

Abstract
We describe the design, synthesis, and characterization of a family of thiol-reactive optical switches for labeling proteins and other biomolecules. Site-selective introduction of photochromic probes within biomolecules is being used as part of a new approach for optical control of biomolecular interactions and activities within cells. The thiol-reactive photochromic probes described in this report include a spironaphthoxazine and five spirobenzopyrans. The location of the thiol-reactive group on the spirobenzopyran is different for each probe; this feature can be used to control the geometry of the optical switch within a bioconjugate. The photochromes undergo rapid and reversible, optically driven transitions between a colorless spiro (SP) state and a brightly colored merocyanine (MC) state. The MC absorption of a spironaphthoxazine conjugate is red shifted by more than 100 nm compared to the equivalent spirobenzopyran, which may be exploited for the independent control of the MC to SP transition for up to two different spironaphthoxazine and spirobenzopyran conjugates within the same sample.
Introduction
The increasing need to understand the molecular basis of cell behavior and physiology is driving the development of new optical probes and microscope techniques to study the interactions and regulation of specific proteins within a cell.1–3 A necessary requirement for these studies is the ability to selectively perturb the interactions or activity of a specific protein in a cell against a background of up to 30 000 other proteins. To this end, we introduced the technique of light-directed activation of “caged” proteins, which has been applied to study protein function within complex molecular and cellular systems.3–7 However, this approach is limited by the photochemistry of the 2-nitrophenyl (caged) group, which functions as a one-way, single-use optical switch. We are improving this technique through the development of new optical switch reagents whose interactions and activities within a bioconjugate can be rapidly, reversibly, and efficiently modulated with light.
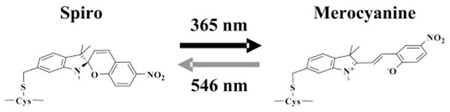
This new approach exploits the properties of photochromic probes that undergo rapid and reversible, light-directed transitions between a colorless, weakly polarized spiro (SP) state and a colorful and highly polarized merocyanine (MC) state8–14 (schematized in Figure 1): excitation of SP at 365 nm light generates MC, while excitation of MC at 546 nm generates SP. These optical switches must be specifically labeled to a biomolecule such that the MC state, but not the SP state, competes against a functional ligand for specific interactions within the conjugate. Optical switching between the MC to SP states using cycles of 365 and 546 nm light is then used to reversibly control the MC–SP states and their interactions within the conjugate and thereby modulate its function.
FIGURE 1.
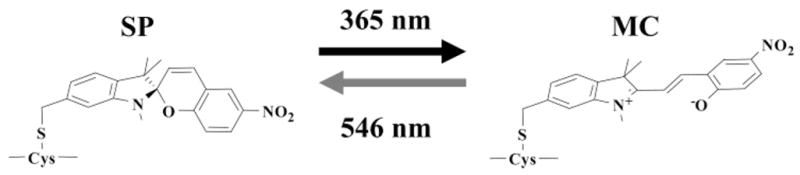
Schematic representation of the reversible optical switching between the SP and MC states of a spirobenzopyran-linked probe (13) to a cysteine residue in a protein.
In this report we describe a family of photochromic probes that harbor maleimide, bromomethyl, or iodo-methyl groups for selective labeling of thiol groups within biomolecules. Previous studies have described photochromic reagents that react with the amino group of the lysine residue in proteins;10,12,13 however, the multiple and randomly labeleld photochromes in these protein conjugates exhibit complex photochemistry and are capable of forming dimers that have different optical properties compared to the monomer and may exhibit irreversible photochemistry.11,13
Results and Discussion
Syntheses for the six thiol-reactive optical switches (3, 6, 9, 12, 13, 17) are summarized in Schemes 1–3. Combinatorial in design, the synthetic approach involves coupling members of a family of indoline derivatives (1, 4, 7, 10) with specific nitrosalicylaldehydes or nitrosonaphthol to yield the four key spirobenzopyrans (2, 5, 8, 11)15,16 or spironaphthoxazin (16).15 Indoline 1 is commercially available (Aldrich). Indoline 4 is prepared from commercially available 2,3,3-trimethyl-3H-indole in two steps. Indolines 7 and 10 are synthesized from compounds 14 and 15, which are generated as a mixture and separated by chromatography, according to Scheme 2. The thiol-reactive spirobenzopyrans (3, 6, 9, 12, 13) are prepared from the corresponding spirobenzopyrans via the halogen exchange reaction, bromination of alcohol, or modified Mitsunobu reaction for the maleimides.17 The yields for the conversion to the thiol-reactive probes are 15% (6), 60% (9), 24% (12), and 55% (13). Compound 3 is easily prepared from commercially available 1 and 3-chloromethyl-5-nitrosalicylaldehyde through compound 2 and is purified only at the final step with a yield of 78%.
