in patients with asymptomatic Brugada syndrome fever and chest pain in Brugada syndrome. Int J Cardiol. 2005;103:338–339. doi: 10.1016/j.ijcard.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 133.Schulze-Bahr E, Eckardt L, Breithardt G, et al. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: Different incidences in familial and sporadic disease. Hum Mutat. 2003;21:651–652. doi: 10.1002/humu.9144. [DOI] [PubMed] [Google Scholar]
- 134.Hong K, Guerchicoff A, Pollevick GD, et al. Cryptic 5′ splice site activation in SCN5A associated with Brugada syndrome. J Mol Cell Cardiol. 2005;38:555–560. doi: 10.1016/j.yjmcc.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 135.Bezzina CR, Shimizu W, Yang P, et al. Common sodium channel promoter haplotype in Asian subjects underlies variability in cardiac conduction. Circulation. 2006;113:338–344. doi: 10.1161/CIRCULATIONAHA.105.580811. [DOI] [PubMed] [Google Scholar]
- 136.Weiss R, Barmada MM, Nguyen T, et al. Clinical and molecular heterogeneity in the Brugada syndrome. A novel gene locus on chromosome 3. Circulation. 2002;105:707–713. doi: 10.1161/hc0602.103618. [DOI] [PubMed] [Google Scholar]
- 137.London B, Sanyal S, Michalec M, et al. AB16-1: A mutation in the glycerol-3-phosphate dehydrogenase 1-like gene (GPD1L) causes Brugada syndrome. Heart Rhythm. 2006;3:S32. (abstract) [Google Scholar]
- 138.Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-Segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Litovsky SH, Antzelevitch C. Rate dependence of action potential duration and refractoriness in canine ventricular endocardium differs from that of epicardium: Role of the transient outward current. J Am Coll Cardiol. 1989;14:1053–1066. doi: 10.1016/0735-1097(89)90490-7. [DOI] [PubMed] [Google Scholar]
- 140.Kilborn MJ, Fedida D. A study of the developmental changes in outward currents of rat ventricular myocytes. J Physiol (Lond) 1990;430:37–60. doi: 10.1113/jphysiol.1990.sp018280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Furukawa Y, Akahane K, Ogiwara Y, Chiba S. K+-channel blocking and anti-muscarinic effects of a novel piperazine derivative, INO 2628, on the isolated dog atrium. Eur J Pharm. 1991;193:217–222. doi: 10.1016/0014-2999(91)90039-s. [DOI] [PubMed] [Google Scholar]
- 142.Lukas A, Antzelevitch C. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc Res. 1996;32:593–603. [PubMed] [Google Scholar]
- 143.Di Diego JM, Antzelevitch C. Pinacidil-induced electrical heterogeneity and extrasystolic activity in canine ventricular tissues. Does activation of ATP-regulated potassium current promote phase 2 reentry? Circulation. 1993;88:1177–1189. doi: 10.1161/01.cir.88.3.1177. [DOI] [PubMed] [Google Scholar]
- 144.Krishnan SC, Antzelevitch C. Flecainide-induced arrhythmia in canine ventricular epicardium. Phase 2 reentry? Circulation. 1993;87:562–572. doi: 10.1161/01.cir.87.2.562. [DOI] [PubMed] [Google Scholar]
- 145.Antzelevitch C, Sicouri S, Lukas A, et al. Clinical implications of electrical heterogeneity in the heart: The electrophysiology and pharmacology of epicardial, M, and endocardial cells. In: Podrid PJ, Kowey PR, editors. Cardiac arrhythmia: Mechanism, diagnosis and management. William & Wilkins; Baltimore, MD: 1995. pp. 88–107. [Google Scholar]
- 146.Brugada R, Brugada J, Antzelevitch C, et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101:510–515. doi: 10.1161/01.cir.101.5.510. [DOI] [PubMed] [Google Scholar]
- 147.Shimizu W, Antzelevitch C, Suyama K, et al. Effect of sodium channel blockers on ST segment, QRS duration, and corrected QT interval in patients with Brugada syndrome. J Cardiovasc Electrophysiol. 2000;11:1320–1329. doi: 10.1046/j.1540-8167.2000.01320.x. [DOI] [PubMed] [Google Scholar]
