

Abstract
Nitric oxide (NO) is responsible for nitrergic neurotransmission in the gut and its release is dependent on its de novo synthesis by nNOS (neuronal nitric oxide synthase). The magnitude of NO synthesis and release during neurotransmission may be related to the fraction of catalytically active nNOS out of a larger pool of inactive nNOS in the nerve terminals. The purpose of the current study was to identify catalytically active and inactive pools of nNOS in the varicosities from mice gut. Enteric varicosities were confirmed as nitrergic by colocalization of nNOS with the nerve varicosity marker synaptophysin. Low temperature SDS PAGE of these varicosity extracts showed 320kD, 250kD and 155kD bands when blotted with anti-nNOS1422–1433 and 320 and 155kD bands when blotted with anti-nNOS1–20 antibodies respectively. The 320kD and 155kD bands represent dimers and monomers of nNOSα; the 250kD and 135kD bands represent dimers and monomers of nNOSβ. Immunoprecipitation with calmodulin (CaM) showed that a portion of nNOSα dimer was bound with CaM. On the other hand, a portion of nNOSα dimer, nNOSβ dimer and all monomers lacked CaM binding. The CaM-lacking nNOS fractions reacted with anti-serine847-phospho-nNOS. In vitro assays of NO production revealed that only the CaM-bound dimeric nNOSα was catalytically active; all other forms were inactive. We suggest that the amount of catalytically active nNOSα dimers may be regulated by serine847 phosphorylation and equilibrium between dimers and monomers of nNOSα.
Keywords: nitric oxide, isoforms of nNOS, serine847-phosphorylated nNOS, enteric nerve varicosities, calmodulin bound nNOS
INTRODUCTION
Nitric oxide (NO) generated by nNOS (neuronal nitric oxide synthase) is responsible for nitrergic inhibitory neurotransmission in the gut (4, 15, 25). However, regulation of nitrergic neurotransmission is not well understood. The classical neurotransmitters, acetylcholine and catecholamines, are preformed and stored in secretory granules in the nerve terminals. The secretory granules exist as a large ‘reserve’ pool and a smaller ‘readily releasable’ pool; the later is docked on the varicosity membrane. During nerve stimulation, propagation of an action potential in the nerve terminal causes influx of calcium into the terminal resulting in a quantal release of the transmitter from the releasable pool (21). Regulation of the ‘readily releasable’ pool of the secretory granules serves as an important determinant of the amount of the neurotransmitters released with each episode of nerve stimulation. On the other hand, NO is a highly diffusible gas and it is not preformed nor stored in secretory granules. nNOS localized to membranes of neural dendrites and motor nerve terminals are the tentative sites of NO generation during retrograde and anterograde nitrergic neurotransmission, respectively (5, 11). It is possible that only a specific fraction of nNOS in the nerve terminal with catalytic activity participates in NO production by the action of calcium influx upon nerve stimulation. We hypothesized that analogous to the control of the amount of release of the classical neurotransmitters, NO synthesis and release may be regulated by the pools of catalytically active and inactive forms of nNOS in the varicosities. However, the nature of these catalytically active and inactive nNOS pools in the nerve terminals is not well known.
Studies have shown that the catalytically active nNOS is a tetramer of two molecules of nNOS associated with two molecules of calmodulin (CaM) (2, 18, 24). In addition, targeting of nNOS to the nerve terminal is also critical in neurotransmission. Some nNOS isoforms possess PDZ binding domains. PDZ is an acronym for postsynaptic density protein (PSD95), Drosophila disc large tumor suppressor (DlgA) and zonula occludens-1 protein (zo-1). PSD95 is anchored to specific regions of the cell membrane. Therefore, protein-protein interactions involving PDZ binding domains allow anchoring of nNOS to the cell membrane via PSD. Studies in hippocampal neurons have shown that the isoforms of nNOS possessing PDZ binding domains are targeted to the complementary PDZ binding domain-containing PSD95 in the dendrites of the hippocampal neurons (19). Similarly, it is possible that PDZ domain-containing nNOS isoforms are targeted to the motor nerve terminals in the gut. Thus, CaM-binding and dimerization of nNOS isoform containing PDZ binding domain may constitute the active nNOS that produces NO from L-arginine during calcium influx caused by action potentials invading the nitrergic nerve terminals (6, 12, 18).
The purpose of the current study was to identify catalytically active and inactive pools of nNOS in varicosities obtained from mice gut. Immunoblots, immunoprecipitation and functional assays were performed to identify: 1) splice variants of nNOS; 2) dimers and monomers in the native extract; 3) CaM-bound and CaM-lacking nNOS fractions; 4) nature of the CaM-lacking nNOS isoforms; and 5) nNOS fractions that were catalytically active and produced NO in an in vitro assay system.
MATERIALS AND METHODS
The experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee of VA Boston Healthcare System.
Antibodies and Chemicals
Details of all the antibodies used in the present study are summarized in table 1. The anti-ser847-phospho-nNOS antibody was raised against 848KRFNSVS854 of human nNOS that recognized serine 852 phosphorylation in the human sequence and corresponded with the sequence 844RFNSVS849 in the mouse nNOS (Santa Cruz). Thus, in mice, this antibody recognizes the serine847-phosphorylated nNOS. All secondary antibodies were obtained from Jackson Immunochemicals and Santa Cruz. Reagents for immunoblot and chemicals used were from BioRad, Wako Chemicals and Sigma. Immunoprecipitation reagents (Protein A/G) were from Roche Molecular Biochemicals and Santa Cruz.
Table 1.
