

Abstract
We discuss a 53 year old woman with systemic lupus erythematosus who presented with vasculitis, hypocomplementemia and nephritis. Although her serum complement 4 (C4) levels were zero, she had four copies of C4 gene. Renal biopsy revealed membranoproliferative glomerulonephritis and the presence of cryoglobulins, detected by electron microscopy, and significant numbers of T cells in the interstitium. Cryoglobulins were considered responsible for the complete consumption of C4 in the serum the levels of which improved gradually after treatment. T cells in the kidney were found to express CD44 and phosphorylated ezrin/radixin/moiesin which explain why they homed to the kidney inappropriately. The contribution of cryoglobulins and T cells in the expression of kidney pathology is discussed.
Case presentation
The patient, a 53-year-old woman, presented in August 2002 with intermittent painless, non-pruritic rashes on her legs during the preceding two years. She also complained of right ankle swelling, pain in her left thigh, right knee and right calf, not associated with activity, that was relieved by ibuprofen. A lower extremity ultrasound showed no evidence of deep venous thrombosis. Lymph node biopsy in the past had shown a reactive process. A skin biopsy two months earlier had shown evidence of small vessel necrotizing vasculitis. Renal function was normal at that time. Antinuclear antibody and rheumatoid factor were present in the serum but anti-cardiolipin, hepatitis B and C antibodies were absent, as were cryoglobulins. She was treated with prednisone and hydroxychloroquine.
In Spring of 2004 skin lesions consistent with the diagnosis of erythema nodosum appeared along with ankle arthritis. Polyclonal hypergammaglobulinemia and trace monoclonal IgM Kappa on serum protein electrophoresis were detected. Treatment with prednisone and hydroxychloroquine, was followed by clinical deterioration. In the winter of 2004, she developed abdominal pain, with negative imaging studies and colonoscopy, and muscle pain. There was no evidence of deep venous thrombosis, a urinalysis was normal, and she was treated with steroids.
In the fall of 2005 she experienced pleuritic chest pain with pericardial effusion, which responded partially to prednisone and non-steroidal anti-inflammatory medications. Later that winter, she again developed skin rashes, fever and chest pain, which was attributed to pericarditis. She had a partial response to prednisone. Over the summer of 2006, the patient continued to experience flares of cutaneous lupus, despite treatment with hydroxychloroquine and prednisone without evidence of kidney involvement.
In September of 2006, she experienced intense abdominal pain. Endoscopy revealed petechial lesions of small and large bowel. Imaging studies revealed paraaortic lymphadenopathy. She was treated with prednisone; the patient had rejected repeated recommendations to initiate cytotoxic drug treatment.
In June 2007, the patient experienced a cutaneous lupus flare, peri-orbital swelling, joint pain lower extremity erythema. The spot urine protein/creatinine ratio was 0.5. Serum creatinine was also elevated at 1.0 mg/dL (baseline 0.5 to 0.7 mg/dL). Subsequently, the patient noticed increased lower extremity swelling and experienced paroxysmal nocturnal dyspnea. Imaging studies excluded the presence of pulmonary emboli but a two-dimensional echocardiogram showed greater mitral valve regurgitation compared to six months earlier. The patient responded to intravenous furosemide. A repeat spot urine protein/creatinine ratio was 4.7 while serum creatinine remained unchanged. Subsequently, serum creatinine peaked at 1.0 mg/dl and spot urine protein/creatinine ratio peaked at 8.4. Microscopic examination of the urine revealed an active sediment with acanthocytes and mixed cellular casts. Serum anti-dsDNA, hepatitis B and C antibodies were negative.
Serum C3 was low while C4 was undetectable on several occasions. Consequently, C4 genotypes were determined by real time PCR and confirmed by genomic Southern blot analysis. The patient was found to have 3 copies of C4A genes and one copy of C4B (1). At the same time flow cytometry on a blood sample taken in October, 2007 revealed that 14% of the patients’ red cells were decorated with C4d, documenting extensive C4 consumption (2;3)
Renal biopsy was performed, and the patient was treated with mycophenolate mofetil and high doses of prednisone. Subsequently, her kidney function normalized, proteinuria resolved and serum C4 increased to 8 mg/dl. The main events in the course of the disease are summarized in Table. I.
Table I.
