

SUMMARY
Necrotizing enterocolitis (NEC) is an inflammatory intestinal disorder that affects 2–5% of all premature infants. Enterobacter sakazakii (ES), a common contaminant in milk-based powdered infant formula, is implicated as a causative agent of sepsis, meningitis, and NEC in newborn infants with high mortality rates. However, the role of ES in the pathogenesis of NEC is not known to date. Here, we demonstrate for the first time that ES is able to induce clinical and histological NEC in newborn rats. ES was found to bind to enterocytes in rat pups at the tips of villi and to intestinal epithelial cells, IEC-6, in culture with no significant invasion. Exposure to ES induced apoptosis and increased the production of interleukin-6 in IEC-6 cells and in the animal model. These data suggest that ES could be a potential pathogen that induces NEC, and triggers intestinal disease by modulating enterocyte intracellular signaling pathways.
Keywords: Necrotizing enterocolitis, intestine, enterocytes, neonatal, bacteria
INTRODUCTION
Enterobacter sakazakii (ES) is an important emerging neonatal pathogen, associated with outbreaks of meningitis, sepsis, and necrotizing enterocolitis (NEC) [1]. ES is prevalent in certain milk-based powdered infant formulas, cereals, chocolate, potato flour, and pasta [2, 3, 4]. The number of ES-related infections has substantially increased during the past ten years due to increased use of infant formula. The United States Food and Drug Administration issued several warnings regarding ES infection in newborns.
NEC is an inflammatory intestinal disorder that affects 2–5% of all premature infants and is associated with high mortality rates. Three principal factors have been implicated in the pathogenesis of the disease: an immature intestinal epithelial barrier, abnormal bacterial-colonization, and formula feeding. Although a variety of bacterial species including Enterobacter, Clostridium, and Staphylococcus have been implicated in the development of NEC, no single pathogen has yet been shown to cause the disease [5]. Enterobacter species have been associated with NEC, and isolated from the blood, skin, cerebrospinal fluid, peritoneal fluid, urine and respiratory tracts of affected infants [6, 7]. Milk-based infant formulas contaminated with ES have been implicated in outbreaks of NEC, suggesting that this pathogen may be important for disease pathogenesis.
The establishment of disease depends upon successful pathogen colonization, and a microbe’s ability to adhere to host surfaces, such as the intestinal epithelial layer [8]. ES adheres to endothelial cells and neoplastic epithelial cell lines [9]. However, ES associations with non-transformed intestinal epithelial cell lines remain poorly understood. Pathogen binding may trigger a number of different host responses, ranging from alteration in intracellular signaling pathways, chemokine and cytokine release, and induction of apoptosis. A variety of inflammatory molecules including tumor necrosis factor alpha (TNF-α), nitric oxide (NO), and interleukin 6 (IL-6) have been implicated in the pathogenesis of NEC [10]. In particular, elevated levels of IL-6 have been found in blood and stool samples of infants with NEC [11]. Apoptosis is a programmed cell death, which under homeostatic conditions exists in harmony with cellular mitosis. Experimental NEC is associated with increased enterocyte apoptosis [12]. We therefore hypothesize that colonization of the intestine by ES may contribute to the pathogenesis of NEC by stimulating release of inflammatory factors and induction of enterocyte apoptosis. The present study examines the causative relationship between ES and NEC in a rat model of the disease.
METHODS
1. Bacterial strains, cells, and reagents
ES (51329), Escherichia coli DH 5-α (EC), and rat intestinal epithelial cells IEC-6 (passages 20–26) were obtained from ATCC (Manassas, VA). IEC-6 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, 1 U/ml insulin, 100 U/ml penicillin G, and 100 U/L streptomycin. Rodent formula (Esbilac) was obtained from PetAg (Hampshire, IL).
2. Transformation of ES with green fluorescent protein (GFP) plasmid
ES were grown for 8 h in Luria Broth (LB) at 37°C, centrifuged to pellet down bacteria, washed, and treated with 0.1 M calcium chloride for 30 min on ice. Then, ES were transformed with a GFP plasmid as described [13]. Colonies were assessed for GFP expression by viewing under ultraviolet light and further cultures were grown from a single colony.
3. ES infection studies using the rat model of NEC
Animal experiments were approved by the IACUC and Biosafety committees of CHLA. NEC was induced in newborn rats by formula feeding/hypoxia [14]. Briefly, timed pregnant rats were purchased from (Harlan, Indianapolis, IN) and induced with oxytocin at term. Six experimental groups were used. A: Twice daily feeding of 0.2 ml clean Esbilac formula (FF); B: Similar feeding protocol to group A, and three times (~5min each) daily hypoxia exposure with 5%O2, 95%N2. (FF+H); C: Once daily feeding of 0.2 ml clean Esbilac formula, and once daily feed of 0.2 ml formula containing a known quantity of ES (FF+ES); D: Similar feeding to group C, and three times daily hypoxia (FF+H+ES); E and F are similar to that of groups C and D, except that ES was replaced with EC and designated as (FF+EC) and (FF+H+EC), respectively. NEC was graded microscopically by a pathologist blinded to groups, from grade 0 (normal) to grade 4 (severe), based on pathologic manifestations including submucosal edema, epithelial sloughing/obliteration, neutrophil infiltration, intestinal perforation, and necrosis [14].