Details of antibodies used in the current study.
| Antibody | Target | Dilution | Usage | Host | Source | Rationale |
|---|---|---|---|---|---|---|
| anti-C-Terminal nNOS1422-1433 | nNOS
(AA 1422-1433) |
1:100 | WB, IP | Rabbit | Alexis | Total nNOS |
| anti-N-Terminal nNOS1-20 | nNOS
(AA 1-20) |
1:100 | WB | Rabbit | Santa Cruz | nNOS containing PDZ domain in N-terminal end |
| anti-serine847-PnNOS | Serine 847-phosphorylated nNOS | 1:50 | WB, IP | Goat | Santa Cruz | nNOS phosphorylated at serine 847 |
| anti-CaM | Calmodulin | 1:100 | IP, WB | Mouse | Upstate | calmodulin-bound nNOS |
| anti-synaptophysin | Synaptophysin | 1:100 | WB, IHC | Mouse | Febgennix | synaptosome |
Note: The appropriate secondaries were use at a ten fold dilution compared to the primary, except for phospho-serine 847-nNOS where a 5-fold dilution (1 in 250) was used compared to the primary.
WB, western blot; IP, immunoprecipitation; IHC, immunohistochemistry.
Tissue Dissection
Adult male C57BL/6j mice (25–30 gm each) (Jackson Laboratories) were euthanized by carbon dioxide (CO2) inhalation in an airtight chamber. Gastrointestinal tissues from three to six mice were pooled for each experiment. For protein interaction studies, six to ten mice were used for preparing each varicosity extract to ensure abundance of proteins of interest. The entire gastrointestinal tract from the stomach to colon was dissected quickly and opened along the antimesenteric border. The gut lumen was cleaned in ice–cold homogenization buffer (0.3M sucrose with 0.1M sodium phosphate and 1 mM EGTA, pH 7.4). Small pieces of intestine were placed in a plastic tube in 10 volumes of the homogenization buffer described earlier and were pulverized into a homogenous extract with a Brinkmann homogenizer and the tube cooled on ice before further processing. The homogenization buffers contained adequate quantities of protease and phosphatase inhibitors (The protease inhibitor was used at a concentration of 1 ml /20 gm wet tissue. The complete gastrointestinal tract from each animal had an average mass of 2.8 gm (n=6 mice). The phosphatase inhibitor was used at a concentration of 1 ml /100 ml of homogenization buffer). The protease inhibitor (P8340, Sigma) contained AEBSF, aprotinin, bestatin, E-64, leupeptin hemisulfate and pepstatin. The phosphatase inhibitor contained cantharidin and microcystin LR (P2850, Sigma) that specifically inhibited serine phosphatases like PP2A and PP1.
Subcellular fractionation
The method used for varicosity isolation is summarized in Figure 1 and was similar to protocols described earlier for varicosities preparation in intestine (10, 27). Samples were centrifuged at 1000 g for 10 min at 4° C to remove undissociated tissue (pellet P1) that was washed once in buffer and the pellet discarded and the combined supernatants was further centrifuged at 4000 g. The pellet P2 obtained after spinning at 4000 g represented the nuclear fraction and the supernatant was the cytoplasmic fraction. This supernatant was subjected to ultra-centrifugation at 25000 rpm at 4°C for 30 minutes. The Pellet P3 obtained was the varicosity fraction, while the supernatant represented the microsomal fraction. Pellet P3 was re-suspended in 400μl of Krebs buffer (111mM NaCl, 26.2mM NaHCO3, 1.2mM NaH2PO4, 4.7mM KCl, 1.8 mM CaCl2,1.2 mM MgCl2, 11mM glucose and gassed with 95% O2 and 5% CO2 to maintain a pH of 7.4) and subjected to further purification. The homogenization buffer contained EGTA to facilitate low levels of calcium during the initial phases of low-velocity centrifugation. The low calcium was deemed necessary for appropriately sealing the varicosity membranes. Furthermore, before the P3 pellet was subjected to ultracentrifugation, it was resuspended in Krebs buffer that was replete with divalent calcium. In all experiments pertaining to studying nNOS phosphorylation, varicosity preparations were made by omitting EGTA in the homogenization buffer. The P3 extract was layered on a 0.8/1.2M sucrose gradient and subjected to sucrose gradient ultracentrifugation at 58,000 rpm for 1hour at 4°C. Intact varicosities that formed a cloudy or ring-like structure at the interface of the two differing sucrose concentrations were carefully collected with a 200 pl pipette tip, diluted in Krebs buffer and centrifuged at 12,000 rpm for 5 minutes at 4°C to pellet down varicosities. Varicosities were stored at −80°C until further experiments.
Figure 1. Protocol for isolation of nerve varicosities from mice gut.

Different velocities of cold ultracentrifugation and sucrose gradient purification were used to obtain varicosities.
Transmission Electron Microscopy
Varicosity samples were fixed immediately after isolation in a fixative composed of 2% paraformaldehyde and 2.5 % glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) at room temperature and after an hour, centrifuged at 12,000 rpm for 5 minutes. The resultant pellet was processed at the Electron Microscopy Core Facility at Harvard Medical School in the following sequence: washed thrice in buffer containing 1% osmium tetroxide/1.5% aq. potassium ferrocyanide for 1 hour at room temperature, in water 3–4 times, with 1% uranyl acetate/maleate buffer for 30 minutes, and again with water thrice. It was then dehydrated using ethyl alcohol (70%) for 15 min, 90% for another 15 min and finally with 100% alcohol for 15 min and repeated one more time. Next, it was treated with propylene oxide for an hour, embedded in a ratio of 1:1 TAAB epon resin at room temperature for 2–3 hours and then transferred to a mold filled with freshly mixed TAAB epon. It was then polymerized at 60°C for 48 hours in an oven. Ultrathin sections (60–80 nm) were cut using a Reichert Ultracuts microtome and observed under a JEOL 1200 EX transmission electron microscope.