- 148.Priori SG, Napolitano C, Gasparini M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation. 2000;102:2509–2515. doi: 10.1161/01.cir.102.20.2509. [DOI] [PubMed] [Google Scholar]
- 149.Goldgran-Toledano D, Sideris G, Kevorkian JP. Overdose of cyclic antidepressants and the Brugada syndrome. N Engl J Med. 2002;346:1591–1592. doi: 10.1056/NEJM200205163462020. [DOI] [PubMed] [Google Scholar]
- 150.Rouleau F, Asfar P, Boulet S, et al. Transient ST segment elevation in right precordial leads induced by psychotropic drugs: Relationship to the Brugada syndrome. J Cardiovasc Electrophysiol. 2001;12:61–65. doi: 10.1046/j.1540-8167.2001.00061.x. [DOI] [PubMed] [Google Scholar]
- 151.Babaliaros VC, Hurst JW. Tricyclic antidepressants and the Brugada syndrome: an example of Brugada waves appearing after the administration of desipramine. Clin Cardiol. 2002;25:395–398. doi: 10.1002/clc.4950250809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Akhtar M, Goldschlager NF. Brugada electrocardiographic pattern due to tricyclic antidepressant overdose. J Electrocardiol. 2006;39:336–339. doi: 10.1016/j.jelectrocard.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 153.Chow BJ, Gollob M, Birnie D. Brugada syndrome precipitated by a tricyclic antidepressant. Heart. 2005;91:651. doi: 10.1136/hrt.2004.049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bolognesi R, Tsialtas D, Vasini P, Conti M, Manca C. Abnormal ventricular repolarization mimicking myocardial infarction after heterocyclic antidepressant overdose. Am J Cardiol. 1997;79:242–245. doi: 10.1016/s0002-9149(96)00727-8. [DOI] [PubMed] [Google Scholar]
- 155.Tada H, Sticherling C, Oral H, Morady F. Brugada syndrome mimicked by tricyclic antidepressant overdose. J Cardiovasc Electrophysiol. 2001;12:275. doi: 10.1046/j.1540-8167.2001.00275.x. [DOI] [PubMed] [Google Scholar]
- 156.Bigwood B, Galler D, Amir N, Smith W. Brugada syndrome following tricyclic antidepressant overdose. Anaesth Intensive Care. 2005;33:266–270. doi: 10.1177/0310057X0503300219. [DOI] [PubMed] [Google Scholar]
- 157.Darbar D, Yang T, Churchwell K, Wilde AA, Roden DM. Unmasking of Brugada syndrome by lithium. Circulation. 2005;112:1527–1531. doi: 10.1161/CIRCULATIONAHA.105.548487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kalla H, Yan GX, Marinchak R. Ventricular fibrillation in a patient with prominent J (Osborn) waves and ST segment elevation in the inferior electrocardiographic leads: A Brugada syndrome variant? J Cardiovasc Electrophysiol. 2000;11:95–98. doi: 10.1111/j.1540-8167.2000.tb00743.x. [DOI] [PubMed] [Google Scholar]
- 159.Yan GX, Lankipalli RS, Burke JF, Musco S, Kowey PR. Ventricular repolarization components on the electrocardiogram: Cellular basis and clinical significance. J Am Coll Cardiol. 2003;42:401–409. doi: 10.1016/s0735-1097(03)00713-7. [DOI] [PubMed] [Google Scholar]
- 160.Aizawa Y, Tamura M, Chinushi M, et al. Idiopathic ventricular fibrillation and bradycardia-dependent intraventricular block. Am Heart J. 1993;126:1473–1474. doi: 10.1016/0002-8703(93)90550-s. [DOI] [PubMed] [Google Scholar]
- 161.Aizawa Y, Tamura M, Chinushi M, et al. An attempt at electrical catheter ablation of the arrhythmogenic area in idiopathic ventricular fibrillation. Am Heart J. 1992;123:257–260. doi: 10.1016/0002-8703(92)90786-u. [DOI] [PubMed] [Google Scholar]
- 162.Bjerregaard P, Gussak I, Kotar Sl, Gessler JE. Recurrent synocope in a patient with prominent J-wave. Am Heart J. 1994;127:1426–1430. doi: 10.1016/0002-8703(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 163.Lukas A, Antzelevitch C. Differences in the electrophysiological response of canine ventricular epicardium and endocardium to ischemia: Role of the transient outward current. Circulation. 1993;88:2903–2915. doi: 10.1161/01.cir.88.6.2903. [DOI] [PubMed] [Google Scholar]
