

Abstract
Purpose
Patients with metastatic or recurrent Ewing’s sarcoma (ESFT) and alveolar rhabdomyosarcoma (AR) have <25% 5-yr. survival in most studies. This study administered a novel immunotherapy regimen aimed at consolidating remission in these patients.
Experimental Design
52 patients with translocation positive, recurrent or metastatic ESFT or AR underwent pre-chemotherapy cell harvest via apheresis for potential receipt of immunotherapy. Following completion of standard multimodal therapy, 30 patients ultimately initiated immunotherapy and were sequentially assigned to three cohorts. All cohorts received autologous T cells, influenza vaccinations and dendritic cells (DCs) pulsed with peptides derived from tumor specific translocation breakpoints and E7, a peptide known to bind HLA-A2. Cohort 1 received moderate dose rhIL-2, cohort 2 received low dose rhIL-2 and cohort 3 did not receive rhIL-2.
Results
All immunotherapy recipients generated influenza specific immune responses, whereas immune responses to the translocation breakpoint peptides occurred in 39%, and only 25% of HLA-A2+ patients developed E7 specific responses. Toxicity was minimal. Intention-to-treat analysis revealed a 31% 5-year OS for all patients apheresed (median potential follow-up 7.3 yrs) with a 43% 5-year OS for patients initiating immunotherapy.
Conclusions
Consolidative immunotherapy is a scientifically based and clinically practical approach for integrating immunotherapy into a multimodal regimen for chemoresponsive cancer. Patients receiving immunotherapy experienced minimal toxicity and favorable survival. The robust influenza immune responses observed suggest that post-chemotherapy immune incompetence will not fundamentally limit this approach. Future studies will seek to increase efficacy by using more immunogenic antigens and more potent DCs.
Keywords: Ewing’s sarcoma family of tumors, rhabdomyosarcoma, dendritic cell vaccine, immunotherapy, rhIL-2
Introduction
Impressive advances in the last 30 years for patients with clinically localized ESFT and AR have led to current survival rates of 60%–70%(1–3). However, several clinical groups continue to fare poorly, and long term toxicity related to therapy is substantial(4). ESFT patients who present with isolated pulmonary metastases have 5-year survival rates approximating 30%, while <20% of ESFT patients with bone or bone marrow involvement at initial diagnosis survive(5–7), and ESFT patients who recur after frontline therapy also have dismal long term survival rates(8, 9). Similarly, 5-yr survival rates among patients with newly diagnosed metastatic AR are <25%(1, 10), and of the 30–40% of patients who present with localized disease but relapse after frontline therapy, most will eventually die of progressive disease(9, 11). Several chemotherapy regimens have shown activity in recurrent AR and ESFT(12–14), but none of these regimens are curative. Therefore, novel therapeutic approaches are needed to improve outcomes for patients with metastatic or recurrent ESFT and AR.
Advances in basic tumor immunology and preliminary evidence for clinical activity of immunotherapy in select settings(15–17) have sustained optimism that immunotherapy will find an established place in cancer therapy. Tumor vaccines are a central component of many cancer immunotherapies, and DC vaccines have shown preliminary activity in some studies(18, 19). However, as single agents, tumor vaccines have been largely unable to induce regression of established tumors(20), which is not surprising given that animal models demonstrate diminished efficacy of T cell mediated immunotherapy with increasing tumor burdens(21, 22). Moreover, effective immunotherapy occurs over several weeks to months(15, 23), a timeline impractical for patients with rapidly growing tumors such as pediatric sarcomas. Therefore, as concluded from an initial trial of tumor vaccination in patients with recurrent pediatric sarcomas(24), tumor vaccinations as single agents undertaken in the setting of recurrent ESFT or AR are unlikely to have meaningful antitumor activity. The current study attempted to circumvent these challenges by delivering immunotherapy as consolidation, during the period of clinical remission often experienced by high-risk ESFT and AR patients following multimodal therapy. Vaccines comprising DCs pulsed with peptides spanning the breakpoint region of the tumor specific translocations present in AR and ESFT were administered with autologous lymphocyte infusions ± rhIL2. This report describes clinical outcomes for all patients apheresed for potential receipt of immunotherapy, clinical outcomes for the 30 patients who ultimately received immunotherapy on this study and immune responses for 23 of 30 immunotherapy recipients from whom post-vaccination lymphocytes were available for analysis. Detailed presentation of the immune reconstitution patterns in these patients was published previously(25).