4. Binding and invasion assays using IEC-6 cells
Various doses (104 to 107 cfu/ml) of ES were added to confluent monolayers of IEC-6 cells separately and incubated for 2–12 h. Media was changed every 1 h (after two-hour incubation) to limit bacterial multiplication. The cells were washed with HBSS medium. Monolayers were solubilized with 0.01% Triton-X 100 for 8 min. The contents were removed, serial dilutions made, plated onto blood agar, and incubated at 37°C overnight. The number of bacterial colonies was recorded and the percent binding calculated based on inoculum size. To determine the invasion of ES into IEC-6 cells, the experiments were performed as described for binding except that the cells were incubated with gentamicin 75 μg/ml for 60 min after completion of initial incubation to kill extracellular bacteria.
5. Cytokine profiling following exposure to ES
IEC-6 cells were grown to confluence and incubated with ES (107 cfu/ml) for 1–6 h. The experimental medium along with bacteria were collected and spun twice at 3, 000 rpm for 10 min to collect the supernatants. Cytokine concentrations in various samples were measured using the Luminex assay [15]. Total RNA was extracted from the infected cells using the RNeasy kit (Qiagen). RT-PCR was performed using equal amounts of cDNA for 30 cycles for IL-6 using forward primer: 5′-GAT GTT GTT GAC AGC CAC TGC CTT-3′ and reverse primer: 5′-TTG GAT GGT CTT GGT CCT TAG CCA-3′.
6. Evaluation of apoptosis in vitro and of intestine in rat pups after ES exposure
IEC-6 cells, grown to 90% confluence in 4-well chamber slides, were infected for varied periods with ES, washed, and fixed with 1% paraformaldehyde. The cells were then stained using the ApopTag Red in situ apoptosis detection kit (Chemicon, Temecula, CA) along with DAPI and visualized using a fluorescent microscope. Additionally, apoptosis of IEC-6 cells infected with ES was assessed by carboxyfluorescein caspase-3 detection kit (ApoLogix, Erie, PA). The number of apoptotic cells per high-powered field was quantified using fluorescent microscopy. Intestinal segments were obtained, placed in 1% paraformaldehyde, and embedded in paraffin. Furthermore, cryosections were also obtained and stained with DAPI. Immunofluorescent microscopy was used to assess the association of GFP-ES with the intestinal epithelial layer. Apoptosis was assessed after staining both paraffin sections and cryosections with ApopTag Red.
7. Assessment of intestinal damage by electron microscopy
Segments of small intestine were obtained from pups subjected to FF+H or FF+H+ ES and washed three times in 0.1 M cacodylate buffer for 15 min each. The intestinal lumen was exposed, post-fixed with 2% osmium tetroxide in 0.1 M cacodylate buffer at 4°C for 1 h, and processed for scanning electron microscopy (SEM). The images were captured on FEI Quanta 200 ESEM under low vacuum conditions.
8. Statistical Analysis
Where appropriate, Man Whitney analysis or Student’s t-test were performed. P values less than or equal to 0.05 were considered significant.
RESULTS
Administration of ES enhances the severity of NEC in a rat model
Previous studies from our laboratory established a reproducible rat model of NEC [10]. In this model, the combination of formula-feeding and hypoxia leads to macro- and microscopic intestinal inflammation similar to human NEC. Findings include segmental small bowel necrosis, edema, neutrophil infiltration, epithelial sloughing, and intestinal perforation/sepsis [16]. Because ES has been implicated as a potential pathogen in human NEC, we investigated whether ES was capable of increasing disease severity in the rat model. A laboratory E. coli strain, DH5-α, was used as a control. The appropriate dose of ES was determined by measuring mortality rates in pups fed formula containing 103, 105, and 107 cfu/dose. Formula containing 103 cfu/dose with hypoxia did not produce a significant increase in disease severity when compared with controls, whereas 107 cfu/dose conferred greater than 95% mortality in rat pups by experimental day 4 (data not shown). However, 105 cfu/dose of ES added to the formula produces clinical symptoms consistent with Grade 3 NEC in the rat pup model by four days (Figure 1A). The rat pups subjected to FF+H+ES developed marked abdominal distention, and abdominal wall discoloration when compared with animals subjected to FF+H (Fig. 1A–D). Abdominal discoloration and distention are commonly found in severe human disease. The FF+H group animals were associated with a mortality of 40%, whereas FF+H+ES subjected pups had 70% mortality. The control group, FF+EC and FF+H+EC, animals did not exhibit a greater mortality than groups FF and FF+H (similar to Fig. 1D). Furthermore, all pups treated with ES were bacteremic at completion of the experiment suggesting systemic disease and loss of intestinal barrier integrity, whereas no bacteremia was detected in the non-ES or EC fed groups (results not shown).
Fig. 1. ES induces increased intestinal injury in the rat model of NEC.

Panel A compares a FF+H+ES (left) treated rat pup with a FF+H treated rat pup (right). The FF+H+ES rat demonstrates increased abdominal girth, abdominal wall discoloration and evidence of clinical peritonitis. The FF+H rat is not distended and only a milk spot is visible through a normal abdominal wall. Panels B and C demonstrate the gross intestinal morphology after four days of FF+H+ES treatment. The intestine has patchy necrosis, with evidence of hemorrhagic intetsine and pneumatosis intestinalis. The controal rat pup has a normal appearing intestine without necrosis, and only stool visible within the lumen (Panel D).