Main events in the evolution of the disease.
| 08/2002 - Cutaneous necrotizing vasculitis (biopsy), arthritis |
| 03/2004 - Erythema nodosum/leukocytoclastic vasculitis, arthritis |
| 12/2004 - Skin lesions, muscle pain |
| 12/2005 - Skin lesions, pericarditis |
| 09/2006 - Possible intestinal vasculitis |
| 06/2007 - Glomerulonephritis |
Kidney biopsy
Histologic examination revealed cortex and medulla containing 43 glomeruli, of which 4 were globally sclerotic. Intact glomeruli showed a membranoproliferative pattern with thickening of peripheral capillary walls and occasional hyalin thrombi. Jones stain showed extensive double contours with minimal spike formation. There was a mild leukocyte infiltration and focal necrosis. No crescents were seen, although the parietal epithelium was focally prominent. There was mild interstitial fibrosis and tubular atrophy. A mild interstitial mononuclear inflammatory infiltrate was seen. Arteries and arterioles showed mild chronic injury with no evidence of active vasculitis.
Immunofluorescence microscopy revealed mesangial and peripheral capillary loop granular staining for IgG (2+), IgA (trace), IgM (2-3+, with scattered glomerular “thrombi”), C3 (trace), kappa (2+), and lambda (1+). Interestingly, C1q was negative. Many glomeruli showed segmental fibrin positivity. Vascular IgG and C3 staining was seen. There were no immune deposits within tubular basement membranes and tissue ANA was not present.
Ultrastructural studies were performed on three intact glomeruli. Two were essentially within normal limits, without any significant electron dense deposits (Figure 1A). The third showed focal foot process effacement, an increase in endocapillary cells, and mesangial interposition (Figure 1B). Extensive subendothelial, and occasional mesangial, deposits, all with the characteristic substructure of cryoglobulins were seen (Figure 1C). No subepithelial deposits were noted. Granular electron dense deposits typical of immune complexes were entirely absent in all three glomeruli. No definite tubuloreticular structures were identified.
Figure 1. Electron microscopy study.
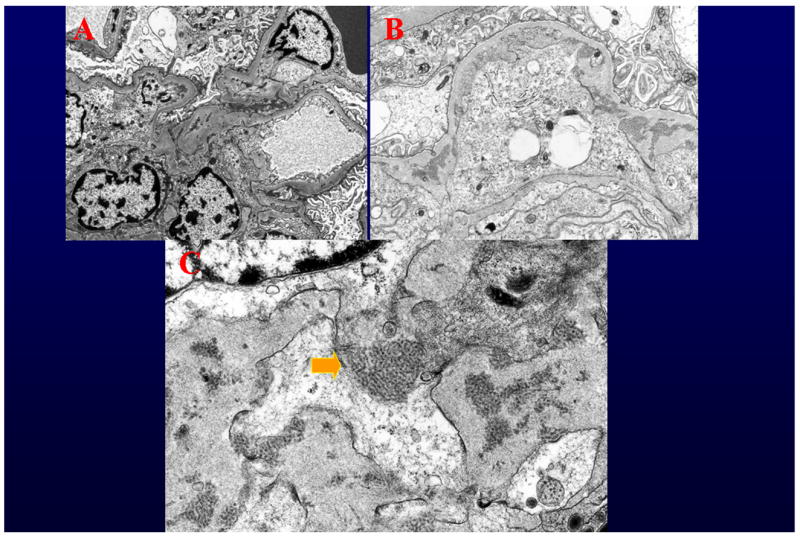
A. Two of the three glomeruli studied by electron microscopy were within normal limits, and showed no deposits (Original magnification 4000X). B. One of the three glomeruli studied by electron microscopy showed mesangial interposition and deposits with substructure (Original magnification 6000X). C. All of the deposits had the characteristic substructure of cryoglobulins. No amorphous granular electron dense deposits were noted. (Original magnification 15000X).
The initial diagnosis rendered was “Lupus Nephritis ISN/RPS Classification Class IV-G (A): Diffuse, active global proliferative glomerulonephritis”. The patient was scored with an Activity Index of 8 and a Chronicity Index of 1. However, after electron microscopy studies, the biopsy was thought to present interpretive difficulties, as the findings were not typical of a “lupus” immune complex glomerulonephritis, as that term is usually understood. All of the deposits noted had the substructure of cryoglobulins – none had the amorphous granular features of typical immune complexes. Furthermore, focal “hyalin thrombi” were seen and IgG deposition was more pronounced in the capillary wall than in the mesangium, and was exceeded by IgM deposition. Interestingly, no nuclear or tubular basement membrane IgG deposition was noted and C1q was entirely negative. Thus, the findings in toto were felt to be far more characteristic of a cryoglobulinemeic glomerulonephritis than a classical “lupus nephritis”. Of course patients with lupus often develop cryoglobulins, and in that sense this glomerulonephritis may indeed have been lupus related. However, in most cases those changes are superimposed on more typical lupus findings. In this case, all the lesions present could be attributed to a “pure” cyroglobulinemic glomerulonephritis.