Materials and Methods
Eligibility
This study was approved by the IRB of the National Cancer Institute and in accordance with an assurance filed with and approved by the Department of Health and Human Services. Patients were eligible for initial enrollment on this study if they had: 1) newly diagnosed metastatic or recurrent AR or ESFT, and 2) tumors expressing a t(2:13) or t(11;22) type 1 or 2 translocation. In addition, patients enrolled with recurrent disease were required to have experienced a chemotherapy-free interval of >12 months for patients > 5 years and > 6 months for patients < 5 years, and to have a CD4+ count which was > 400 cells/µl in order to assure that the T cell harvest obtained via apheresis would be sufficient. Documentation of the t(11;22) and t(2:13) translocations was made by RT-PCR analysis of fresh or frozen tumor tissue at the time of initial diagnosis or tumor recurrence as previously described(26). Following apheresis for cell harvest, all patients received cytoreductive therapy comprising chemotherapy, radiation therapy and/or surgery as determined by the patients’ referring medical teams (Table 1 and Supplemental Table 1). Upon completion of cytoreductive therapy, the following eligibility criteria were required in order to initiate immunotherapy: acceptable performance status (ECOG 0,1 or 2), acceptable vital organ function and no requirement for continued cytoreductive therapy during the period of immunotherapy. Tumor response to cytoreductive therapy did not, in and of itself, impact eligibility to receive immunotherapy as two patients with progressive disease initiated immunotherapy (Table 1). Because a prolonged period of time elapsed between apheresis and immunotherapy (Fig 1), informed consent was obtained prior to apheresis, then again prior to initiation of immunotherapy.
Table 1.
Patient Characteristics
| Characteristics | No Immunotherapy | Immunotherapy | ||
|---|---|---|---|---|
| No. | % | No. | % | |
| No. of patients | 22 | 30 | ||
| Male | 14 | 64% | 17 | 57% |
| Female | 8 | 36% | 13 | 43% |
| Age# (Years) | ||||
| Median | 20 | 16 | ||
| Range | 1 – 36 | 3 – 39 | ||
| Diagnosis | ||||
| ESFT | 19 | 86% | 20 | 67% |
| AR | 3 | 14% | 10 | 33% |
| Disease Status at Enrollment | ||||
| New dx w/ metastasis | 17 | 77% | 16 | 53% |
| Late recurrence | 4 | 18% | 14 | 47% |
| R1 | 1 | 5% | 9 | 30% |
| R2 | 2 | 10% | 2 | 7% |
| R3+ | 1 | 5% | 3 | 10% |
| Cytoreductive therapy* | ||||
| Transplant (BMT/SCT) | n/a | n/a | 5 | 17% |
| Chemotherapy | n/a | n/a | 30 | 100% |
| Radiation | n/a | n/a | 14 | 47% |
| Surgery | n/a | n/a | 7 | 23% |
| Status at Initiation of Immunotherapy | ||||
| Complete Response | n/a | n/a | 17 | 57% |
| Partial Response | n/a | n/a | 11 | 37% |
| Progressive Disease | n/a | n/a | 2 | 6% |
| Status at last followup | ||||
| Alive | 3 | 14% | 12 | 40% |
| Dead | 19 | 86% | 18 | 60% |
| Potential Follow Up (Years)¤ | ||||
| Median | 5.8 | 7.8 | ||
| Range | 5.4–8.7 | 5.1–9.4 | ||
Abbreviations: ESFT, Ewing sarcoma family of
Age at time of apheresis
Given between apheresis and immunotherapy
for surviving patients
Figure 1.