Fluorescence Microscopy
After isolation, varicosity samples were fixed immediately in a fixative (Histochoice, Amresco Inc) and incubated overnight at 4° C. This solution was then spun down and the resultant pellet was used for immunostaining. The varicosity pellet was washed with Tween/PBS thrice for 15 min, and then further washed with 70% alcohol for at least 10 min, followed with a quick wash with PBS/Tween. The pellet was resuspended in a primary antibody solution (nNOS1422–1433 and synaptophysin, respectively) and incubated overnight at 4°C in a shaking incubator. Next, the samples were spun and varicosity pellet resuspended in washing solution PBS/Tween for 5 min, after which it was spun down and an appropriate secondary antibody solution (donkey anti-rabbit Texas Red and goat anti-rat Cy5, respectively) added. The pellet was incubated with secondary antibody under shaking conditions for 2 hours, spun down and washed with PBS/Tween for 5 min. The samples were mounted on clean glass slips and observed with a fluorescent microscope (Olympus).
Preparation of the varicosity extracts for Western Blots
Isolated varicosities were centrifuged at 12,000 rpm for 5 min at 4° C after addition of Cellytic buffer (Sigma) to obtain proteins in solution. The extracts were processed at low temperature (4° C) or heat treated at 37 ° C for 10 minutes. For the low temperature processing, 60–80 pg of protein in Laemmli buffer (0.125M Tris/HCl (pH 6.8), 4% (w/v) SDS, 10% (v/v) 2-mercaptoethanol, 20% (w/v) glycerol and 0.02% bromophenol blue) at 4° C was used for SDS-PAGE. The low temperature process was used to identify nNOS dimers and monomers in the native state as low temperature is known to prevent monomerization of nNOS dimers (18). Heat treated samples were processed as follows: protein was treated with Laemmli buffer for 10 minutes at 37 ° C, and immediately subjected to electrophoresis. 35 pl of protein samples were loaded into each lane during electrophoresis.
SDS-PAGE of varicosity extracts
Electrophoresis was carried out using Biorad mini-protean II system or a Wako gel casting system. Preliminary experiments were carried out in 4–20% gradient polyacrylamide gels and confirmatory gels were run by separating proteins in 7.5% gels that provided efficient resolution of the higher molecular weight proteins examined in the current study. Pilot experiments were performed for standardization of the type of gel, the run-time, the concentrations of the primary and secondary antibodies and the incubation times.
Total protein concentrations were measured using Bradford method at 595λ optical density. For almost every experiment, 60μg of protein was loaded for low temperature SDS/PAGE. The samples were subjected to SDS/PAGE for a variable period of time (between 2 and 5 hrs, depending on the migration of the MW marker) at 90 V in cold-room at 4°C. For all electrophoresis, Precision Plus (BioRad) molecular weight marker was used to identify migration patterns. All experiments were performed with appropriate positive controls and loading controls were evaluated for homogeneity of results. Additionally, for each experiment, at least three different sets of varicosity extracts were used (each extract from a set of 3–10 mice) and electrophoresis was always repeated greater than thrice to confirm the findings.
After electrophoresis, the separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes (BioRad) overnight at 30 V at 4°C and the efficiency of immunoblotting was checked using Ponceau staining. Blots (membranes with separated proteins) were washed with Tris-buffered saline (Biorad) with Tween (TBST) for 10 minutes and blocked for 1 h in 5% nonfat milk/TBST (unless otherwise mentioned) at room temperature. The primary antibody was added in the blocking solution at differing dilutions (see table 1) and the blot was probed with this primary antibody on a rocking platform overnight at 4° C. For experiments probing phospho-nNOS, the blocking solution used was 5% bovine serum albumin (BSA) and the same solution was used for diluting the antibodies. Milk was avoided as the blocking solution to reduce background because milk proteins are abundantly phosphorylated. The blot was subsequently washed with washing buffer for an hour and an appropriate secondary antibody conjugated to chemiluminescent horse radish peroxidase (HRP) was added at a five-ten-fold dilution to the primary (see table 1) and incubated for an hour on a rocking platform at room temperature. After washing the blot with TBST thrice for 20 min each/wash, immunolabeled blots were developed using a premixed chemiluminescent developer ECL reagent (UpState). Blots were exposed to a chemiluminescent detection film in the dark for variable time periods and processed in an X-Ray developer machine (Kodak X-OMAT 2000A).
Immunoprecipitation studies
Solubilized varicosity extracts in Cellytic buffer (Sigma) were centrifuged and the supernatant was diluted to a starting protein concentration of 1mg/ml. The buffer used resembled composition of the standard RIPA buffer and contained protease inhibitors and 50mM Tris-HCl pH 7.5 (5ml), 150mM NaCl (3.75ml), 1% Nonidet P40 and 0.5% sodium deoxycholate (2.5ml). 50μl of a homogenous suspension of Protein G-agarose (A/G) was added to 1 ml varicosity extract (1 mg protein/ml) and incubated overnight at 40% speed at 2–8° C on a rotating rocker (Glas-Col, Terre Haute). Varicosity extracts with beads were centrifuged at 12,000g for 20 seconds in a microfuge. After the supernatant was transferred to fresh tubes, 10 pg of the specific antibody was added and gently rocked overnight at 2–8°C. Antibodies used for immunoprecipitation were anti-nNOS1422–1433 that detected all forms of nNOS and anti-CaM antibody that detected association of calmodulin with nNOS. After the complexes were collected by centrifugation at 12,000g for 20seconds in a microfuge, the supernatant was carefully aspirated and used for further experiments. The beads were washed thrice with 1 ml of lysis buffer and spun at 12000 g for 2 min each time and 2X loading buffer (the Laemmli buffer) was added to this protein A/G beads on ice to isolate protein from the beads. Immunoprecipitated protein was subjected to low temperature SDS PAGE and probed with different antibodies. In order to identify nNOS isoforms in CaM-lacking nNOS, the supernatant after CaM immunoprecipitation was removed and concentrated using a centrifugal filter device at 4000 g for 30 min in a swinging-bucket type ultracentrifuge (Sorvall RT) at 4° C. The filter devices had low-binding Ultracel membranes with a nominal molecular weight limit of 50 kD (less than twice the size of the least anticipated molecular weight in the current set of experiments) (Amicon Ultra-4, Millipore). The retentate (containing proteins with MWs>50kD) was subjected to further electrophoresis and hybridized with serine847-phospho-nNOS antibody.