Characteristic of T cells infiltrating the kidney
A dense lymphoid infiltrate was demonstrated in large areas of the kidney biopsy. Immunophenotype revealed that it was composed primarily of T cells (Figure 2). Staining with specific antibodies demonstrated that the T cells expressed the hyaluronic acid-binding molecule CD44, primarily the v3 and v6 isoforms (Figure 3). Engagement of CD44 by its ligand, hyluronic acid, results in phosphorylation of ezrin, radixin, and moesin (ERM) ptotein complex. Accordingly, the presence of phosphorylated ERM (pERM) should be observed in infiltrating T cells (Figure 4), activated through CD44 (4). Indeed, CD3+ pERM+ cells were noted in the kidney tissues (Figure 4). Interestingly, FoxP3 – the transcription factor associated with regulatory T cells – could not be detected in the examined tissue (Figure 4). In summary, the kidney had a dense T cell infiltrate in which inflammatory activated cells were abundant, but lacking anti-inflammatory regulatory T cells.
Figure 2. T cells form a dense infiltrate in the kidney.
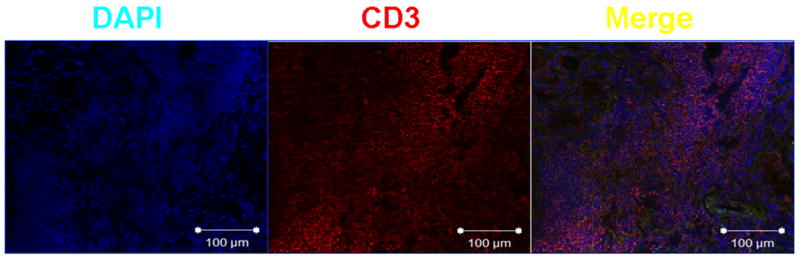
Frozen sections of the kidney biopsy were fixed with ice-cold acetone and incubated with mouse anti-human CD3 (1:100). After thorough washing, sections were incubated with goat anti-mouse IgG labeled with Texas Red (1:50). Finally, nuclei were stained by brief incubation with DAPI (4',6-diamidino-2-phenylindole; 0.5 μg/mL) (4). Slides were scanned in a Nikon Eclipse Ti confocal microscope; images were analyzed with EZ-C1 v. 3.6 software.
Figure 3. Kidney infiltrating T cells express CD44v3 and CD44v6.
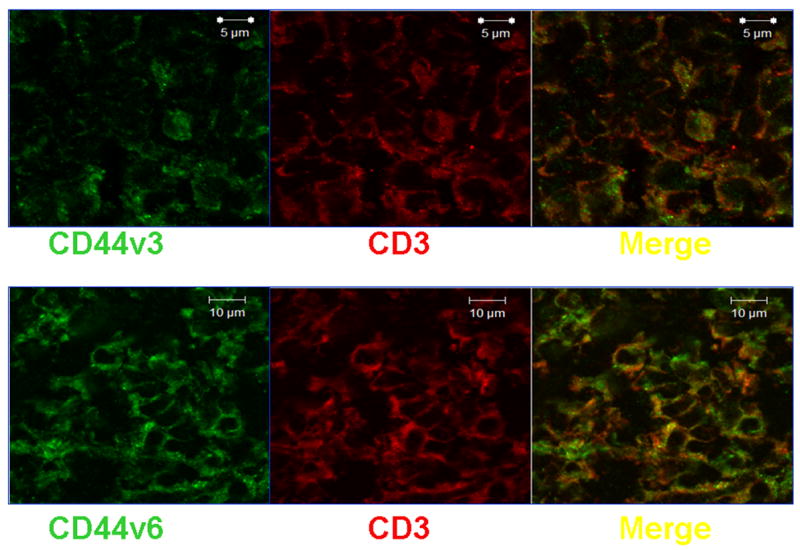
Sections were fixed and stained with mouse anti-human CD3 and either rabbit anti-human CD44v3 or rabbit anti-human CD44v6 (1:100). After thorough washing, sections were incubated with goat anti-mouse IgG labeled with Texas Red and goat anti-rabbit IgG labeled with Alexa Fluor 488 (1:50).
Figure 4. Phosphorylated ERM proteins are detectable in infiltrating T cells; FoxP3 is absent.