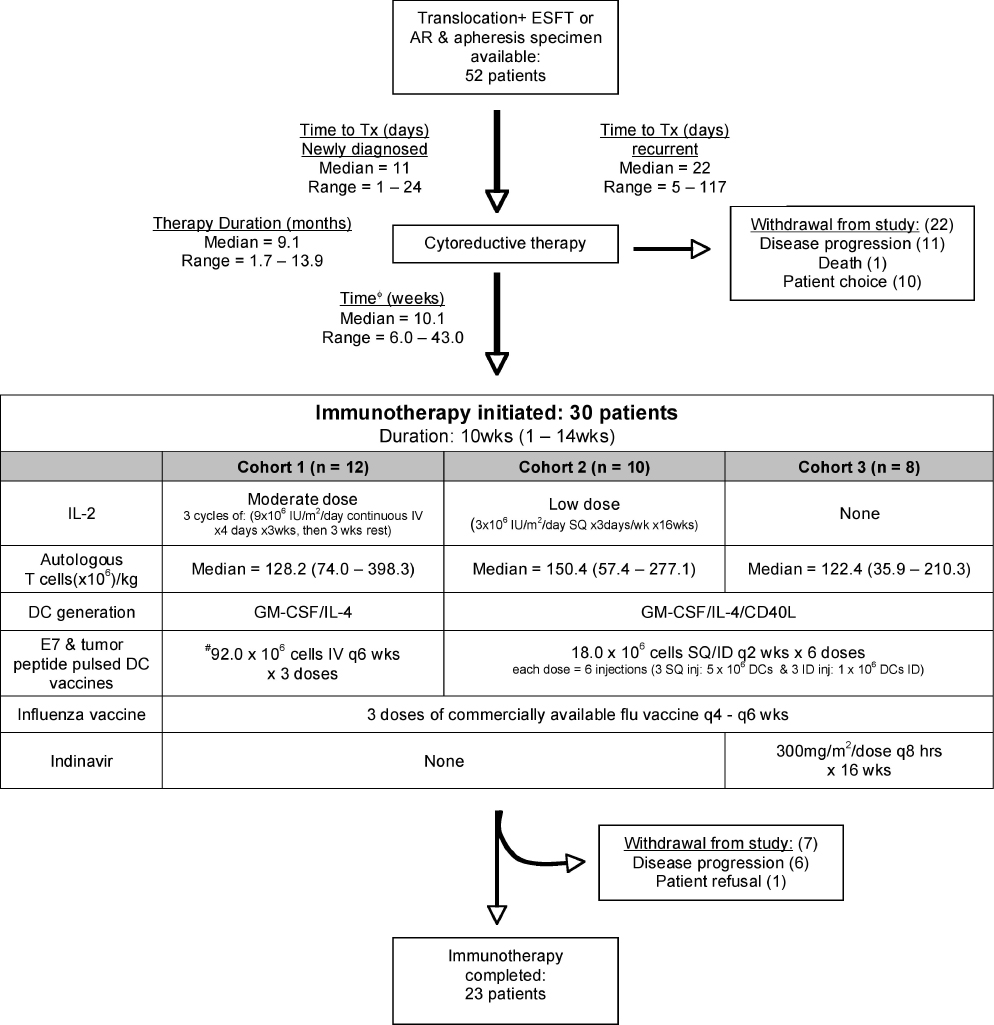
Schema for Consolidative Immunotherapy. *Time to Tx = time from diagnosis/recurrence to cytoreductive therapy initiation. ˙Time from cytoreductive therapy completion to immunotherapy initiation. #Mean shown, range: 4.2 – 143.0 × 106 cells
Cell Harvest and Immunotherapy
Approximately 3 blood volumes were processed via apheresis, then separated into lymphocyte and monocyte fractions via countercurrent centrifugal elutriation as previously described(27), then cryopreserved. Fifty million cells from each fraction were assessed for tumor contamination by nested RT-PCR for the tumor specific translocation as described previously (sensitivity: 1 tumor cell/106 cells)(28). See Supplemental Table 2 for primer sequences. Details of the immunotherapy administered to each cohort is shown in Figure 1. Briefly, all cohorts received three influenza vaccinations and similar doses of autologous T cells. Cohorts 1 and 2 received rhIL-2 (moderate and low dose respectively) whereas cohort 3 did not receive rhIL-2 but received oral indinavir. To generate the DC vaccines, cryopreserved monocytes were thawed, placed into culture for 5–7 days at 37°C using 10% autologous plasma or human AB serum, rhIL-4 (2000 U/ml) and rhGM-CSF (2000 U/ml) as previously described(29). In cohorts 2 and 3, CD40 ligand (1µg/ml) was added during the last 24 hours. On the day of vaccine administration, 50% of the cells were co-cultured with 10µM of the appropriate tumor derived breakpoint peptide and 50% were co-cultured with the control HPV16 E7 peptide (Table 2) for 2hr, then washed, pooled, and irradiated (2,500 cGy) prior to injection.
Table 2.
Vaccine Peptide Sequences
| Vaccine Peptide | Peptide Sequence |
|---|---|
| EF-1 (EWS/FLI-1)a | SSSYGQQN/PSYDSVRRGA |
| EF-2 (EWS/FLI-2)a | SSSYGQ/QSSLLAYNT |
| PXFK (PAX3/FKHR)b | TIGNGLSPQ/NSIRHNLSL |
| HPV16E7 | MLDLQPETT-MET-9-THR |
t(11;22)(q24;q12) Ewing’s Sarcoma Family of Tumors (ESFT)
t(2;13)(q35;q14) Alveolar Rhabdomyosarcoma (AR)
Immune Response Assessment