Histological examination of intestine from day-4 control FF-animals revealed intact villus architecture (Fig. 2A), whereas villus tip sloughing and blunting was observed in both FF+H and FF+ES pups (Fig. 2B and C). In contrast, histology grading of FF+H+ES pups revealed greater intestinal injury and inflammation when compared with FF+H animals (p = 0.02, Fig. 2D). Pups subjected to FF+H+EC or FF+EC showed no significant intestinal damage when compared to controls (Fig. E and F). Although, supplementation of ES increased intestinal injury, no statistically significant difference was observed with ES-feeding alone (p = 0.078 when comparing FF+H)(Fig. 2G). Rat pups fed with EC demonstrated no significant increase in pathology scoring from baseline. ES-treated groups had more cellular infiltrates, demonstrated by a larger number of neutrophils within the body of the villus and epithelial sloughing.
Fig. 2. Histology of intestine treated with or without ES.
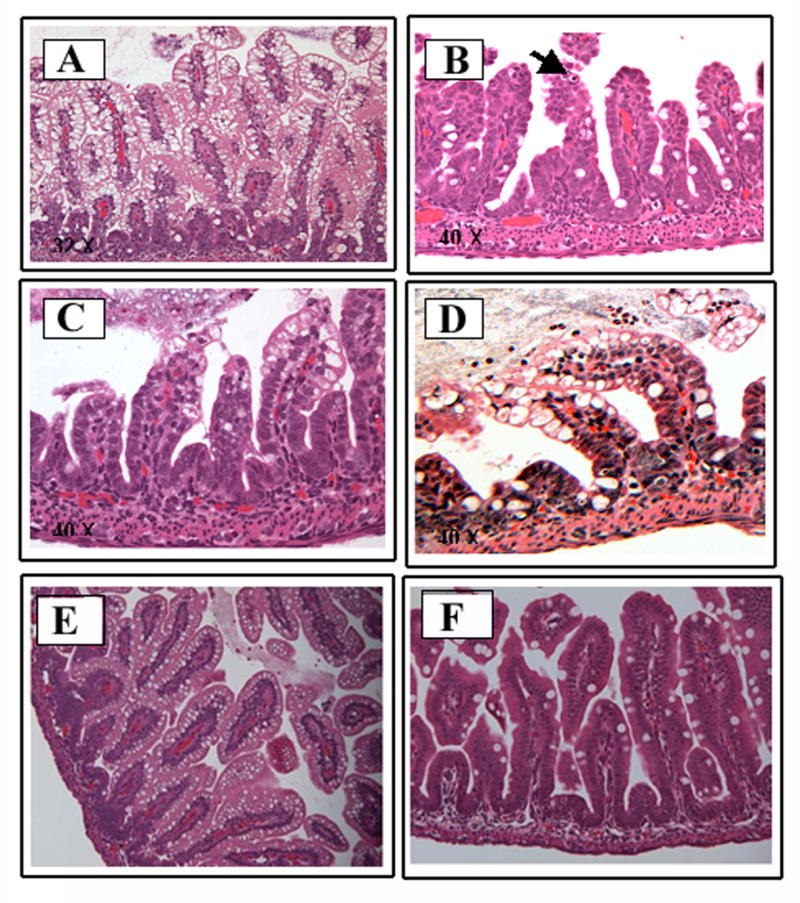

The parafin embedded intestine sections from each rat group are representative sections from several experiments. The sections were stained with hematoxylin and eosin at day 4. Panel A shows the preserved architecture of villi after four days of FF treatment. Panel B reveals neutrophils in the villus tip (see arrow) and alteration in villus structure in group FF+ES. Panel C demonstrates villus tip sloughing and villus blunting seen in a rat pup after four days of FF+H treatment. Panel D demonstrates the greatest degree of derrangement with luminal neutrophils, loss of normal villus architecture, and sloughing in rat pups treated with FF+H+ES. Panel E shows the preserved architecture in a rat pup subjected to FF+EC treatment whereas Panel F only demonstrates mild goblet cell hyperplasia in the setting of FF+H+EC. Histologic scoring of FF+H and FF+H+ES subjected rat pup intestine sections, which is statistically significant (p = 0.020), was shown in G. The difference between groups FF and group FF+H+ES is also statistically significant (p = 0.027).
Electron microscopy of intestine revealed significant damage to villi in ES treated rat pups
To further observe the morphological changes in villi architecture after ES-infection, SEM was performed. As shown in Fig. 3A and B, FF+H intestine showed intact villi, without a significant number of attached bacteria. In contrast, ES feeding of rat pups greatly increases intestinal injury and villus disruption (Fig. 3D). A large number of bacteria were attached to the infected villi with enterocyte blebbing and formation of gaps in the epithelium (Fig. 3C and E). Higher magnification photographs showed that bacteria were present at the site of bleb rupture (Fig. 3F), suggesting that ES directly damages the epithelial cells of villi tips.