SCHEME 1. Reaction Scheme Employed for the Synthesis of the Five Thiol-Reactive Spirobenzopyrans (3, 6, 9, 12, and 13) Described in This Papera.

a Reagents: (a) 3-chloromethyl-5-nitrosalicylaldehyde, THF; (b) NaI, acetone; (c) 5-nitrosalicylaldehyde, EtOH; (d) PPh3, DIAD, maleimide, neopentyl alcohol, THF; (e) PPh3, CBr4, THF.
SCHEME 3. Reaction Scheme Employed for the Synthesis of the Thiol-Reactive Spironaphthoxazine (17)a.
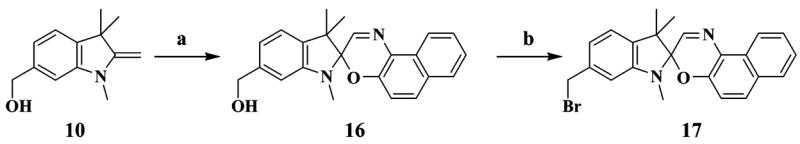
a Reagents: (a) 1-nitroso-2-naphthol, EtOH; (b) PPh3, CBr4, THF.
SCHEME 2. Preparation of Indoline Derivativesa.
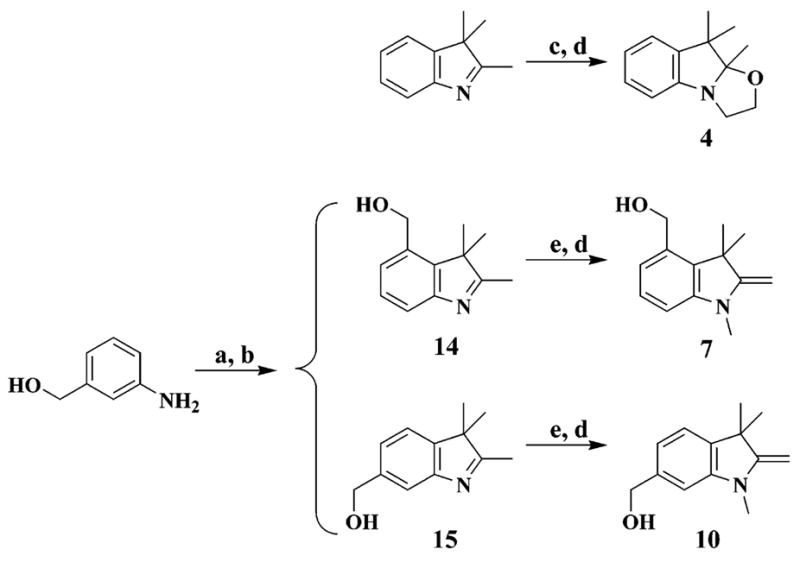
a Reagents: (a) NaNO2, SnCl2·H2O, HCl; (b) 3-methyl-2-butanone, H2SO4, EtOH; (c) 2-iodoethanol, CH3CN; (d) KOH, H2O; (e) MeI, CH2Cl2.
The thiol-reactive spironaphthoxazine (17) is prepared via a same method as compound 13 with a yield of 15% (Scheme 3).