Immune responses (T cell proliferation, cytokine production, and cytolysis) in response to influenza, the immunizing tumor translocation breakpoint peptide and the E7 peptide were measured using cells obtained at the time of initial apheresis, following cytoreductive therapy prior to initiation of immunotherapy and at several time points during and following completion of immunotherapy. Briefly, autologous peptide pulsed DCs were incubated with the immunizing peptides or whole influenza, then added to PBMC at 1:10 – 1:10,000 stimulator:effector ratios, and proliferation was measured via 3H incorporation. Cytolytic T cell responses were quantified using a Cr51 assay and cytokine (IFN· + IL-5) levels were measured in culture supernatants. Positive response criteria, determined prior to initiation of the study, were: 1) for cytolytic activity, a percent specific lysis of at least 10% as this value was greater than 2 SD of the percent specific lysis measured from a pool of 6 normal donors, 2) for cytokine assays, > 2-fold increase compared to no-peptide controls and 3) for proliferation assays, a stimulation index of > 3.0. Anti-influenza A antibody responses were performed by Mayo Medical Lab (Rochester, MN).
Statistics
This study used a Phase II trial design, with alpha=0.10 and 90% power to detect a targeted 25% immune response rate to the breakpoint peptide with a goal to enroll a total of 24 patients. Tumor histologies were combined for determination of response rates. Based upon interim immune response and immune reconstitution endpoints, the protocol was sequentially revised leading to 3 cohorts. The immune response data for cohorts 1, 2 and 3 were combined after the data were determined to be sufficiently similar by the Kruskal-Wallis test (results not shown). Statistical analyses included construction of Kaplan-Meier survival curves with two-tailed log rank tests, and Wilcoxon signed rank tests to test the significance of paired differences in immune response data over time. Survival data is calculated from the date of diagnosis for patients enrolled with newly diagnosed metastatic disease and from the date of the last recurrence detection prior to enrollment on this study for patients with recurrent disease.