Quantification of blots
After developing, the blots were scanned with unaltered luminosity so that the optical density remained unaffected. The peak intensity of a user-identified band was evaluated using the NIH program ImageJ. The band intensity was expressed as arbitrary units and mean expression was obtained by averaging intensity values of 3–9 different lanes for each experiment, each lane representing gut varicosity extract from 3–10 mice.
Functional studies of in vitro Nitric Oxide Production
NO production by various fractions of nNOS (using varicosity extracts and immunoprecipitated samples with specific anibodies) was measured by using DAF-2 (diaminofluorescein). This dye is highly sensitive to nitric oxide and binds to it to form a fluorescent product. This product can be detected at 495/515 nm in a spectrofluorometer (BioRad). Varicosity pellets were dissolved in a buffer composed of 50 mM TrisHCl, pH 7.4, and protease inhibitors. 100μl of this sample was mixed with 100μl of assay buffer. The assay buffer consists of 55mM N-2 hydroxyethylpiperazine-N-2-ethane-sulfonic acid (Hepes), 20mM Tris-HCl, pH 7.4, 2mM L-Arginine, 0.8 mM dithiothreitol (DTT), 0.8 μM NADPH, 1mM MgCl2, 1mM CaCl2, 0.5μM calmodulin, 0.8 μM riboflavin monophosphate (FMN), 6 μM (6R)-5,6,7,8-tetrahydrobiopterin (BH4), 0.8 μM flavin-adenine dinucleotide (FAD), and protease inhibitors. DAF-2 (10 μM) was also added to this reaction mixture. The assay was performed in different experimental conditions using nNOS inhibitors L-NAME and 7-NI and by omitting L-arginine in the buffer. Additionally, heat treated samples were also assayed for nitric oxide production. Nitric oxide donor, Nor-3 was used for plotting known nitric oxide levels as standard graph and this was used to calculate values of unknown biological samples (13).
Statistical Analysis
Expression values of enzymes or enzymatic activities were represented as means ± SEM. Comparison of means was performed using t statistics using MS Excel.
RESULTS
Identification of nitrergic varicosities
The identity of varicosities in the enriched fraction was confirmed using transmission electron microscopy. Figure 2A shows transmission electron microscopic appearance of varicosities containing secretory granules and diffuse cellular contents. Average size of varicosities ranged between 0.5μ and 2μ. The majority (~90 %) of varicosities showed secretory vesicles with a granular appearance. Figure 2B shows phase contrast image of the varicosity preparation mounted on a glass slide. Varicosities immunostained with anti-nNOS1422–1433 confirmed their nitrergic identity (Figure 2C). nNOS and synaptophysin were colocalized indicating that the isolated structures containing nNOS were neural varicosities (Figure 2D). The nNOS/synaptophysin colocalization was also confirmed by electrophoresis of the varicosity extracts and by immunoblotting with anti-nNOS1–20 and anti-synaptophysin antibodies (Figure 2E).
Figure 2. Identification of varicosities isolated from the gut.

(A) Transmission electron microscopy demonstrated ~2μ varicosity with dense granules and diffuse cellular contents. (B) Phase contrast appearance of smears of varicosities laid out on clean glass-slips. Many enteric varicosities were immunopositive for nNOS (pseudocolored red) while others were nNOS negative (C) and colocalized with synaptophysin (pseudocolored blue at the rim of the varicosities) (D), a synaptic marker, when varicosity smears were examined under fluorescence microscopy. nNOS/synaptophysin colocalization was also demonstrated by immunoblotting (E).
Anti-nNOS1422–1433 antibody reactive nNOS
Immunoblots probed with anti-nNOS1422–1433 antibody that is specific for the C-terminal end of nNOS identified all isoforms of nNOS because the C-terminal regions of all isoforms of nNOS are identical. The C-terminal antibody identified 3 nNOS bands of approximately 320 kD, 250 kD and 155 kD on low temperature SDS PAGE (Figure 3). The relative proportions of nNOS 320, 250, 155 and 135kD bands in cold-processed SDS PAGE were determined densitometrically as 1:0.57±0.44:1.1±0.99:0. The relative proportion of 320kD and 250kD bands of 1:0.57 indicated that 320kD nNOS made up the bulk of nNOS dimer in the varicosities and the 250kD fraction was only about half of the 320kD fraction.
Figure 3. Identification of different nNOS forms in varicosity extracts.

Left Panel; Varicosity extracts under non-denaturing conditions (on ice) immunoblotted with anti-nNOS1422–1433 antibody showed dimers and monomer at 320, 250 and 155kD. Heat treated (37° C) extract probed with the same antibody detected monomers at 155 and 135kD. Right Panel; The nNOSα splice variant was identified in the varicosity extract by anti-nNOS1–20 antibody in the cold condition that detected a 320kD dimer and a 155kD monomer band. Heat treated extract probed with anti-nNOS1–20 antibody detected only the monomer at 155kD. Note that both splice variants of nNOS, nNOSα and nNOSβ, are present in the gut nerve terminals.
The warm (37° C) SDS in the Laemmli buffer showed no 320kD and 250kD bands and were replaced by 155kD and 135kD nNOS bands (Figure 3). These observations are consistent with the fact that the 320 kD and 250 kD bands in low temperature SDS-PAGE were dimers that appear as 155kD and 135 kD monomers respectively on heat treatment.
anti-nNOS1–20 antibody reactive nNOS
In order to identify the nNOS bands containing PDZ domain, the Western blots were probed with anti-nNOS1–20 antibody (specific for N-terminal end of nNOS) that reacts with the PDZ binding site of nNOS. The N-terminal antibody identified 320 and 155kD bands on low temperature SDS PAGE and only 155 kD nNOS band on heat treated SDS-PAGE (Figure 3). The N-terminal nNOS antibody did not recognize the 250kD and 135kD bands that represented nNOS lacking the PDZ binding domain. The relative proportion of nNOS1–20 reactive nNOS was 1:1.8±1 for the 320 kD dimer and 155 kD monomer respectively on cold temperature SDS-PAGE. These observations suggest that over half of nNOSα in the varicosities in the resting state exist as monomers.