The six optical switch reagents efficiently label cysteine residues in proteins; in this study we describe spirobenzopyran conjugates of BSA and the spironaphthoxazine conjugate of G-actin. BSA is often used as a model protein to test new thiol-reactive probes21 (although BSA harbors 35 cysteine residues, all but Cys-34 engage in disulfide bond formation);22 consequently, most studies show that BSA can only be labeled with a single thiol-reactive probe at residue Cys-34.21,23,24
The MC state of the five spirobenzopyrans has a ground-state dipole moment of 20 D.18,19 To the best of our knowledge, this is the largest dipole moment of all commonly used optical probes; we suggest that this property underlies the strong dipolar interactions between the MC probe and polar groups on the protein. The MC absorption spectra of the five spirobenzopyrans BSA conjugates are shown in Figure 2A and the spironaphthoxazine–G-actin conjugate is shown in Figure 2B. The most red-shifted MC–BSA conjugate is seen for compound 6 (Figure 2A, curve d; 544.5 nm), while the absorption maximum progresses to higher energy within the BSA conjugates of 12 (c, 534 nm); 9 (b, 520) nm; 3 (e, 515.5 nm); 13 (a, 503.5 nm). Since the five spirobenzopyran reagents harbor their thiol-reactive group at different sites on a common photochromic scaffold, the MC dipole moment should adopt a different geometry, and interactions, within each BSA conjugate, which is reflected in their absorption maxima. This demonstration satisfies an important criterion in our approach for optical switching of biomolecular function in cells; thus, in its most optimal form, optical switching will be realized when a specific and essential dipolar interaction between the conjugate and its binding partner is inhibited by a competing interaction involving the highly polarized MC state, but not by the weakly polar SP state.
FIGURE 2.
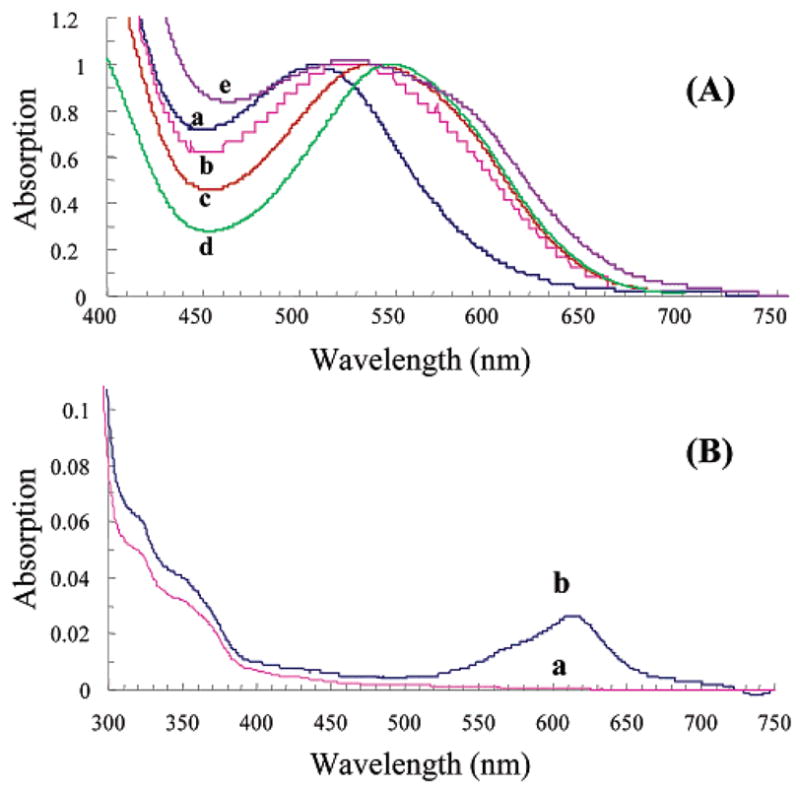
(A) Normalized absorption spectra for the lowest energy transition of the MC state of the five thiol-reactive probes attached to the reactive cysteine residue in BSA (Cys-34). The letters a–e refer to compounds 13, 9, 12, 6, and 3, respectively. (B) Absorption spectra of the spironaphthoxazine (17) labeled G-actin (Cys-374) in the SP-state (a) and MC state (b). The MC state was generated by irradiating a 50% glycerol solution of a with 365 nm light for 10 s.
A demonstration of optical switching between the SP and MC states of 17 in propylene glycol is shown in the absorption spectra in Figures 3B. While the SP states for 17 and the equivalent spirobenzopyran (13) exhibit similar absorption maxima (Figure 3A,B), the MC state of 17 is red-shifted by almost 100 nm at 614 nm compared to compound 13. A similar shift is found in the BSA conjugates of 13 (Figure 2A) and 17 (Figure 2B). The red shift is most likely due to the additional phenyl ring of 17. The demonstration that MC absorption spectra of spirobenzopyran and spironaphthoxazine conjugates exhibit large spectral shifts is significant because it provides an opportunity to independently control the MC states of spirobenzopyran and naphthoxazine conjugates within the same sample.