Results
Patient characteristics, feasibility and toxicity
Between 1997 and 2003, fifty-two patients were enrolled and underwent apheresis for potential receipt of immunotherapy. Because accrual of this rare population required recruitment from long distances, one concern was that the travel required to perform pre-cytoreductive therapy apheresis for cell harvest would result in undue treatment delays, especially for newly diagnosed patients. However, newly diagnosed patients with metastatic disease initiated frontline therapy with minimal delay from the time of diagnosis (median 11 days, range 1 – 24 days). The median time for patients with recurrent disease between recurrence detection and initiation of chemotherapy was 22 (range 5 – 117) days (Fig 1). Apheresis was well tolerated, and adequate harvests meeting phenotypic, viability and sterility standards established as release criteria were obtained from all patients. Following apheresis, all patients received cytoreductive therapy as dictated by their referring medical teams. Of 52 patients initially enrolled and apheresed, 22 did not initiate immunotherapy due to: progressive disease resulting in an unacceptable performance status (n= 11), death due to complications of therapy (n= 1), or patient choice (n= 10).
The immunotherapy regimen was well tolerated (grade 4 thrombocytopenia n=1; grade 3 neutropenia n=6, diarrhea n=2, elevated total bilirubin n=1, abdominal pain n=1, skin rash n=3). Four patients required dose adjustment or discontinuation of rhIL-2. Of the 30 patients in whom immunotherapy was initiated, 23 (77%) completed the full course, whereas 7 patients did not due to disease progression (n=6) or patient choice (n=1). Thus, of patients with metastatic or recurrent ESFT and AR who do not develop progressive disease during frontline cytoreductive therapy, a majority will tolerate a complex immunotherapy regimen without undue toxicity.
Immune responses
We have demonstrated previously that dose intensive chemotherapy for AR and ESFT induces profound lymphopenia(30), T cell subset alterations(31), and relative expansion of the CD4+CD25+ regulatory T cell subset(25), which could potentially limit responsiveness to vaccination. To determine whether patients treated on this trial could respond to a vaccine known to be effective in a healthy population, immune responses were measured following three sequential influenza vaccines administered during the same period as the peptide pulsed DC vaccines. Despite profound CD4+ depletion at the initiation of immunotherapy(25), all patients analyzed demonstrated immunity to influenza within 6 months of completing cytoreductive therapy. As shown in Fig. 2, increased IFN· production in response to influenza occurred within 1.5 months of initiating immunotherapy (p=0.05), followed by increases in influenza-specific T cell proliferation (p=0.05) and humoral immunity (p=0.006) within 4.5 months of initiating immunotherapy. Most patients also showed cytolytic responses to influenza (data not shown). Thus, patients rendered lymphopenic by dose intensive cytoreductive therapy for cancer who receive autologous lymphocyte infusions ± rhIL2 retain a robust capacity to respond to vaccination, as evidenced by the brisk influenza-directed immune responses.
Figure 2.