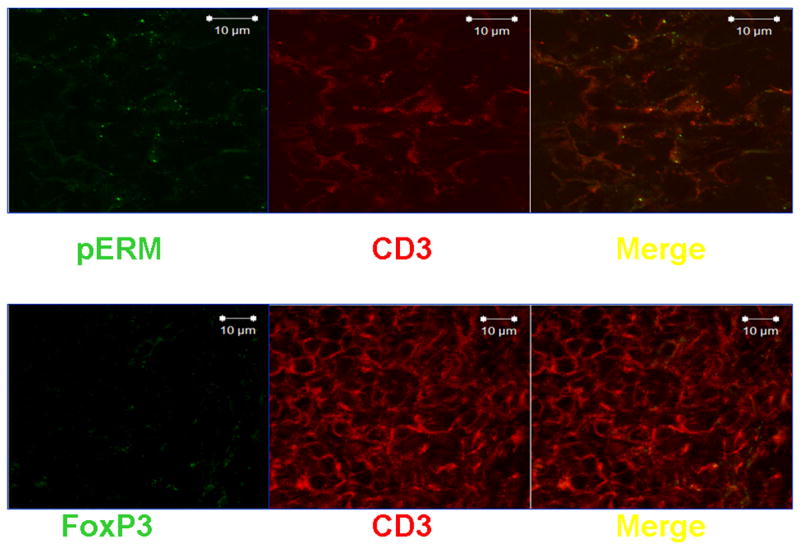
Sections were fixed and stained with mouse anti-human CD3 and either rabbit anti-human pERM or rabbit anti-human FoxP3 (1:100). After thorough washing, sections were incubated with goat anti-mouse IgG labeled with Texas Red and goat anti-rabbit IgG labeled with Alexa Fluor 488 (1:50).
Role of T cells in tissue pathology
T cells play a pivotal role in the pathogenesis of SLE (5). Although their contribution to the auto-antibody response is well documented, their participation as direct mediators of tissue damage is less well recognized. In particular, lupus nephritis is commonly regarded as an antibody-mediated condition. Nevertheless, accumulated evidence indicates that antibodies are not entirely responsible for kidney injury. Interstitial inflammation correlates with the degree of renal damage and predicts progression to renal failure in SLE patients (6;7). Moreover, therapies that target T cells, or diminish T cell infiltration into the kidneys, have been shown to diminish nephritis in mice with lupus-like diseases (8;9). Using an elegant approach, Chan et al demonstrated that the lack of antibodies does not prevent nephritis in a genetically modified lupus-prone mouse (MRL/lpr) whose B cells could not secrete antibodies. Although mice lacked glomerular immune complex deposition, interstitial and perivascular infiltrates were still observed (10). Thus, although antibodies play an extremely important role in lupus nephritis –particularly in the glomerular aspect of the disease – their presence does not account for the whole spectrum of pathogenic mechanisms involved.
Peripheral blood T cells from SLE patients have a number of characteristics that grant them pathogenic capacity (5). Specifically, they have been shown to express higher levels of CD44 and to have an enhanced migration capacity (4). T cells that infiltrate affected kidneys of patients with SLE exhibit some of the phenotypic markers evident in peripheral blood. There is an augmented expression of CD44, as well as enhanced phosphorylation of the CD44-associated proteins ezrin, radixin, and moesin (ERM) (4). Thus, circulating T cells in SLE patients have an abnormally increased capacity to migrate to tissues and to infiltrate them, enhancing the local inflammatory response. This notion has been explored as well in affected skin of SLE patients. Similar to the findings in kidney biopsies, T cell infiltrates are a common finding in several types of SLE skin manifestations, particularly discoid lupus, were high numbers of CD4+ and CD8+ T cells are found in affected tissue (11).
The concept that T cells may act as local inflammatory mediators, and play a role not limited to driving a B cell autoimmune response is clearly exemplified in this case. Further, the finding of CD44+ and pERM+ cells, along with the absence of regulatory T cells, suggests that the observed T cells play a significant role in end-organ damage, producing cytokines and chemokines and orchestrating a chronic pathologic autoimmune response.
Hypocomplementemia in SLE
The complement system encompasses approximately 30 proteins that have immunologic activity both individually and through the formation of the membrane attack complex. Complement can be activated through 3 pathways: the classical pathway, where IgM and IgG bind to antigen and form an immune-complex that can bind C1q; the alternative pathway, where lipopolysaccharides in microbial cell surfaces activate C3; and the lectin pathway, where mannan-binding lectin binds to microbial ligands activating proteases related to C1r and C1s.