FIGURE 3.
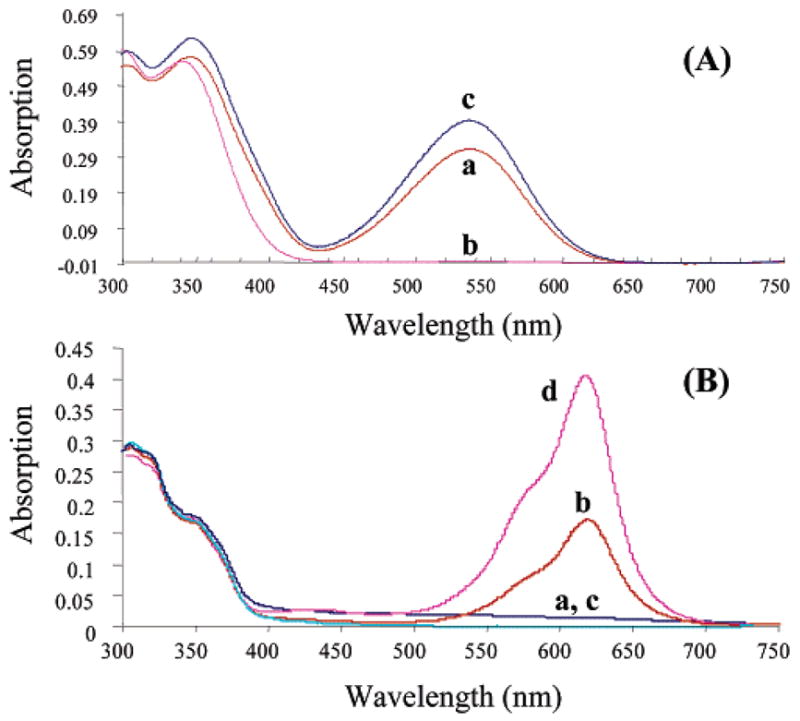
Absorption spectra of compound 13 (A) and 17 (B) in propylene glycol at 0 °C. (A) The ability to optically control the MC to SP states of spirobenzopyrans is shown in the spectrum of a 10 μM solution of 13 following a 10 s irradiation using 365 nm light (a), followed by 10 s of 546 nm light (b), and 10 s of 365 nm light (c). (B) The ability to optically control the MC to SP states of spironaphthozaxine (17) is shown in the spectrum of a 10 μM solution of 17 following a 10 s irradiation using 546 nm light (a), followed by 10 s of 365 nm light (b), 10 s of 546 nm light (c) and finally 10 s of 365 nm light (d). The difference in the absorption value for the MC state between switch cycles observed in Figure 3A is caused by slight differences in the method of illumination, while in Figure 3B, the different MC absorption values result from the rapid, thermally driven MC to SP transition (time constant of 6 s) and simply reflect a difference in the period between the UV illumination of the SP state and the absorption measurement.
Experimental Section
General Procedure for the Synthesis of Thiol-Reactive Optical Switches. 6′-(Hydroxymethyl)spirobenzopyran (11)
A solution of 6-(hydroxymethyl)-2,3,3-trimethyl-3H-indole (15) (50 mg, 0.27 mmol) and CH3I (100 ul, 1.6 mmol) in CH2Cl2 (2 mL) was refluxed for 12 h. The reaction mixture was filtered, and the filtrate was dissolved in 0.5 N NaOH and stirred for 15 min. After extraction with CH2Cl2, the extract was evaporated to afford oil 10 as a crude product. With 5-nitrosalicylaldehyde (50 mg, 0.30 mmol), 10 was refluxed in EtOH (2 mL) for 2 h, and then the reaction mixture was evaporated and subjected to column chromatography (silica gel; eluent, EtOAc) to afford 11 (70 mg, 75% based on 15): MS-(EI) 352 (M+, 10), 337 (4), 83 (100); HRMS(EI) M+352.1434 (calcd 352.1423); 1H NMR (CDC13) 1.19 (s, 3H), 1.30 (s, 3H), 2.76 (s, 3H), 4.70 (s, 2H), 5.87 (d, J = 10.5 Hz, 1H), 6.62 (s, 1H), 6.77 (d, J = 8.7 Hz, 1H), 6.94 (d, J = 10.5 Hz, 1H), 7.07 (d, J = 7.2 Hz, 1H), 8.01 (d, J = 2.4, 1H), 8.02 (dd, J = 2.4, 8.7 Hz, 1H).