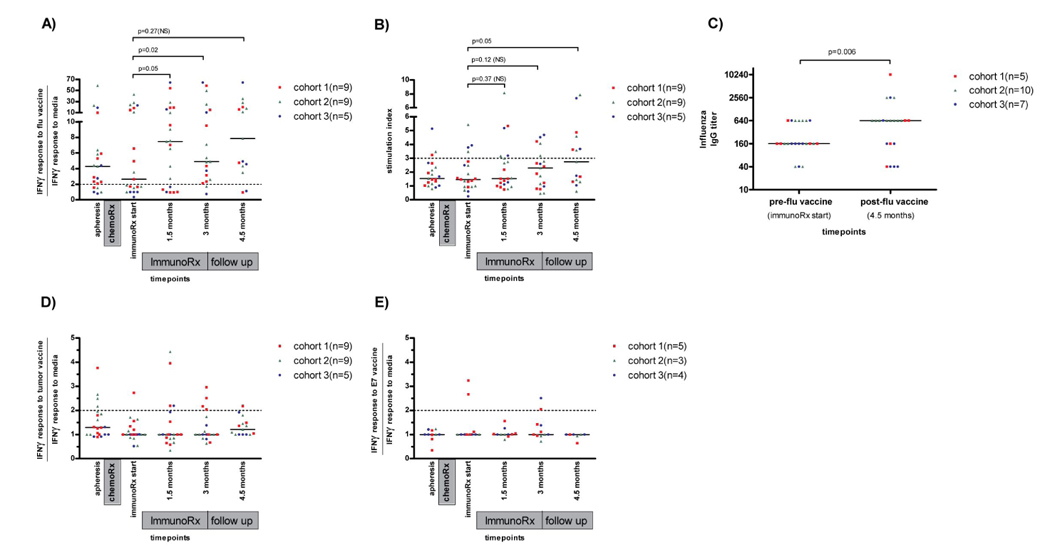
Immune Responses to influenza (A–C), tumor peptide DC vaccines (D) and E7 peptide DC vaccine shown for HLA-A2+ patients only (E). IFN·: interferon gamma; chemoRx: chemotherapy; immunoRx: immunotherapy; Dashed line shows threshold of positivity determined prior to protocol initiation.
With regard to immune responses to the tumor translocation breakpoint peptides, 5/23 patients (22%) had measurable immune responses prior to initiation of immunotherapy (Fig 2D), demonstrated by IFN· production following exposure to the tumor-specific peptide in vitro. Following DC vaccination, 2 of those 5 patients maintained immune responses, and 7 patients generated new positive responses. Therefore, 9 of the 23 patients (39%) showed measurable immune responses to the vaccinating peptide (Figure 2D), mostly limited to IFN· production. Importantly, in most patients with immune reactivity toward the immunizing peptide, the responses were transient, dropping below the threshold of positivity at the time of the next analysis (6 weeks later). We detected no difference in response rate to the three tumor breakpoint derived peptides used for vaccination. Because it was likely that the breakpoint peptides used in this study would not bind to all HLA alleles(32) and because tumor associated tolerance could limit immune reactivity toward tumor associated peptides, the HLA-A2 binding HPV16-derived peptide E7 was used as a control. Only 3/12 (25%) HLA-A2+ patients enrolled on this study generated immune responses to E7. Thus, inadequate HLA binding of the tumor breakpoint peptides cannot fully explain the low immune response rate observed since immune responsiveness was also poor following E7 peptide pulsed DC vaccination in HLA-A2+ patients where binding could be assured.
Clinical outcomes
The 5-year OS for all patients who underwent apheresis following initial enrollment on this study is 31% (3A, median f/u of surviving patients = 7.6 yrs) with improved survival in patients initiating immunotherapy in both the ESFT and AR cohorts (3B, 3C). Significant survival differences were not observed between histologic subtypes (3C), however patients with recurrent disease had improved overall survival compared to those with primary metastatic disease (Fig 3D), although they had similar event free survival (Fig 3E). With regard parameters that correlated with survival in the cohort that initiated immunotherapy, we observed with no significant survival differences based upon whether patients had a partial response or complete response to cytoreductive therapy (data not shown), evidence for tumor contamination in the lymphocyte product administered (3F), differences in immunotherapy administered in cohorts 1–3 (3G), immune responses measured to the tumor vaccine (3H) or total CD4+ counts at baseline, or post-cytoreductive therapy or the frequency of CD4+CD25hi cells at these timepoints (Supplemental Table 3). Further information regarding disease sites, cytoreductive therapy received and status at last follow-up for the 30 patients who initiated immunotherapy is shown in Supplemental Table 1 and detailed information regarding the long term survivors in immunotherapy recipients in shown in Table 3.
Figure 3.