Calmodulin association with nNOS isoforms
CaM association of nNOS is critical for its catalytical activity. To identify nNOS fractions that were associated with calmodulin in enteric nerve varicosities, immunoprecipitation was performed using anti-nNOS 1422–1433 antibody and the immunoblots were probed with an anti-CaM antibody. Anti-CaM antibody detected a 320kD band in the immunoprecipitate (Figure 4). Heat treatment showed a band at 17kD representing the calmodulin dissociated from its bound state with nNOS (Figure 4). The 250kD and 155kD in the varicosity extracts were not found to be associated with CaM.
Figure 4. CaM-associated and CaM-lacking fractions of nNOS.
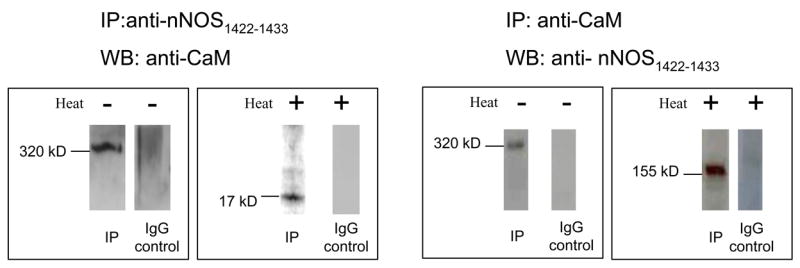
Left panel shows nNOS1422–1433 immunoprecipitate probed with anti-calmodulin antibody. The 320kD band shows that calmodulin is bound to the nNOS immunoprecipate. Heating the immunoprecipitate sample dissociated the calmodulin that was bound to nNOS, and was detected as a 17kD band. Right panel shows calmodulin immunoprecipitate probed with anti-nNOS1422–1433 antibody. In the native state of the nerve terminals in gut, calmodulin binds only to nNOS dimer at 320kD. Note that calmodulin does not associate with the nNOSβ dimer or the nNOSα monomer.
To further confirm the above findings, we performed a reverse experiment using varicosity-immunoprecipitate with calmodulin and probing the IP with anti-nNOS1422–1433 antibody. This process revealed only the 320 kD band on Western blotting. Neither the 250kD nor 155kD bands were identified in the CaM immunoprecipitate at low temperature SDS PAGE (Figure 4). CaM-immunoprecipitate on heat treated SDS-PAGE showed a 155kD band that presumably represented the monomer of the 320 kD nNOS (Figure 4).
Identification of serine 847-phosphorylated nNOS
One of the important mechanisms of regulation of CaM-binding to nNOS is phosphorylation of the nNOS enzyme at serine 847 that inhibits CaM binding. Therefore we examined the presence of ser847-phosphorylated nNOS isoforms in the native varicosities in the gut in the presence of inhibitors of serine phosphatases (PP2A). Immunoblots of varicosity extracts probed with serine 847-phosphorylated nNOS antibody revealed 320kD, 250kD and 155kD bands at low temperature SDS PAGE. Heat treatment of varicosity protein extracts with Laemmli buffer at 37 C demonstrated only the 155kD band (Figure 5).
Figure 5. Phosphorylation patterns of nNOS in gut nerve terminals.

Left panel shows phosphorylation states of nNOS in varicosity extracts. In native condition, portions of nNOS α and β dimers and nNOSα monomers were phosphorylated. Imunoblots of non-denatured varicosities extracts revealed bands at 320, 250 and 155kD when probed with serine-847-phospho-nNOS, and mild heat treatment showed prominent band at 155kD. Right panel shows phosphorylation states of calmodulin-bound and calmodulin lacking nNOS. Calmodulin immunoprecipitates was probed with anti-phospho-serine 847-nNOS antibody. No signal was detected. These experiments showed that calmodulin-bound nNOS was not phosphorylated at serine 847. Floating fraction obtained after calmodulin immunprecipitation was probed with anti-phospho-serine 847-nNOS antibody. The floating fraction obtained after precipitating varicosities extracts with anti-calmodulin antibody represented the pool of nNOS that did not bind to calmodulin. This fraction was concentrated using a membrane dialyzer at high centrifugation speeds and the retentate was probed with phospho-serine-847-nNOS antibody. Under non-denaturing conditions (4° C, SDS-PAGE) 320, 250 and 155kD bands were visualized, representing ser847-phospho-nNOS α dimer, β dimer and a monomer respectively. Mild heat treatment (37° C, 10 min) showed a 155kD band. IP with mouse IgG served as negative controls.
Serine847 phosphorylation state of calmodulin-bound and calmodulin-lacking nNOS
In order to determine serine847 phosphorylation state of nNOS isoforms of CaM-bound and CaM-lacking fractions in the varicosities, varicosity extracts were immunoprecipitated with anti-calmodulin antibody and the supernatant was concentrated using a membrane dialyzer at high speed and this retentate (containing proteins >50kDs) was subsequently hybridized with serine 847-phospho-nNOS antibody. CaM-immunoprecipitated nNOS showed no ser847 phosphorylated nNOS (Figure 5). On the other hand, the supernatant that was obtained after the initial immunoprecipitate with the calmodulin antibody, representing the calmodulin-lacking nNOS fraction, revealed bands at 320, 250 and 155kD when probed with the serine 847-phospho-nNOS antibody (Figure 5). These results suggest that the CaM-lacking nNOS contain nNOS isoforms that were phosphorylated at ser847. The ser847 phospho-nNOS fraction in the supernatant after CaM-IP showed relative proportions of 320kD, 250kD and 155kD of 1:1.08±1.26:1.79±1.2 (n=3, each set of varicosity samples were prepared from 6 mice).