6′-(Maleimidomethyl)spirobenzopyran (12)
To a dry THF (1 mL) solution of PPh3 (25 mg, 95 μmol), was added DIAD (18 ul, 95 μmol) over 2 min at − 78 °C, and the reaction mixture was stirred for 5 min. To this solution was added 11 (34 mg, 97 μmol) in dry THF (0.3 mL) over 2 min, and the mixture was stirred for 5 min. Neopentyl alcohol (4 mg, 4 μmol) and maleimide (9 mg, 9 μmol) were added sequentially to the reaction mixture as solids. After being stirred for 5 min, the reaction mixture was allowed to warm to rt and stirred for an additional 1 h. The reaction mixture was concentrated and then applied to preparative TLC (silica gel; hexane/EtOAc = 1:1) to afford 12 (10 mg, 24%): MS(EI) 431(M+, 10), 416 (3), 268 (8), 83 (100); HRMS(EI) M+ 431.1497 (calcd 431.1481); 1H NMR (CDCl3) δ 1.61 (s, 3H), 1.27 (s, 3H), 2.74 (s, 3H), 5.84 (d, J = 10.2 Hz, 1H), 6.54 (s, 1H), 6.73 (s, 2H), 6.78 (d, J = 8.6 Hz, 1H), 6.88 (d, J = 7.6 Hz, 1H), 6.92 (d, J = 10.2 Hz, 1H), 7.02 (d, J = 7.6 Hz, 1H), 8.01 (d, J = 2.4, 1H), 8.03 (dd, J = 2.4, 8.6 Hz, 1H).
6′-(Bromomethyl)spirobenzopyran (13)
To a THF solution (1.5 mL) of 11 (28 mg, 78 μmol) and CBr4, (53 mg, 160 μmol) was added dropwise a THF solution (0.5 mL) of Ph3P (42 mg, 160 μmol) at 0 C. The reaction mixture was stirred at 0° for 30 min and at rt overnight. After evaporation of the reaction mixture, the residue was subjected to column chromatography (silica gel; eluent, hexane/EtOAc = 5:1) to afford 13 (18 mg, 55%) with recovered 11 (10 mg, 36%): MS(EI) 416 (1), 414 (M+, 1), 335 (2); HRMS(EI): M+ 414.0577 (calcd 414.0579); 1H NMR (CDC13) 1.19 (s, 3H), 1.29 (s, 3H), 2.76 (s, 3H), 4.52 (d, J = 10.5 Hz, 1H), 4.56 (d, J = 10.5 Hz, 1H), 5.86 (d, J = 10.2 Hz, 1H), 6.58 (d, J = 1.3 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 6.92 (dd, J = 1.3, 7.3 Hz, 1H), 6.94 (d, J = 10.3 Hz, 1H), 7.04 (d, J = 7.3 Hz, 1H), 8.01 (d, J = 3.2, 1H), 8.04 (dd, J = 3.2, 8.4 Hz, 1H).
Preparation of Protein Conjugates Using Thiol-Reactive Photochromes
A 20 μM solution of BSA (1 mL; Sigma) was reacted with 100 μM each thiol-reactive photochrome (3, 6, 9, 12, 13, and 17) for 1 h at room temperature in the dark. The protein was centrifuged for 10 min at 2000g at 4 °C and applied to a Bio-Rad PD-10 column equilibrated in G-buffer containing 1 mM DTT. The concentration of the SP state within each BSA conjugate was determined from the value of the absorption maximum using an extinction coefficient of 35 000 M−1 cm−1 at 350 nm.15 The G-actin conjugate of 17 was prepared using a standard protocol.20 Transitions between the SP and MC states for each photochrome were triggered in 1 mL absorption cuvettes using 365 and 546 nm light from a 100 W Hg-arc lamp attached to a Zeiss Axiovert 35 fluorescence microscope.
Supplementary Material
Experimental procedures and characterization data for compounds described in this manuscript and copies of 1H NMR spectra for purified compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported in part by a grant awarded to G.M. (NIH R01 HL069970-01). NSF (CHE-9208463 and CHE-9629688) and NIH (1 S10 RR0 8389-01) are greatly acknowledged for NMR spectrometry instrumentation.