Complement deficiencies may be either hereditary or acquired. Hereditary deficiencies of C1q, C1r, C1s, C4, and C2 are involved in the susceptibility for the development of SLE. Seventy-five to 90% of patients with a homozygous deficiency of C1 or C4 develop SLE or a lupus-like syndrome. There is also increased susceptibility to infections with most complement deficiencies and of glomerulonephritis with C2 deficiency. The net result of complement deficiencies includes impaired handling of immune-complexes, decreased clearance of apoptotic material, and aberrant B cell tolerance induction (12), all relevant mechanisms in the setting of SLE.
Acquired deficiencies of complement proteins (such as C1q, C3, C4, and CR1) in SLE may be due to auto-antibodies directed against a specific complement component, or to immune complex-mediated activation and consumption (13). The acquired complement dysfunction in SLE seems to be more evident during a disease flare, where low C1q and C4 levels may be useful in monitoring disease activity (14). In a recent study examining the prevalence of hypocomplementemia in 597 patients with SLE, 62% of patients were found to have either low CH50, C3, or C4 values. Furthermore, a possible deficiency of C1q or C2 in two patients was suggested by finding undetectable levels of CH50 but normal C3 and C4 levels (14).
Although deficiencies of the various complement components share common disease associations, we will focus on C4 deficiency, which was considered in our patient under discussion. The human C4 gene is located at the class III region of the major histocompatibility complex on the short arm of chromosome 6. The complement component C4 has two isoforms, C4A and C4B, and the genes coding for such isoforms are highly polymorphic. The presence of C4 null alleles, especially C4A null, has been associated with the development of SLE (1). In the general population, studies have reported a complete deficiency of both C4A and C4B in 26 individuals from 18 families of diverse racial backgrounds (15;16). Over half of these individuals were eventually diagnosed with SLE. In contrast to idiopathic SLE, the age of diagnosis in this study population encompassed a wider range (from 2 to 41 years) and the female-to-male ratio was close to 1:1, whereas idiopathic SLE has a predominant female predilection between the ages of 15 and 45 years. Among the remaining 11 subjects with inherited C4 deficiency who were not diagnosed with SLE, 5 had skin findings typically seen in lupus such as photosensitive lesions or discoid lupus, and 6 had renal disease including mesangioproliferative glomerulonephritis, membranous nephropathy, and recurrent hematuria. Interestingly, patients with inherited C4 deficiency have an antibody profile characterized by low titer or absent antinuclear antibodies but often have detectable anti-Ro antibodies (13). Thus, there are phenotypic similarities but also demographic and serological differences among patients with inherited complement deficiency and acquired complement deficiency associated with SLE.
The renal histopathology in our patient under discussion was in part suggestive of cryoglobulinemia, though cryoglobulins were not detected in the patient’s serum with repeated testing. It is known that up to 80% of cases of mixed cryoglobulinemia may have low C4 levels and previous reports evaluating autoimmune diseases and cryoglobulins have described the association between RA and SLE and the presence of cryoglobulins (14;17;18). In these studies, the prevalence of cryoglobulins range from 26–67% in patients with SLE. In one particular cohort (14), hypocomplementemia was significantly associated with the presence of cryoglobulins, cutaneous vasculitis, female gender, anti-dsDNA antibodies, and nephropathy. However, in a logistic regression analysis, nephropathy but not cryoglobulins remained as a significant independent variable associated with hypocomplementemia. Though the numbers of patients in some of these studies were small, the presence of C1aAB, C3, and C4 in cryoglobulins of some SLE patients suggested a possible role of cryoblobulins in complement activation in SLE (19). Thus, in our particular case hypocomplementemia may be multifactorial, and mechanisms that need to be considered are: inherited or acquired complement deficiency, underlying nephropathy, or low titer of cryoglobulins that may have not been detected by laboratory assay. The presence of C3d on the red cell surface of the patient’s red blood cells suggests that C4 is produced and consumed (3). Such a finding should obviate the need for C4 genotyping.