Fig. 3. Scanning electron microscopy of rat intestine with or without infection with ES.

Rat pups were subjected to FF+H treatment without (A and B) or with ES (C to F) as described in Methods. Small pieces of intestine samples were prepared for SEM. Bacteria are shown by arrows. Magnification: A. ~300X; B. 3400X; C. 5000X; D. 580X E. 3200X; and F. 9600X.
ES adheres to IEC-6 cells in vitro and to enterocytes in newborn rats
Infection of rat pups show that ES interacts and damages intestinal epithelial cells, and this could be responsible for the onset of NEC. To understand the mechanisms involved in ES-induced intestinal epithelial cell damage, we developed an in vitro model of ES-infection using IEC-6 cells in culture. Initially, we determined the effect of inoculum size on ES-binding to IEC-6 cells after 2 h of infection. ES bound to IEC-6 intestinal cells at a frequency of 0.5% at an inoculum of 104 cfu/well and increased to 2.5% when 107 cfu/well bacteria were added (Fig. 4A). Subsequent increases in inoculum sizes did not result in higher binding. Of note, the invasion of ES into IEC-6 cells was very low at a frequency of 0.01%, suggesting that ES could not efficiently invade intestinal epithelial cells. Since we observed optimal binding and invasion with 107 cfu, time course experiments were performed with 107 cfu over a period of 6h. As shown in Fig. 4B, the binding of ES to IEC-6 increased significantly by four-fold at 6 h when compared to 2 h post-infection, after which time the number of ES bound to the cells reached a plateau. However, the invasion was increased by only two-fold (0.01% at 2 h versus 0.02% at 6 h). Light microscopy analysis of ES-binding to IEC-6 cells revealed that the bacteria bound diffusively to enterocytes, which was similar to the pattern seen in SEM (Fig. 4C). To examine whether ES binds to enterocytes in vivo, we examined cryosections of intestine obtained from GFP-ES-fed rat pups. The sections were stained with DAPI to view villus structure. GFP-ES was associated mostly with the villus surface in FF+H+ES treated rats (Fig. 4C). Interestingly, the association was seen only in the distal small intestine when compared to proximal areas, suggesting that there may be specific receptors expressed in the distal small intestine for ES binding. Notably, distal small intestine is the intestinal region most frequently affected in both human and experimental NEC [14].
Fig. 4. Binding to and invasion of ES into IEC-6 cells.

Confluent monolayers of IEC-6 cells were infected either with different inoculum sizes (104 to 107 cfu/well) of ES for 2 h (A) or with an inoculum size of 107 cfu/well for varying periods (B). Total cell associated (binding) or total intracellular bacteria (invasion) were determined as described in Methods. The experiments were performed at least five times and the data represent an average ± SD and expressed as relative binding or invasion taking either 6 h treatment with ES or 107 inoculum of ES as 100%. In some experiments, ES infected IEC-6 cells were stained with DAPI and observed the bacteria using transmitted light. Similarly, rat pups were infected with GFP-labeled ES to establish in vivo epithelial association of ES. Cryosections of intestines were stained with DAPI and examined with a fluorescence microscope (C).
ES induces apoptosis in both IEC-6 cells and enterocytes in newborn rats
SEM studies demonstrated that administration of ES caused damage to the tips of villi, suggesting that the bacterium might kill the cells it attaches to. To examine whether ES interaction with IEC-6 cells induces apoptosis, IEC-6 cells were stained with ApoTag after incubating the cells with 107 cfu/well for 0–12 h. This method identifies apoptotic cells by staining fragmented DNA, which is late stage in apoptosis. The number of apoptotic IEC-6 cells increased with increasing time of ES infection (Fig. 5A). In addition, caspase-3 activation, an early marker of apoptosis was also examined. Quantification of apoptotic cells suggested that apoptosis reached statistical significance by 4 h, and appeared to plateau at 6 h post-infection, at which point 70% of IEC-6 demonstrated signs of apoptosis (Fig. 5B, p < 0.001). Furthermore, staining of GFP-ES infected hypoxic rat pup intestinal segments with ApoTag kit revealed a greater degree of apoptosis in enterocytes when compared to controls (Fig. 5C). Apoptosis is mainly at the tip in the control sample, whereas it is everywhere in the villus epithelium upon infection with ES. Apoptotic changes were almost entirely absent in the FF group (data not shown). These results indicate that ES induces apoptosis in intestinal epithelial cells both in vitro and in vivo.
Fig. 5. Induction of apoptosis in intestinal epithelial cells by ES.

IEC-6 cells were grown to 90% confluence in eight-well chamber slides and incubated with ES for varying periods. The cells were washed, fixed, and treated with TUNEL-RED reagent, and viewed under a fluorescence microscope (A). In separate experiments, infected cells were washed, released from plates with medium containing EGTA/1% BSA, stained with TUNEL-RED kit, and the number of apoptotic cells were counted (B). In addition, cryosections of intestines from rat pups subjected to FF+H or FF+H+ES treatment were also stained with DAPI and Apo Logix kit (C).