- 164.Thomsen PE, Joergensen RM, Kanters JK, et al. Phase 2 reentry in man. Heart Rhythm. 2005;2:797–803. doi: 10.1016/j.hrthm.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 165.Antzelevitch C. In vivo human demonstration of phase 2 reentry. Heart Rhythm. 2005;2:804–806. doi: 10.1016/j.hrthm.2005.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Litovsky SH, Antzelevitch C. Differences in the electrophysiological response of canine ventricular subendocardium and sub-epicardium to acetylcholine and isoproterenol. A direct effect of acetylcholine in ventricular myocardium. Circ Res. 1990;67:615–627. doi: 10.1161/01.res.67.3.615. [DOI] [PubMed] [Google Scholar]
- 167.Krishnan SC, Antzelevitch C. Sodium channel block produces opposite electrophysiological effects in canine ventricular epicardium and endocardium. Circ Res. 1991;69:277–291. doi: 10.1161/01.res.69.2.277. [DOI] [PubMed] [Google Scholar]
- 168.Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol. 1996;27:1061–1070. doi: 10.1016/0735-1097(95)00613-3. [DOI] [PubMed] [Google Scholar]
- 169.Tsuchiya T, Ashikaga K, Honda T, Arita M. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 2002;13:698–701. doi: 10.1046/j.1540-8167.2002.00698.x. [DOI] [PubMed] [Google Scholar]
- 170.Fish JM, Antzelevitch C. Role of sodium and calcium channel block in unmasking the Brugada syndrome. Heart Rhythm. 2004;1:210–217. doi: 10.1016/j.hrthm.2004.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tukkie R, Sogaard P, Vleugels J, De Groot IK, Wilde AA, Tan HL. Delay in right ventricular activation contributes to Brugada syndrome. Circulation. 2004;105:1272–1277. doi: 10.1161/01.CIR.0000118467.53182.D1. [DOI] [PubMed] [Google Scholar]
- 172.Antzelevitch C, Fish J, Di Diego JM. Cellular mechanisms underlying the Brugada syndrome. In: Antzelevitch C, Brugada P, Brugada J, Brugada R, editors. The Brugada syndrome: From bench to bedside. Blackwell Futura; Oxford: 2004. pp. 52–77. [Google Scholar]
- 173.Antzelevitch C. The Brugada syndrome: Ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol. 2001;12:268–272. doi: 10.1046/j.1540-8167.2001.00268.x. [DOI] [PubMed] [Google Scholar]
- 174.Antzelevitch C. The Brugada syndrome: Diagnostic criteria and cellular mechanisms. Eur Heart J. 2001;22:356–363. doi: 10.1053/euhj.2000.2461. [DOI] [PubMed] [Google Scholar]
- 175.Gussak I, Antzelevitch C, Bjerregaard P, Towbin JA, Chaitman BR. The Brugada syndrome: Clinical, electrophysiologic and genetic aspects. J Am Coll Cardiol. 1999;33:5–15. doi: 10.1016/s0735-1097(98)00528-2. [DOI] [PubMed] [Google Scholar]
- 176.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST segment elevation. Circulation. 1999;100:1660–1666. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 177.Shimizu W, Aiba T, Kamakura S. Mechanisms of disease: Current understanding and future challenges in Brugada syndrome. Nat Clin Pract Cardiovasc Med. 2005;2:408–414. doi: 10.1038/ncpcardio0268. [DOI] [PubMed] [Google Scholar]
- 178.Antzelevitch C, Brugada P, Brugada J, et al. Brugada syndrome. A decade of progress Circ Res. 2002;91:1114–1119. doi: 10.1161/01.res.0000046046.53721.90. [DOI] [PubMed] [Google Scholar]
- 179.Kurita T, Shimizu W, Inagaki M, et al. The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. J Am Coll Cardiol. 2002;40:330–334. doi: 10.1016/s0735-1097(02)01964-2. [DOI] [PubMed] [Google Scholar]
- 180.Pertsov AM, Davidenko JM, Salomonsz R, Baxter WT, Jalife J. Spiral waves of excitation underlie reentrant activity in isolated cardiac muscle. Circ Res. 1993;72:631–650. doi: 10.1161/01.res.72.3.631. [DOI] [PubMed] [Google Scholar]