Nitric oxide (NO) production by different nNOS isoforms
As shown in figure 6, immunoprecipitates of varicosity extracts with CaM showed that the fraction of CaM-bound nNOS actively produced nitric oxide in an in vitro assay (49.19 ± 1.47 picomole/30min/mg protein). This nitric oxide production was significantly suppressed after pharmacologic treatment with L-NAME (p<0.05), a drug that inhibits all forms of nNOS enzyme. The nitric oxide production was also significantly suppressed by 7-nitroindazole (p<0.05), a drug that inhibits nNOS enzyme by inhibiting BH4 and consequently disrupting nNOS dimer formation. Nitric oxide was not produced when L-arginine was removed from the assay mixture. These studies show that CaM-associated nNOS was catalytically active. In contrast, precipitates of varicosity extracts with serine-847-phosphorylated-nNOS antibodies (i.e., serine-phosphorylated nNOS) showed very low levels of NO production (9.60 ± 1.45 pMole/30min/mg protein). The nNOS inhibitors L-NAME and 7-NI did not further inhibit NO production in these samples (p>0.05). Additionally, the floating fraction obtained after CaM-IP of the varicosity extract showed scant or no production of nitric oxide in the in vitro assay, showing that the CaM-lacking nNOS in the supernatant could not generate nitric oxide. These data indicate that serine-847-phosphorylated nNOS was catalytically inactive. Heat treated varicosity samples (37° C) did not show any detectable level of NO during the functional assay, suggesting that monomers of nNOS were ineffective in NO synthesis.
Figure 6. Quantification of nitric oxide production by different nNOS fractions in mice nerve terminals in vitro.

Histograms represent activity profiles of in vitro NO synthesis as assessed by a fluorimetric assay using DAF-2 in whole varicosity extracts, calmodulin IP and phospho-serine-847-nNOS IP samples. For each of the categories, the nitric oxide assays were performed in quadruplicates. A nitric oxide donor, nor-3, was used to calculate the standard graph from which nitric oxide production in unknown biological samples were computed (shown on the top). Note the robust production of nitric oxide by the CaM-IPs as compared to ser847-P-nNOS-IPs. Also note that heat (37° C) and omission of L-arginine from the samples resulted in total absence of in vitro NO synthesis.
DISCUSSION
This study shows that in the enteric varicosity extracts: 1) nNOS was present as 320kD, 250kD, 155kD and 135 kD bands; 2) These bands represented dimers and monomers of α and β isoforms of nNOS respectively; 3) 320kD nNOSα dimer existed in both CaM-bound and CaM-free forms; 4) CaM-associated nNOSα dimer was catalytically active, while the CaM-lacking nNOSα dimer was catalytically inactive and was phosphorylated at serine 847; 5) 155kD nNOSα monomer and 250kD nNOSβ dimers lacked CaM and were also catalytically inactive.
Various isoforms of nNOS, namely α, β, γ, μ and nNOS2 isoforms, are produced by the post-transcriptional splicing of nNOS mRNA (22, 23). The dimers of nNOS a, β, γ, μ and nNOS2 are known to have approximate molecular weights of 320, 250, 250, 330 and 288kDs respectively, and their monomers have molecular weights of approximately 155, 135, 125, 165 and 144kDs respectively (2). Because of this proximity, approximate determinations of molecular weights of the bands do not adequately distinguish various isoforms of nNOS. N-terminal nNOS antibody was used to distinguish nNOS isoforms that contain complementary PDZ binding domain from those lacking it. Hence, the 320kD band could be nNOSα, nNOSμ or nNOS-2 that possess PDZ binding domain. On the other hand, the 250kD band could be either nNOSβ or nNOSγ that lack the PDZ binding domain (1).
However, studies of tissue localization, mRNA expression and functional assay helped to identify the nature of these nNOS isoforms. Tissue localization studies have shown that nNOSα is localized to neurons and nNOSμ is localized to striated and cardiac muscles (2, 14). A detailed study of nNOS mRNA splice variants and proteins in gut nerve-muscle preparation have shown that both nNOS mRNA and protein for nNOSα are localized in the gut, whilst there was no expression of nNOSμ (22). These studies supported the view that the 320kD band visualized in the current study was not nNOSμ. On the other hand, nNOS-2 has no enzymatic activity (2). However, the 320 band in our studies was catalytically active in an in vitro assay, suggesting that it was not nNOS-2. Therefore, by exclusion, it appears that the 320kD band represented nNOSα. The 250kD band did not react with the N-terminal nNOS antibody and therefore represented PDZ domain-lacking nNOSβ or nNOSγ isoforms. Saur et. al. have shown that while mRNA for nNOSβ is present in certain regions of the gut, mRNA for nNOSγ is never expressed (22, 23). Therefore, it appears that the 250kD fraction represented nNOSβ.
Since nNOSα contains a PDZ-binding domain, nNOSα is the likely candidate enzyme involved in nitrergic neurotransmission because of its ability to localize at the cell membrane (2). Localization to the varicosity membrane places it in close proximity to calcium channels and hence may allow prompt response to calcium influx on activation of the nerve terminal. The critical role of nNOSα in nitrergic neurotransmission is also supported by functional studies. For example, it has been shown that loss of nNOSα in genetically engineered mice lacking exon 2 of the nNOS gene (8) shows loss of nitrergic slow inhibitory junction potentials in the gut (15) and phenotypic abnormalities like pyloric stenosis consistent with the loss of nitrergic neurotransmission (16, 26). Moreover, the phenotypic changes due to loss of nNOSα in exon2 nNOS knockout mice are similar to loss of total nNOS activity due to deletion of exon 6 of nNOS (7, 8).