Induction of pro-inflammatory cytokine response in intestinal epithelial cells by ES
Cytokines activate local and systemic inflammatory responses that may contribute to the pathogenesis of NEC. It is possible that ES increases NEC by stimulating epithelial inflammatory cytokine expression. To examine the production of cytokines in intestinal epithelial cells we collected supernatants from IEC-6 cells treated with ES and assayed for cytokine production. Exposure to ES leads to a time-dependent increase in IL-6 production after 2 h of infection, when compared with FF+H treated controls (p = 0.018) (Fig. 6A). Of note, IL-1α was also detected after 2 h of infection however, there was no apparent correlation with ES dose (data not shown). Other cytokines including interferon-γ, tumor necrosis factor-α, IL-2, IL-4, IL-10 and IL-12 were also assayed but failed to meet detectable levels. IL-6 production reached maximal levels within 12 h of post-infection. In addition, the expression of IL-6 was also determined at mRNA level by RT-PCR. ES-treated IEC-6 cells showed elevated levels of IL-6 mRNA expression after 60 min (Fig. 6B). RPS-17, which was used as a control showed no significant difference between various time points. We further assessed whether ES induces IL-6 production in neonatal rats. The animals were subjected to FF+H+ES, and serum samples were collected to evaluate IL-6 levels. In agreement with IEC-6 cells data, rat pups that received FF+H+ES had higher levels of IL-6 following four days of treatment when compared to animals subjected to FF+H treatment alone (Fig. 6C). Taken together, these data suggest that IL-6 may be an important inducible cytokine in the pathogenesis of ES-induced NEC.
Fig. 6. Expression of IL-6 in IEC-6 cells and in rat pups infected with ES.
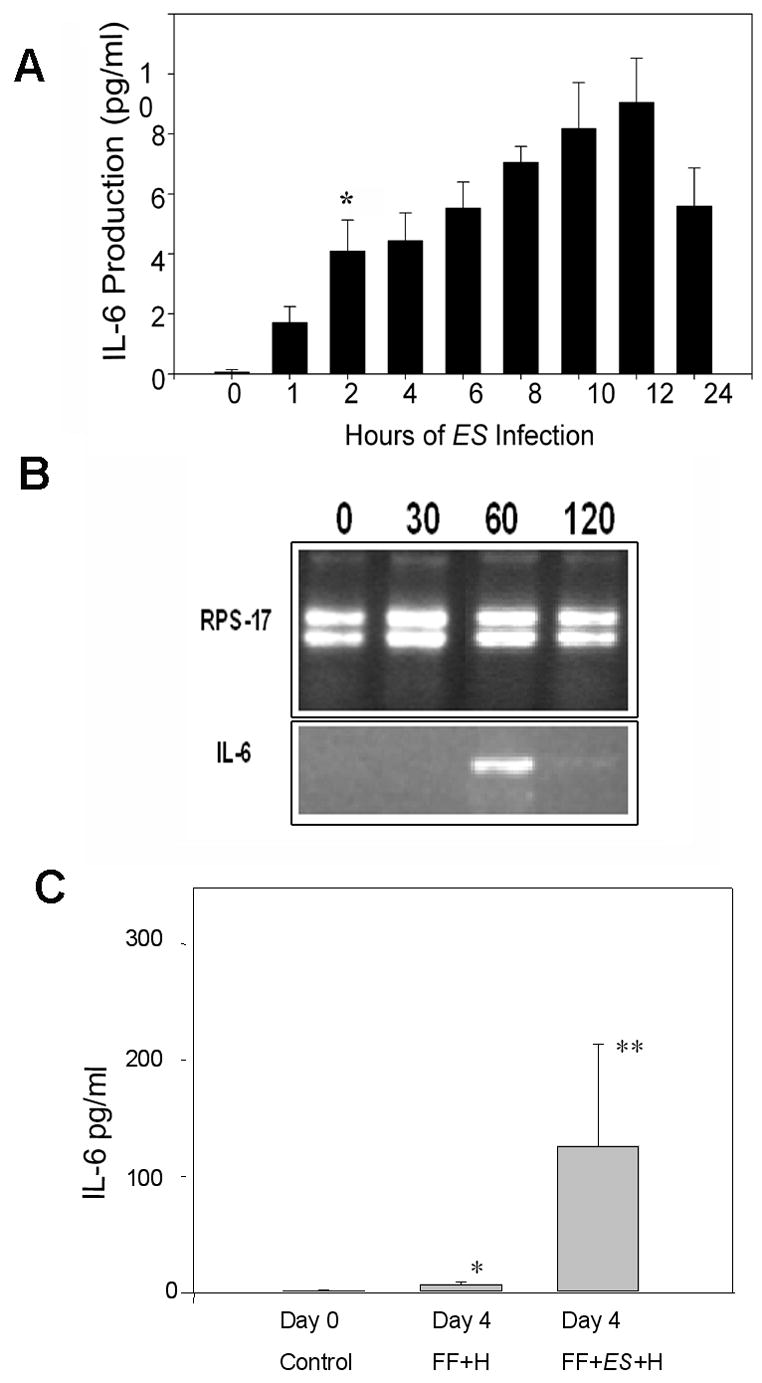
Confluent monolayers of IEC-6 cells were infected with ES for varying periods and the cytokines present in the supernatants were determined by Luminex assay. Results are representative of three independent experiments performed in triplicate (A). Total RNA was extracted from the cells and RT-PCR was performed for 30 cycles on normalized cDNA using IL-6 specific primers. RPS-17 primers were used as internal controls (B). In addition, serum samples were collected from day 4 rat pups subjected to various treatments and IL-6 production was determined by Luminex assay. The production of IL-6 from animals subjected to FF+H+ES treatment is significantly greater than the controls pups (*p<0.05 and **p<0.001).