- 181.Asano Y, Davidenko JM, Baxter WT, Gray RA, Jalife J. Optical mapping of drug-induced polymorphic arrhythmias and torsade de pointes in the isolated rabbit heart. J Am Coll Cardiol. 1997;29:831–842. doi: 10.1016/s0735-1097(96)00588-8. [DOI] [PubMed] [Google Scholar]
- 182.Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- 183.Donger C, Denjoy I, Berthet M, et al. KVLQT1 C-terminal missense mutation causes a forme fruste long-QT syndrome. Circulation. 1997;96:2778–2781. doi: 10.1161/01.cir.96.9.2778. [DOI] [PubMed] [Google Scholar]
- 184.Napolitano C, Schwartz PJ, Brown AM, et al. Evidence for a cardiac ion channel mutation underlying drug-induced QT prolongation and life-threatening arrhythmias. J Cardiovasc Electrophysiol. 2000;11:691–696. doi: 10.1111/j.1540-8167.2000.tb00033.x. [DOI] [PubMed] [Google Scholar]
- 185.Yang P, Kanki H, Drolet B, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation. 2002;105:1943–1948. doi: 10.1161/01.cir.0000014448.19052.4c. [DOI] [PubMed] [Google Scholar]
- 186.Roden DM. Long QT syndrome: Reduced repolarization reserve and the genetic link. J Intern Med. 2006;259:59–69. doi: 10.1111/j.1365-2796.2005.01589.x. [DOI] [PubMed] [Google Scholar]
- 187.Abbott GW, Sesti F, Splawski I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- 188.Splawski I, Timothy KW, Tateyama M, et al. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297:1333–1336. doi: 10.1126/science.1073569. [DOI] [PubMed] [Google Scholar]
- 189.Pollevick GD, Oliva A, Viskin S, Carrier T, Guerchicoff A, Antzelevitch C. Genetic predisposition to post-myocardial infarction long QT intervals and torsade de pointes. Heart Rhythm. 2007;4:S121. (abstract) [Google Scholar]
- 190.Crotti L, Lundquist AL, Insolia R, et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation. 2005;112:1251–1258. doi: 10.1161/CIRCULATIONAHA.105.549071. [DOI] [PubMed] [Google Scholar]
- 191.Anson BD, Ackerman MJ, Tester DJ, et al. Molecular and functional characterization of common polymorphisms in HERG (KCNH2) potassium channels. Am J Physiol Heart Circ Physiol. 2004;286:H2434–H2441. doi: 10.1152/ajpheart.00891.2003. [DOI] [PubMed] [Google Scholar]
- 192.Paavonen KJ, Chapman H, Laitinen PJ, et al. Functional characterization of the common amino acid 897 polymorphism of the cardiac potassium channel KCNH2 (HERG) Cardiovasc Res. 2003;59:603–611. doi: 10.1016/s0008-6363(03)00458-9. [DOI] [PubMed] [Google Scholar]
- 193.Bezzina CR, Verkerk AO, Busjahn A, et al. A common polymorphism in KCNH2 (HERG) hastens cardiac repolarization. Cardiovasc Res. 2003;59:27–36. doi: 10.1016/s0008-6363(03)00342-0. [DOI] [PubMed] [Google Scholar]
- 194.Ford GA, Wood SM, Daly AK. CYP2D6 and CYP2C19 genotypes of patients with terodiline cardiotoxicity identified through the yellow card system. Br J Clin Pharmacol. 2000;50:77–80. doi: 10.1046/j.1365-2125.2000.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Roden DM. Pharmacogenetics and drug-induced arrhythmias. Cardiovasc Res. 2001;50:224–231. doi: 10.1016/s0008-6363(00)00302-3. [DOI] [PubMed] [Google Scholar]
- 196.Krishnan SC, Josephson ME. ST segment elevation induced by class IC antiarrhythmic agents: underlying electrophysiologic mechanisms and insights into drug-induced proarrhythmia. J Cardiovasc Electrophysiol. 1998;9:1167–1172. doi: 10.1111/j.1540-8167.1998.tb00088.x. [DOI] [PubMed] [Google Scholar]
- 197.Fujiki A, Usui M, Nagasawa H, Mizumaki K, Hayashi H, Inoue H. ST segment elevation in the right precordial leads induced with class IC antiarrhythmic drugs: Insight into the mechanism of Brugada syndrome. J Cardiovasc Electrophysiol. 1999;10:214–218. doi: 10.1111/j.1540-8167.1999.tb00662.x. [DOI] [PubMed] [Google Scholar]