Role of cryoglobulins in lupus nephritis
The hallmark of SLE is the prolific production of autoantibodies, some of which may mediate pathogenic effects while others constitute an immunologic smoke-screen. The formation of immune complexes either in the circulation or in situ in the organ-targets is an attractive mechanism to explain some of the pathologic effects of SLE. The kidney in particular, which represent the major target of disease pathology in SLE, offers a large surface for the local deposition of immune complexes or the targeting of molecules by circulating autoantibodies. Much attention has been paid to the role of anti-dsDNA antibodies in the development of kidney pathology in SLE. Indeed studies have shown repeatedly that anti-dsDNA antibodies deposit in the kidneys of both SLE patients and murine lupus models; but the question remains whether these antibodies are already bound with their cognate antigens in immune complexes when they enter the kidneys or they bind in situ to their targets. In the first scenario, DNA antigens in the circulation bind anti-dsDNA antibodies and the complexes deposit on the basement membrane of the kidney and promote inflammation via complement activation. The circulating complexes that contain DNA may also promote kidney pathology not simply by depositing but also by promoting the production of IFN-α, an important cytokine in SLE. Indeed it has been shown that DNA containing complexes can stimulate the production of IFN-αby plasmacytoid dendritic cells in a TLR9/FcRγII dependent manner (19–23).
Anti-dsDNA antibodies may also cause kidney pathology by binding to local antigens either through charge interactions or cross-reactivity. It has been proposed DNA or nucleosomal material may be trapped in the glomerular membrane via electrostatic interactions (24). Subsequently, anti-DNA (or anti-nucleosome) antibodies bind to this embedded material and activate complement locally. Anti-dsDNA antibodies may alternatively interact with other molecules in the kidney. Indeed, a subset of pathogenic anti-dsDNA has been shown to bind to α-actinin, a molecule that is found in podocytes (25). The interaction of α-actinin with anti-dsDNA antibodies therefore may result in impaired function of podocytes and instigation of proteinuria.
An interesting, yet still controversial, role in lupus nephritis for another autoantibody binding to the complement component C1q has been proposed (26–28). Anti-C1q antibodies show a strong correlation with the presence of lupus nephritis. The precise role of these autoantibodies is still unclear: the binding of C1q-anti-C1q on deposited complexes may result in the more potent local activation of complement and/or the activation via the FcRγof inflammatory cells.
In the present case, evidence from the kidney biopsy suggested the presence of circulating immune complexes, similar to cryoglobulins, that deposited in the kidney. It is of interest the fact that cryoglobulins were never detected in the peripheral blood suggesting that all produced cryoglobulins were trapped in the kidney or that cryglobulins were elevated in the peripheral blood only for a limited period of time during which no appropriate detection test was performed. Patients with SLE have been reported to have cryoglobulins anywhere from 16–83%. In a large series of 122 patients with SLE, 25% had evidence of cryoglobulinemia (29). 21% of the patients with SLE and cryoglobulinemia were infected with HCV as compared to 5% of the control SLE patients. Distinctive laboratory features of the SLE patients with cryoglobulins were the higher rate of rheumatoid factor presence (42% vs. 15% in SLE patients without cryoglobulinemia), and low CH50 (present in 84% vs. 45%). There was no apparent difference in the clinical manifestations between the two groups besides the higher prevalence of cutaneous vasculitis in the patients with cryoglobulinemia. In conclusion, circulating and/or in situ formation of immune complexes may play a role in lupus nephritis by inducing complement activation, activating cells via the FcRγ receptor and augmenting the production of IFN-α.
Conclusions
The patient that we discuss here presents an interesting list of diagnostic considerations and significant insights in the pathogenesis of glomerulonephritis. Although the patient presented sufficient criteria for the diagnosis of SLE and the clinical manifestations could have been attributed to typical lupus nephritis, the electron microscopy study of the kidney tissue demonstrated typical feature of cryoglobulin deposition. It should be noted that repeated tests failed to detect circulating cryoglobulins suggesting that they were either present a low amounts continuously adsorbed to the kidney or they were produced only for a short period of time which was missed during testing. The case urges the performance of kidney biopsy in patients with clinical picture of SLE in order to obtain a complete picture of the underlying pathology even if treatment may not differ.
Another interesting feature of our patient is the complete lack of complement 4 prior to treatment. Although we proceeded to determine the number of complement 4 copies (in the context of additional ongoing studies) and found to be normal, the presence of complement 4d fragment deposited on the surface of red cells clearly demonstrates extensive consumption of complement probably by the deposited cryoglobulins in the kidney and other tissues. Search for complement fragments deposited on the surface of red cells may be used to determine the extent of ongoing complement activation.