DISCUSSION
Although bacterial colonization is broadly regarded as a key step in the pathogenesis of NEC, there is paucity of information regarding the roles of individual bacterial species. Several reports have suggested that NEC may be associated with ES; a common contaminant of infant formula [1, 17, 18]. Here, we demonstrated for the first time that ES is capable of inducing the clinical and morphologic changes of NEC in a rat pup model. Infection with ES was confirmed in all surviving rat pups, as evidenced by the association of GFP-labeled ES within the intestine and in cultures of the intestinal contents of pups fed with ES (results not shown). The enhanced pathogenesis of ES on the compromised intestine shown here has particular relevance to the newborn infants who develop NEC. Premature infants exhibit a relative intestinal immune deficiency such as decreased production of local anti-bacterial products [19], decreased immunoglobulin levels such as IgA [20], and altered immune cell production [21], all of which may contribute to a compromised intestinal epithelial layer. In addition a subgroup of newborn infants at increased risk for developing NEC have underlying congenital defects, such as pulmonary or cardiac anomalies [22]. These defects may result in global hypoxia and thus lead to intestinal hypoxia. Intestinal epithelial hypoxia has been previously shown to diminish epithelial barrier integrity, increase intestinal permeability and allow for increased bacterial translocation [23]. Therefore in accordance with our animal findings, the effects of ES may be especially enhanced in the premature or hypoxic newborn.
A primary function of the intestine is to protect the internal environment from the external world, and regulation of enterocyte apoptosis is crucial to healthy maintenance of the gut barrier [24]. In fact, enterocytes are amongst the most rapidly proliferating cells in the body, and mitotic activity is matched by apoptosis, to maintain normal form and function. Dysregulated enterocyte apoptosis has been associated with several intestinal disorders [25]. Moreover, certain pathogens are capable of increasing epithelial apoptosis, thereby contributing to disease pathogenesis [26]. However, when there is an imbalance between normal rates of apoptosis and restitution, this may result in decreased barrier integrity allowing for bacterial translocation and the development of systemic sepsis. In agreement with this concept, our studies demonstrate that ES induces significant apoptosis in IEC-6 cells in vitro and in an animal model of NEC, particularly in combination with hypoxia. However, non-pathogenic E. coli did not increase disease severity indicating some specific traits of ES are responsible for the pathogenesis. Although the impact of enterocyte apoptosis on intestinal barrier integrity was not specifically addressed in our studies at this point, other investigators have clearly documented a direct relationship between increased enterocyte apoptosis and the loss of barrier integrity [24]. Our SEM studies revealed intestinal epithelial disarray, blebbing and holes in the intestinal mucosa in FF+H+ES rat pups, suggesting a necrotic pattern. Therefore, induction of apoptosis in NEC by bacterial insult or by local inflammatory cytokines may progress to necrosis after prolong infection resulting in loss of intestinal barrier integrity. Furthermore, all rat pups subjected to FF+H+ES treatment developed bacteremia after four days, suggesting that intestinal translocation of bacteria occurred [27]. Some pathogens are able to enter the systemic circulation through direct invasion of the intestinal epithelium [28]. However, the absence of invasion in our in vitro studies leads us to speculate that ES induces intestinal damage by binding to the external surface of enterocytes and inducing apoptosis/necrosis. However, the precise mechanisms of ES interaction with intestinal epithelial cells, requires further elucidation.
Pathogen-associated molecular pattern (PAMP) molecules, including LPS, lipoproteins, glycolipids, peptidoglycans, fatty acids, and nucleic acids of bacteria often interact with various pattern recognition receptors known as toll-like receptors (TLRs) [29]. Different TLR family members have been identified and are expressed in a variety of cell types including macrophages, dendritic cells, and enterocytes [30]. Since LPS is an outer membrane virulence factor of ES, it is possible that ES interacts with enterocytes through LPS mediated binding to TLR4. Previous studies have demonstrated that LPS facilitates bacterial attachment to Caco-2 cells and increases bacterial translocation [31]. Similarly, we have previously demonstrated that IEC-6 cells express TLR4 [32]. TLR expression may vary based on intestinal location and with postnatal maturation, thereby providing a greater opportunity for ES to bind to certain portions of the intestine and alter the host’s response to commensal or pathogenic microorganisms. Indeed, in mouse models of colitis and NEC, TLR-4 knockout mice have reduced inflammatory infiltrates have been detected when compared to wild-type mice [33]. Studies are in progress to identify the importance of TLRs in ES intestinal epithelial cells injury. Alterations in cytokine production, including elevated levels of IL-6 have been associated with human NEC, and may be useful clinical markers of disease severity [11, 34, 35]. Our studies revealed that IL-6 expression and production were elevated in intestinal epithelial cell cultures following exposure to ES. Increased levels of IL-6 were found in the serum of rat pups following ES-formula feeding, however it’s role in ES-pathogenesis remains to be defined. Thus, the capability of ES to trigger apoptosis and initiate a pro-inflammatory response may be fundamental to the pathogenesis of ES-induced NEC.