- 198.Gasparini M, Priori SG, Mantica M, et al. Flecainide test in Brugada syndrome: A reproducible but risky tool. PACE. 2003;26:338–341. doi: 10.1046/j.1460-9592.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- 199.Takenaka S, Emori T, Koyama S, Morita H, Fukushima K, Ohe T. Asymptomatic form of Brugada syndrome. PACE. 1999;22:1261–1263. doi: 10.1111/j.1540-8159.1999.tb00612.x. [DOI] [PubMed] [Google Scholar]
- 200.Shimizu W, Aiba T, Kurita T, Kamakura S. Paradoxic abbreviation of repolarization in epicardium of the right ventricular outflow tract during augmentation of Brugada-type ST segment elevation. J Cardiovasc Electrophysiol. 2001;12:1418–1421. doi: 10.1046/j.1540-8167.2001.01418.x. [DOI] [PubMed] [Google Scholar]
- 201.Matana A, Goldner V, Stanic K, Mavric Z, Zaputovic L, Matana Z. Unmasking effect of propafenone on the concealed form of the Brugada phenomenon. PACE. 2000;23:416–418. doi: 10.1111/j.1540-8159.2000.tb06774.x. [DOI] [PubMed] [Google Scholar]
- 202.Rolf S, Bruns HJ, Wichter T, et al. The ajmaline challenge in Brugada syndrome: Diagnostic impact, safety, and recommended protocol. Eur Heart J. 2003;24:1104–1112. doi: 10.1016/s0195-668x(03)00195-7. [DOI] [PubMed] [Google Scholar]
- 203.Wilde AA, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Circulation. 2002;106:2514–2519. doi: 10.1161/01.cir.0000034169.45752.4a. [DOI] [PubMed] [Google Scholar]
- 204.Tada H, Nogami A, Shimizu W, et al. ST segment and T wave alternans in a patient with Brugada syndrome. PACE. 2000;23:413–415. doi: 10.1111/j.1540-8159.2000.tb06773.x. [DOI] [PubMed] [Google Scholar]
- 205.Sarkozy A, Caenepeel A, Geelen P, Peytchev P, de Zutter M, Brugada P. Cibenzoline induced Brugada ECG pattern. Europace. 2005;7:537–539. doi: 10.1016/j.eupc.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 206.Aouate P, Clerc J, Viard P, Seoud J. Propranolol intoxication revealing a Brugada syndrome. J Cardiovasc Electrophysiol. 2005;16:348–351. doi: 10.1046/j.1540-8167.2005.40564.x. [DOI] [PubMed] [Google Scholar]
- 207.Matsuo K, Shimizu W, Kurita T, Inagaki M, Aihara N, Kamakura S. Dynamic changes of 12-lead electrocardiograms in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 1998;9:508–512. doi: 10.1111/j.1540-8167.1998.tb01843.x. [DOI] [PubMed] [Google Scholar]
- 208.Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89:1363–1372. doi: 10.1136/heart.89.11.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Pastor A, Nunez A, Cantale C, Cosio FG. Asymptomatic Brugada syndrome case unmasked during dimenhydrinate infusion. J Cardiovasc Electrophysiol. 2001;12:1192–1194. doi: 10.1046/j.1540-8167.2001.01192.x. [DOI] [PubMed] [Google Scholar]
- 210.Lopez-Barbeito B, Lluis M, Delgado V, et al. Diphenhydramine overdose and Brugada sign. PACE. 2005;28:730–732. doi: 10.1111/j.1540-8159.2005.00154.x. [DOI] [PubMed] [Google Scholar]
- 211.Ortega-Carnicer J, Bertos-Polo J, Gutierrez-Tirado C. Aborted sudden death, transient Brugada pattern, and wide QRS dysrrhythmias after massive cocaine ingestion. J Electrocardiol. 2001;34:345–349. doi: 10.1054/jelc.2001.26318. [DOI] [PubMed] [Google Scholar]
- 212.Littmann L, Monroe MH, Svenson RH. Brugada-type electrocardiographic pattern induced by cocaine. Mayo Clin Proc. 2000;75:845–849. doi: 10.4065/75.8.845. [DOI] [PubMed] [Google Scholar]
- 213.Shimizu W. Acquired forms of the Brugada syndrome. J Electrocardiol. 2005;38 (suppl):22–25. doi: 10.1016/j.jelectrocard.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 214.Antzelevitch C, Brugada P, Brugada J, Brugada R. The Brugada syndrome. From cell to bedside. Curr Probl Cardiol. 2005;30:9–54. doi: 10.1016/j.cpcardiol.2004.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]