Calcium-calmodulin (Ca-CaM) binding to nNOS is critical for its enzymatic activity (19). The nNOS molecule contains an autoinhibitory hinge loop that links its C-terminal reductase domain to the N-terminal oxygenase domain and inhibits enzyme catalysis (2). Ca-CaM binds to a site (724–757 amino acids of nNOSα) that is adjacent to this inhibitory loop (amino acids spanning 831–872). The binding of Ca-CaM to this site displaces the inhibitory loop and forms a bridge between oxygenase and reductase domains and promotes the flow of electrons from the reductase domain to oxygenase domain which is imperative in increasing the rate of enzyme catalysis. NO formation is critically dependent upon this inter-subunit electron transfer (17, 18) and consequently, calcium-activated CaM-nNOS tetramer is necessary for catalytic activity of nNOS.
This study showed that the gut varicosities contained both CaM-bound and CaM-lacking nNOSα fractions. It further documented that CaM-associated nNOS was catalytically active and produced NO in an in vitro assay system, while CaM-lacking nNOSα was inactive. The regulation of CaM-associated and CaM-lacking fractions of nNOS may be of critical importance in the regulation of the amount of NO synthesis and release during nitrergic neurotransmission in the gut. One of the important mechanisms determining whether calmodulin binds to nNOS and activates it is its state of phosphorylation of serine at position 847 (3, 19). An important finding of the current study was that a fraction of nNOS in the gut nerve terminals is serine847 phosphorylated. This observation is similar to our previous preliminary report of ser847 phosphorylated nNOS using a different anti-ser847-phospho-nNOS antibody (NP847) (20). Since serine847 is present within the autoinhibitory loop of the nNOS enzyme, phosphorylation of serine 847 of nNOSα prevents Ca-CaM binding and prevents displacement of the inhibitory loop even in the presence of high concentrations of Ca-CaM and keeps the enzyme inactive (19). Phosphorylation is a rapid cellular event and serine847 dephosphorylation/phosphorylation of nNOSα in the enteric nerve terminals may facilitate a rapid turnover between active and inactive enzyme forms.
The present study documented using an in vitro assay that while calmodulin-bound nNOSα actively synthesized nitric oxide, serine847-phosphorylated nNOS was not calmodulin-bound and lacked the ability to synthesize nitric oxide. Thus, in the gut varicosities, serine847 phosphorylation of nNOSα may be an important mechanism for regulation of pools of inactive and active nNOS isoforms. In addition to CaM-lacking nNOSα dimer, the varicosities were also found to contain CaM-lacking nNOSα monomers and nNOSβ isoforms, all of which were ser847 phosphorylated and were catalytically inactive. Thus, while nNOSβ and nNOSα monomers do not contribute to the enzymatic activity, they add to the total nNOS present in the gut nerve terminals. Function of the nNOSβ isoform in nitrergic neurotransmission, if any, is not known. However, it has been suggested that this splice variant of nNOS may participate in residual penile erection in mice lacking nNOSα (9).
The equilibrium between the catalytically active nNOSα dimer and catalytically inactive nNOSα monomer has been reported to correlate with functional nitrergic responses in the stomach (6). In endogenous system, this equilibrium is regulated by BH4, heme and L-arginine (12). The dimer:monomer equilibrium of nNOSα may also regulate nitrergic neurotransmission.
In summary, the present study showed that in mice gastrointestinal nerve terminals, nNOSα existed as unphosphorylated, CaM-associated and serine 847-phosphorylated, but CaM-deficient dimers. The CaM-associated nNOSα dimer was catalytically active and yielded NO in vitro. On the other hand, the serine 847-phosphorylated nNOSα dimer and monomers of nNOSα were catalytically inactive. During calcium influx in the nerve varicosities in the gut, only the catalytically active nNOSα (the enzyme dimer that is bound to calmodulin and not phosphorylated at its serine 847) can serve as an immediate source for catalysis of nitric oxide production. The measurement of total nNOS in nerve terminals may not provide adequate information on the state of nitrergic neurotransmission. The present study is the first attempt to address the regulation of nitrergic neurotransmission in the gut nerve terminals, the precise sites of nitric oxide synthesis during anterograde inhibitory neurotransmission in the gastrointestinal tract. Regulation of the equilibrium between catalytically active and inactive nNOSα pools in the enteric varicosities may be critically important in regulating nitrergic neurotransmission. Further studies of time-dependent changes in the chemical nature of different nNOS isoforms after nerve stimulation are needed to test this hypothesis.
Acknowledgments
This work was supported by a NIDDK grant (DK 062867) and a Merit Review Award from the Office of Research and Development, Medical Research Services, Department of Veterans Affairs. We thank Ms. Louise Trakimas of the Electron Microscopy Core Facility at the Harvard Medical School Division of Cell Biology and Dr. Hemant Thatte for help with imaging studies, and Dr. Maryrose Sullivan for helpful suggestions.