In summary, oral administration of ES increases epithelial injury in experimental NEC. Our study supports the notion that ES might be a potential pathogen associated with human NEC. Furthermore, ES likely elicits epithelial damage through enterocyte association without direct invasion. ES may cause barrier damage by inducing enterocyte apoptosis, and by stimulating expression of the inflammatory cytokine IL-6. Further understanding the role of ES in the pathogenesis of NEC will provide clues for developing new therapeutic strategies to prevent this disease.
Acknowledgments
This research was supported in part by NIH/NIAID grants AI 40567 (NVP) and AI 49473 (HRF); and a research fellowship award from the Surgical Infection Society (CJH).
We are grateful for the technical assistance of Xioru Zhang and Jin Wang. We wish to thank John Hardy, City of Hope, Duarte, CA for his assistance with electron microscopy.
Footnotes
The authors do not have a commercial or other association that might pose a conflict of interest.
Part of the information contained herein was presented at the Surgical Infection Society, April 2007, Toronto, Ontario, Canada.
References
- 1.Mullane NR, Iversen C, Healy B, et al. Enterobacter sakazakii an emerging bacterial pathogen with implications for infant health. Minerva Pediatr. 2007;59:137–48. [PubMed] [Google Scholar]
- 2.Biering G, Karlsson S, Clark NC, Jonsdottir KE, Ludvigsson P, Steingrimsson O. Three cases of neonatal meningitis caused by Enterobacter sakazakii in powdered milk. J Clin Microbiol. 1989;27:2054–6. doi: 10.1128/jcm.27.9.2054-2056.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simmons BP, Gelfand MS, Haas M, Metts L, Ferguson J. Enterobacter sakazakii infections in neonates associated with intrinsic contamination of a powdered infant formula. Infect Control Hosp Epidemiol. 1989;10:398–401. doi: 10.1086/646060. [DOI] [PubMed] [Google Scholar]
- 4.Kandhai MC, Reij MW, Gorris LG, Guillaume-Gentil O, van Schothorst M. Occurrence of Enterobacter sakazakii in food production environments and households. Lancet. 2004;363:39–40. doi: 10.1016/S0140-6736(03)15169-0. [DOI] [PubMed] [Google Scholar]
- 5.Peter CS, Feuerhahn M, Bohnhorst B, et al. Necrotising enterocolitis: is there a relationship to specific pathogens? Eur J Pediatr. 1999;158:67–70. doi: 10.1007/s004310051012. [DOI] [PubMed] [Google Scholar]
- 6.Millar MR, MacKay P, Levene M, Langdale V, Martin C. Enterobacteriaceae and neonatal necrotising enterocolitis. Arch Dis Child. 1992;67:53–6. doi: 10.1136/adc.67.1_spec_no.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powell J, Bureau MA, Pare C, Gaildry ML, Cabana D, Patriquin H. Necrotizing enterocolitis. Epidemic following an outbreak of Enterobacter cloacae type 3305573 in a neonatal intensive care unit. Am J Dis Child. 1980;134:1152–4. [PubMed] [Google Scholar]
- 8.Klemm P, Schembri MA. Bacterial adhesins: function and structure. Int J Med Microbiol. 2000;290:27–35. doi: 10.1016/S1438-4221(00)80102-2. [DOI] [PubMed] [Google Scholar]
- 9.Mange JP, Stephan R, Borel N, et al. Adhesive properties of Enterobacter sakazakii to human epithelial and brain microvascular endothelial cells. BMC Microbiol. 2006;6:58. doi: 10.1186/1471-2180-6-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ford H, Watkins S, Reblock K, Rowe M. The role of inflammatory cytokines and nitric oxide in the pathogenesis of necrotizing enterocolitis. J Pediatr Surg. 1997;32:275–82. doi: 10.1016/s0022-3468(97)90194-9. [DOI] [PubMed] [Google Scholar]
- 11.Duffy LC, Zielezny MA, Carrion V, et al. Concordance of bacterial cultures with endotoxin and interleukin-6 in necrotizing enterocolitis. Dig Dis Sci. 1997;42:359–65. doi: 10.1023/a:1018826204819. [DOI] [PubMed] [Google Scholar]
- 12.Levine AD. Apoptosis: implications for inflammatory bowel disease. Inflamm Bowel Dis. 2000;6:191–205. doi: 10.1097/00054725-200008000-00006. [DOI] [PubMed] [Google Scholar]
- 13.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–5. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 14.Nadler EP, Dickinson E, Knisely A, et al. Expression of inducible nitric oxide synthase and interleukin-12 in experimental necrotizing enterocolitis. J Surg Res. 2000;92:71–7. doi: 10.1006/jsre.2000.5877. [DOI] [PubMed] [Google Scholar]