References
- 1.Zhang J, Campbell RE, Ting AY, Tsien RY. Nat Rev Mol Cell Biol. 2002;3:906–918. doi: 10.1038/nrm976. [DOI] [PubMed] [Google Scholar]
- 2.Yan Y, Marriott G. Curr Opin Chem Biol. 2003;7:635–640. doi: 10.1016/j.cbpa.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 3.Roy P, Rajfur Z, Jones D, Marriott G, Loew L, Jacobson K. J Cell Biol. 2001;153:1035–1048. doi: 10.1083/jcb.153.5.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marriott G, Miyata H, Kinosita K., Jr Biochem Int. 1992;26:943–951. [PubMed] [Google Scholar]
- 5.Marriott G, Heidecker M. Biochemistry. 1996;35:3170–3174. doi: 10.1021/bi952207o. [DOI] [PubMed] [Google Scholar]
- 6.Cambridge SB, Davis RL, Minden JS. Science. 1997;277:825–828. doi: 10.1126/science.277.5327.825. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS. Science. 2004;304:743–746. doi: 10.1126/science.1094561. [DOI] [PubMed] [Google Scholar]
- 8.Inouye M. Mol Cryst Liq Cryst A. 1994;246:169–172. [Google Scholar]
- 9.Giordano L, Jovin TM, Irie M, Jares-Erijman EA. J Am Chem Soc. 2002;124:7481–7489. doi: 10.1021/ja016969k. [DOI] [PubMed] [Google Scholar]
- 10.Medintz IL, Trammell SA, Mattoussi H, Mauro JM. J Am Chem Soc. 2004;126:30–31. doi: 10.1021/ja037970h. [DOI] [PubMed] [Google Scholar]
- 11.Angelini N, Corrias B, Fissi A, Pieroni O, Lenci F. Biophys J. 1998;74:2601–2610. doi: 10.1016/S0006-3495(98)77966-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song L, Jares-Erijman EA, Jovin TM. J Photochem Photobiol, A. 2002;150:177–185. [Google Scholar]
- 13.Willner I, Rubin S, Wonner J, Effenberger F, Baeuerle P. J Am Chem Soc. 1992;114:3150–3151. [Google Scholar]
- 14.Raymo FM, Giordani S, White AJP, Williams DJ. J Org Chem. 2003;68:4158–4169. doi: 10.1021/jo0340455. [DOI] [PubMed] [Google Scholar]
- 15.Inouye M, Ueno M, Tsuchiya K, Nakayama N, Konishi T, Kitao T. J Org Chem. 1992;57:5377–5383. [Google Scholar]
- 16.Tanaka M, Nakamura M, Salhin MAA, Ikeda T, Kamada K, Ando H, Shibutani Y, Kimura K. J Org Chem. 2001;66:1533–1537. doi: 10.1021/jo0013756. [DOI] [PubMed] [Google Scholar]
- 17.Walker MA. J Org Chem. 1995;60:5352–5355. [Google Scholar]
- 18.Bletz M, Pfeifer-Fukumura U, Kolb U, Baumann W. J Phys Chem A. 2002;106:2232–2236. [Google Scholar]
- 19.Gorner H. Phys Chem Chem Phys. 2001;3:416–423. [Google Scholar]
- 20.Marriott G, Zechel K, Jovin TM. Biochemistry. 1988;27:6214–6220. doi: 10.1021/bi00417a004. [DOI] [PubMed] [Google Scholar]
- 21.Pedersen AO, Jacobsen J. Eur J Biochem. 1980;106:291–295. doi: 10.1111/j.1432-1033.1980.tb06022.x. [DOI] [PubMed] [Google Scholar]
- 22.He XM, Carter DC. Nature. 1992;358:209–215. doi: 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- 23.Marriott G, Jovin TM, Yan-Marriott Y. Anal Chem. 1994;66:1490–94. [Google Scholar]
- 24.Narazaki R, Maruyama T, Otagiri M. Biochim Biophys Acta. 1997;1338:275–281. doi: 10.1016/s0167-4838(96)00221-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for compounds described in this manuscript and copies of 1H NMR spectra for purified compounds. This material is available free of charge via the Internet at http://pubs.acs.org.