References
- 1.Abdelmoity A, Padre RC, Burzynski KE, Stull JT, Lau KS. Neuronal nitric oxide synthase localizes through multiple structural motifs to the sarcolemma in mouse myotubes. FEBS Lett. 2000;482:65–70. doi: 10.1016/s0014-5793(00)02038-x. [DOI] [PubMed] [Google Scholar]
- 2.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bredt DS, Ferris CD, Snyder SH. Nitric oxide synthase regulatory sites. Phosphorylation by cyclic AMP-dependent protein kinase, protein kinase C, and calcium/calmodulin protein kinase; identification of flavin and calmodulin binding sites. J Biol Chem. 1992;267:10976–10981. [PubMed] [Google Scholar]
- 4.Bult H, Boeckxstaens GE, Pelckmans PA, Jordaens FH, Van Maercke YM, Herman AG. Nitric oxide as an inhibitory non-adrenergic non-cholinergic neurotransmitter. Nature. 1990;345:346–347. doi: 10.1038/345346a0. [DOI] [PubMed] [Google Scholar]
- 5.Esplugues JV. NO as a signalling molecule in the nervous system. Br J Pharmacol. 2002;135:1079–1095. doi: 10.1038/sj.bjp.0704569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gangula PR, Maner WL, Micci MA, Garfield RE, Pasricha PJ. Diabetes induces sex-dependent changes in neuronal nitric oxide synthase dimerization and function in the rat gastric antrum. Am J Physiol Gastrointest Liver Physiol. 2007;292:G725–733. doi: 10.1152/ajpgi.00406.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gyurko R, Leupen S, Huang PL. Deletion of exon 6 of the neuronal nitric oxide synthase gene in mice results in hypogonadism and infertility. Endocrinology. 2002;143:2767–2774. doi: 10.1210/endo.143.7.8921. [DOI] [PubMed] [Google Scholar]
- 8.Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- 9.Hurt KJ, Sezen SF, Champion HC, Crone JK, Palese MA, Huang PL, Sawa A, Luo X, Musicki B, Snyder SH, Burnett AL. Alternatively spliced neuronal nitric oxide synthase mediates penile erection. Proc Natl Acad Sci U S A. 2006;103:3440–3443. doi: 10.1073/pnas.0511326103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jonakait GM, Gintzler AR, Gershon MD. Isolation of axonal varicosities (autonomic varicosities) from the enteric nervous system. J Neurochem. 1979;32:1387–1400. doi: 10.1111/j.1471-4159.1979.tb11076.x. [DOI] [PubMed] [Google Scholar]
- 11.Kiss JP, Vizi ES. Nitric oxide: a novel link between synaptic and nonsynaptic transmission. Trends Neurosci. 2001;24:211–215. doi: 10.1016/s0166-2236(00)01745-8. [DOI] [PubMed] [Google Scholar]
- 12.Klatt P, Schmidt K, Lehner D, Glatter O, Bachinger HP, Mayer B. Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and L-arginine in the formation of an SDS-resistant dimer. Embo J. 1995;14:3687–3695. doi: 10.1002/j.1460-2075.1995.tb00038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leikert JF, Rathel TR, Muller C, Vollmar AM, Dirsch VM. Reliable in vitro measurement of nitric oxide released from endothelial cells using low concentrations of the fluorescent probe 4,5-diaminofluorescein. FEBS Lett. 2001;506:131–134. doi: 10.1016/s0014-5793(01)02901-5. [DOI] [PubMed] [Google Scholar]
- 14.Lin CS, Lau A, Bakircioglu E, Tu R, Wu F, Week S, Nunes L, Lue TF. Analysis of neuronal nitric oxide synthase isoform expression and identification of human nNOS-mu. Biochem Biophys Res Commun. 1998;253:388–394. doi: 10.1006/bbrc.1998.9658. [DOI] [PubMed] [Google Scholar]
- 15.Mashimo H, He XD, Huang PL, Fishman MC, Goyal RK. Neuronal constitutive nitric oxide synthase is involved in murine enteric inhibitory neurotransmission. J Clin Invest. 1996;98:8–13. doi: 10.1172/JCI118781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mashimo H, Kjellin A, Goyal RK. Gastric stasis in neuronal nitric oxide synthase-deficient knockout mice. Gastroenterology. 2000;119:766–773. doi: 10.1053/gast.2000.16509. [DOI] [PubMed] [Google Scholar]
- 17.Panda K, Adak S, Aulak KS, Santolini J, McDonald JF, Stuehr DJ. Distinct influence of N-terminal elements on neuronal nitric-oxide synthase structure and catalysis. J Biol Chem. 2003;278:37122–37131. doi: 10.1074/jbc.M304456200. [DOI] [PubMed] [Google Scholar]
- 18.Panda K, Ghosh S, Stuehr DJ. Calmodulin activates intersubunit electron transfer in the neuronal nitric-oxide synthase dimer. J Biol Chem. 2001;276:23349–23356. doi: 10.1074/jbc.M100687200. [DOI] [PubMed] [Google Scholar]
- 19.Rameau GA, Chiu LY, Ziff EB. Bidirectional regulation of neuronal nitric-oxide synthase phosphorylation at serine 847 by the N-methyl-D-aspartate receptor. J Biol Chem. 2004;279:14307–14314. doi: 10.1074/jbc.M311103200. [DOI] [PubMed] [Google Scholar]
- 20.Rao YM, Watanabe Y, Goyal RK. Identification of pools of Calmodulin bound and Calmodulin lacking forms of nNOS in the Nerve Terminals of Gut. Gastroenterology. 2005;128:A611. [Google Scholar]
- 21.Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- 22.Saur D, Neuhuber WL, Gengenbach B, Huber A, Schusdziarra V, Allescher HD. Site-specific gene expression of nNOS variants in distinct functional regions of rat gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 2002;282:G349–358. doi: 10.1152/ajpgi.00226.2001. [DOI] [PubMed] [Google Scholar]
- 23.Saur D, Paehge H, Schusdziarra V, Allescher HD. Distinct expression of splice variants of neuronal nitric oxide synthase in the human gastrointestinal tract. Gastroenterology. 2000;118:849–858. doi: 10.1016/s0016-5085(00)70171-5. [DOI] [PubMed] [Google Scholar]
- 24.Su Z, Blazing MA, Fan D, George SE. The calmodulin-nitric oxide synthase interaction. Critical role of the calmodulin latch domain in enzyme activation. J Biol Chem. 1995;270:29117–29122. doi: 10.1074/jbc.270.49.29117. [DOI] [PubMed] [Google Scholar]
- 25.Van Geldre LA, Lefebvre RA. Interaction of NO and VIP in gastrointestinal smooth muscle relaxation. Curr Pharm Des. 2004;10:2483–2497. doi: 10.2174/1381612043383890. [DOI] [PubMed] [Google Scholar]
- 26.Watkins CC, Sawa A, Jaffrey S, Blackshaw S, Barrow RK, Snyder SH, Ferris CD. Insulin restores neuronal nitric oxide synthase expression and function that is lost in diabetic gastropathy. J Clin Invest. 2000;106:373–384. doi: 10.1172/JCI8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.White TD, Leslie RA. Depolarization-induced release of adenosine 5′-triphosphate from isolated varicosities derived from the myenteric plexus of the guinea pig small intestine. J Neurosci. 1982;2:206–215. doi: 10.1523/JNEUROSCI.02-02-00206.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]