- 15.Seideman J, Peritt D. A novel monoclonal antibody screening method using the Luminex-100 microsphere system. J Immunol Methods. 2002;267:165–71. doi: 10.1016/s0022-1759(02)00168-0. [DOI] [PubMed] [Google Scholar]
- 16.Musemeche C, Caplan M, Hsueh W, Sun X, Kelly A. Experimental necrotizing enterocolitis: the role of polymorphonuclear neutrophils. J Pediatr Surg. 1991;26:1047–9. doi: 10.1016/0022-3468(91)90671-f. discussion 1049–50. [DOI] [PubMed] [Google Scholar]
- 17.van Acker J, de Smet F, Muyldermans G, Bougatef A, Naessens A, Lauwers S. Outbreak of necrotizing enterocolitis associated with Enterobacter sakazakii in powdered milk formula. J Clin Microbiol. 2001;39:293–7. doi: 10.1128/JCM.39.1.293-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drudy D, Mullane NR, Quinn T, Wall PG, Fanning S. Enterobacter sakazakii: an emerging pathogen in powdered infant formula. Clin Infect Dis. 2006;42:996–1002. doi: 10.1086/501019. [DOI] [PubMed] [Google Scholar]
- 19.Salzman NH, Polin RA, Harris MC, et al. Enteric defensin expression in necrotizing enterocolitis. Pediatr Res. 1998;44:20–6. doi: 10.1203/00006450-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Mayer L. Mucosal immunity. Pediatrics. 2003;111:1595–600. [PubMed] [Google Scholar]
- 21.Hannet I, Erkeller-Yuksel F, Lydyard P, Deneys V, DeBruyere M. Developmental and maturational changes in human blood lymphocyte subpopulations. Immunol Today. 1992;13:215–218. doi: 10.1016/0167-5699(92)90157-3. [DOI] [PubMed] [Google Scholar]
- 22.Bolisetty S, Lui K, Oei J, Wojtulewicz J. A regional study of underlying congenital diseases in term neonates with necrotizing enterocolitis. Acta Paediatr. 2000;89:1226–30. doi: 10.1080/080352500750027619. [DOI] [PubMed] [Google Scholar]
- 23.Xu DZ, Lu Q, Kubicka R, Deitch EA. The effect of hypoxia/reoxygenation on the cellular function of intestinal epithelial cells. J Trauma. 1999;46:280–5. doi: 10.1097/00005373-199902000-00014. [DOI] [PubMed] [Google Scholar]
- 24.Sun Z, Wang X, Wallen R, et al. The influence of apoptosis on intestinal barrier integrity in rats. Scand J Gastroenterol. 1998;33:415–22. doi: 10.1080/00365529850171053. [DOI] [PubMed] [Google Scholar]
- 25.Brunner T, Mueller C. Apoptosis in disease: about shortage and excess. Essays Biochem. 2003;39:119–30. doi: 10.1042/bse0390119. [DOI] [PubMed] [Google Scholar]
- 26.Zychlinsky A, Sansonetti P. Perspectives series: host/pathogen interactions. Apoptosis in bacterial pathogenesis. J Clin Invest. 1997;100:493–5. doi: 10.1172/JCI119557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porras M, Martin MT, Yang PC, Jury J, Perdue MH, Vergara P. Correlation between cyclical epithelial barrier dysfunction and bacterial translocation in the relapses of intestinal inflammation. Inflamm Bowel Dis. 2006;12:843–52. doi: 10.1097/01.mib.0000231571.88806.62. [DOI] [PubMed] [Google Scholar]
- 28.Kim BY, Kang J, Kim KS. Invasion processes of pathogenic Escherichia coli. Int J Med Microbiol. 2005;295:463–70. doi: 10.1016/j.ijmm.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 29.Takeda K, Akira S. Toll receptors and pathogen resistance. Cell Microbiol. 2003;5:143–53. doi: 10.1046/j.1462-5822.2003.00264.x. [DOI] [PubMed] [Google Scholar]
- 30.Takeda K. Evolution and integration of innate immune recognition systems: the Toll-like receptors. J Endotoxin Res. 2005;11:51–5. doi: 10.1179/096805105225006687. [DOI] [PubMed] [Google Scholar]
- 31.Deitch EA, Berg R, Specian R. Endotoxin promotes the translocation of bacteria from the gut. Arch Surg. 1987;122:185–90. doi: 10.1001/archsurg.1987.01400140067008. [DOI] [PubMed] [Google Scholar]
- 32.Neal MD, Leaphart C, Levy R, et al. Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J Immunol. 2006;176:3070–9. doi: 10.4049/jimmunol.176.5.3070. [DOI] [PubMed] [Google Scholar]
- 33.Fukata M, Michelsen KS, Eri R, et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1055–65. doi: 10.1152/ajpgi.00328.2004. [DOI] [PubMed] [Google Scholar]
- 34.Hsueh W, Caplan MS, Sun X, Tan X, MacKendrick W, Gonzalez-Crussi F. Platelet-activating factor, tumor necrosis factor, hypoxia and necrotizing enterocolitis. Acta Paediatr Suppl. 1994;396:11–7. doi: 10.1111/j.1651-2227.1994.tb13234.x. [DOI] [PubMed] [Google Scholar]
- 35.Morecroft JA, Spitz L, Hamilton PA, Holmes SJ. Plasma cytokine levels in necrotizing enterocolitis. Acta Paediatr Suppl. 1994;396:18–20. doi: 10.1111/j.1651-2227.1994.tb13235.x. [DOI] [PubMed] [Google Scholar]