Finally, we established the molecular features of the T cells that they were found in the kidney interstitium. They were found to express the adhesion molecule CD44 and its signaling partner pERM. Cells with similar features are found in the peripheral blood of patients with lupus (4) and its has been proposed that their presence in the periphery precedes the inappropriate homing to tissues.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Wu YL, Savelli SL, Yang Y, Zhou B, Rovin B, Birmingham DJ, Nagaraja HN, Hebert LA, Yu CY. Sensitive and specific real-time PCR assays to accurately determine copy number variations (CNVs) of human complement C4A, C4B, C4-long, C4-short and RCCX modules: Elucidation of C4 CNVs in 50 consanguineous subjects with defined HLA genotypes. J Immunol. 2007;179:3012–3025. doi: 10.4049/jimmunol.179.5.3012. [DOI] [PubMed] [Google Scholar]
- 2.Tsokos GC. Exploring complement activation to develop biomarkers for systemic lupus erythematosus. Arthritis Rheum. 2004;50:3404–3407. doi: 10.1002/art.20602. [DOI] [PubMed] [Google Scholar]
- 3.Manzi S, Navratil JS, Ruffing MJ, Liu CC, Danchenko N, Nilson SE, Krishnaswami S, King DE, Kao AH, Ahearn JM. Measurement of erythrocyte C4d and complement receptor 1 in systemic lupus erythematosus. Arthritis Rheum. 2004;50:3596–3604. doi: 10.1002/art.20561. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Harada T, Juang YT, Kyttaris VC, Wang Y, Zidanic M, Tung K, Tsokos GC. Phosphorylated ERM Is Responsible for Increased T Cell Polarization, Adhesion, and Migration in Patients with Systemic Lupus Erythematosus. J Immunol. 2007;178:1938–1947. doi: 10.4049/jimmunol.178.3.1938. [DOI] [PubMed] [Google Scholar]
- 5.Crispin JC, Kyttaris VC, Juang YT, Tsokos GC. Signaling and gene transcription aberrations dictate systemic lupus erythematosus T cell phenotype. Trends Immunol. 2008 doi: 10.1016/j.it.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Abe S, Amagasaki Y, Iyori S, Konishi K, Kato E, Sakaguchi H. Significance of tubulointerstitial lesions in biopsy specimens of glomerulonephritic patients. Am J Nephrol. 1989;9:30–37. doi: 10.1159/000167931. [DOI] [PubMed] [Google Scholar]
- 7.Austin HA, III, Boumpas DT, Vaughan EM, Balow JE. Predicting renal outcomes in severe lupus nephritis: contributions of clinical and histologic data. Kidney Int. 1994;45:544–550. doi: 10.1038/ki.1994.70. [DOI] [PubMed] [Google Scholar]
- 8.Gilkeson GS, Spurney R, Coffman TM, Kurlander R, Ruiz P, Pisetsky DS. Effect of anti-CD4 antibody treatment on inflammatory arthritis in MRL-lpr/lpr mice. Clin Immunol Immunopathol. 1992;64:166–172. doi: 10.1016/0090-1229(92)90195-t. [DOI] [PubMed] [Google Scholar]
- 9.id-Peralta L, Mathian A, Tran T, Delbos L, Durand-Gasselin I, Berrebi D, Peuchmaur M, Couderc J, Emilie D, Koutouzov S. Leukocytes and the kidney contribute to interstitial inflammation in lupus nephritis. Kidney Int. 2008;73:172–180. doi: 10.1038/sj.ki.5002625. [DOI] [PubMed] [Google Scholar]
- 10.Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189:1639–1648. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hasan T, Stephansson E, Ranki A. Distribution of naive and memory T-cells in photoprovoked and spontaneous skin lesions of discoid lupus erythematosus and polymorphous light eruption. Acta Derm Venereol. 1999;79:437–442. doi: 10.1080/000155599750009861. [DOI] [PubMed] [Google Scholar]
- 12.Truedsson L, Bengtsson AA, Sturfelt G. Complement deficiencies and systemic lupus erythematosus. Autoimmunity. 2007;40:560–566. doi: 10.1080/08916930701510673. [DOI] [PubMed] [Google Scholar]
- 13.Tsokos GC, Gordon C, Smolen J, editors. Systemic Lupus Erythematosus: A Companion to Rheumatology. Elsevier; Amsterdam: 2007. pp. 1–608. [Google Scholar]
- 14.Ramos-Casals M, Campoamor MT, Chamorro A, Salvador G, Segura S, Botero JC, Yague J, Cervera R, Ingelmo M, Font J. Hypocomplementemia in systemic lupus erythematosus and primary antiphospholipid syndrome: prevalence and clinical significance in 667 patients. Lupus. 2004;13:777–783. doi: 10.1191/0961203304lu1080oa. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Lhotta K, Chung EK, Eder P, Neumair F, Yu CY. Complete complement components C4A and C4B deficiencies in human kidney diseases and systemic lupus erythematosus. J Immunol. 2004;173:2803–2814. doi: 10.4049/jimmunol.173.4.2803. [DOI] [PubMed] [Google Scholar]
- 16.Hauptmann G, Tappeiner G, Schifferli JA. Inherited deficiency of the fourth component of human complement. Immunodefic Rev. 1988;1:3–22. [PubMed] [Google Scholar]
- 17.Erhardt CC, Mumford P, Maini RN. Differences in immunochemical characteristics of cryoglobulins in rheumatoid arthritis and systemic lupus erythematosus and their complement binding properties. Ann Rheum Dis. 1984;43:451–456. doi: 10.1136/ard.43.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sikander F, Salgaonkar DS, Joshi VR. Cryoglobulin studies in systemic lupus erythematosus. J Postgrad Med. 1989;35:139–143. [PubMed] [Google Scholar]
- 19.Datta SK. Production of pathogenic antibodies: cognate interactions between autoimmune T and B cells. Lupus. 1998;7:591–596. doi: 10.1191/096120398678920703. [DOI] [PubMed] [Google Scholar]
- 20.Vlahakos DV, Foster MH, Adams S, Katz M, Ucci AA, Barrett KJ, Datta SK, Madaio MP. Anti-DNA antibodies form immune deposits at distinct glomerular and vascular sites. Kidney Int. 1992;41:1690–1700. doi: 10.1038/ki.1992.242. [DOI] [PubMed] [Google Scholar]
- 21.Wener MH, Mannik M, Schwartz MM, Lewis EJ. Relationship between renal pathology and the size of circulating immune complexes in patients with systemic lupus erythematosus. Medicine (Baltimore) 1987;66:85–97. doi: 10.1097/00005792-198703000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Wener MH, Mannik M. Mechanisms of immune deposit formation in renal glomeruli. Springer Semin Immunopathol. 1986;9:219–235. doi: 10.1007/BF02099023. [DOI] [PubMed] [Google Scholar]
- 23.Wener MH, Mannik M. Selective losses of large immune complexes during density gradient ultracentrifugation and an approach for prevention of these losses. J Immunol Methods. 1985;84:1–10. doi: 10.1016/0022-1759(85)90409-0. [DOI] [PubMed] [Google Scholar]
- 24.Lefkowith JB, Kiehl M, Rubenstein J, DiValerio R, Bernstein K, Kahl L, Rubin RL, Gourley M. Heterogeneity and clinical significance of glomerular-binding antibodies in systemic lupus erythematosus. J Clin Invest. 1996;98:1373–1380. doi: 10.1172/JCI118924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mason LJ, Ravirajan CT, Rahman A, Putterman C, Isenberg DA. Is alpha-actinin a target for pathogenic anti-DNA antibodies in lupus nephritis? Arthritis Rheum. 2004;50:866–870. doi: 10.1002/art.20103. [DOI] [PubMed] [Google Scholar]
- 26.Coremans IE, Spronk PE, Bootsma H, Daha MR, van der Voort EA, Kater L, Breedveld FC, Kallenberg CG. Changes in antibodies to C1q predict renal relapses in systemic lupus erythematosus. Am J Kidney Dis. 1995;26:595–601. doi: 10.1016/0272-6386(95)90595-2. [DOI] [PubMed] [Google Scholar]
- 27.Siegert CE, Daha MR, Tseng CM, Coremans IE, van Es LA, Breedveld FC. Predictive value of IgG autoantibodies against C1q for nephritis in systemic lupus erythematosus. Ann Rheum Dis. 1993;52:851–856. doi: 10.1136/ard.52.12.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trouw LA, Groeneveld TW, Seelen MA, Duijs JM, Bajema IM, Prins FA, Kishore U, Salant DJ, Verbeek JS, van KC, Daha MR. Anti-C1q autoantibodies deposit in glomeruli but are only pathogenic in combination with glomerular C1q-containing immune complexes. J Clin Invest. 2004;114:679–688. doi: 10.1172/JCI21075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Carrasco M, Ramos-Casals M, Cervera R, Trejo O, Yague J, Siso A, Jimenez S, De La RG, Font J, Ingelmo M. Cryoglobulinemia in systemic lupus erythematosus: prevalence and clinical characteristics in a series of 122 patients. Semin Arthritis Rheum. 2001;30:366–373. doi: 10.1053/sarh.2001.20265. [DOI] [PubMed] [Google Scholar]