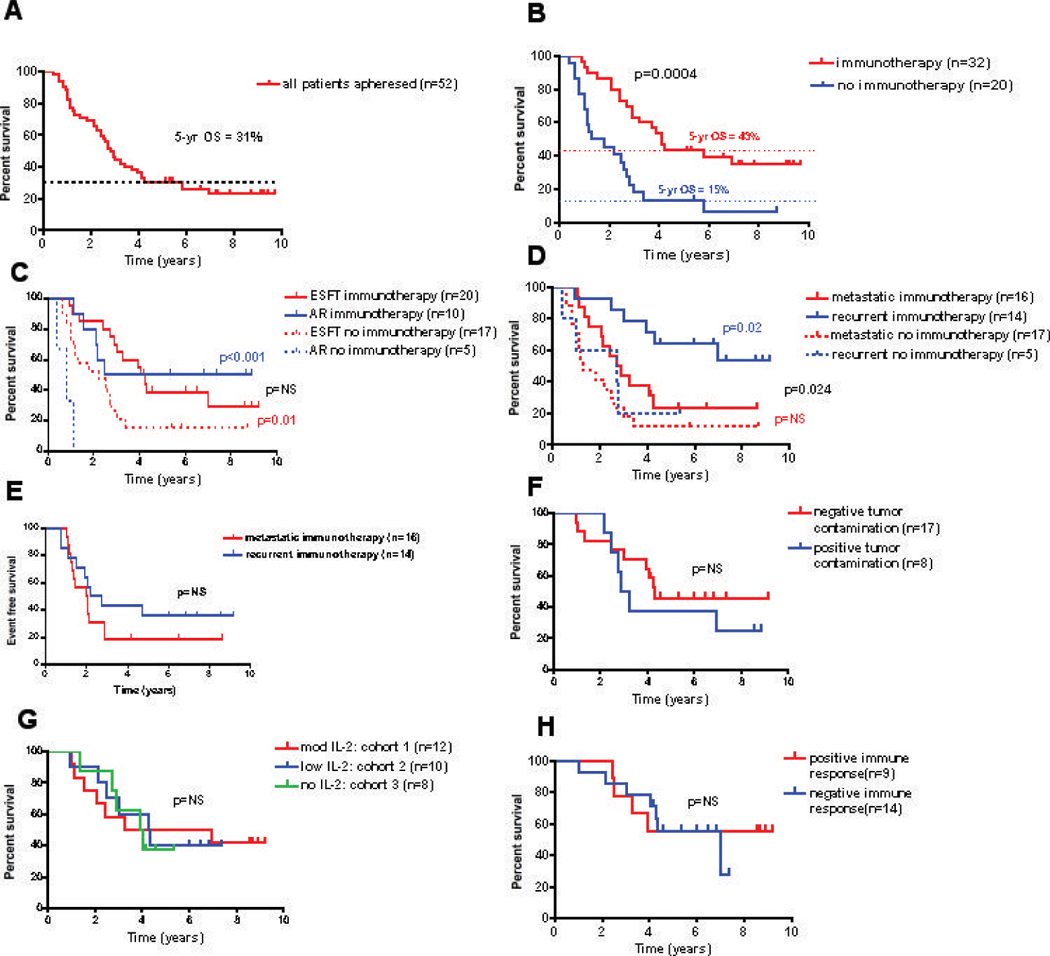
Clinical Outcomes. Kaplan-Meier survival curves: (A) all patients who underwent apheresis (B) patients who initiated immunotherapy vs. those who did not (C) by diagnosis (D) by disease status at the time of apheresis (F) by tumor contamination of the autologous T cells (G) by immunotherapy cohort (H) by immune response to the immunizing peptide and (E) event free survival by disease status at enrollment.
Table 3.
Characteristics of surviving patients
| pt # | age | sex | diagnosis | year of initial dx | year of recurrence | primary site | metastasis or recurrence at enrollment | Cytoreductive therapy | status post frontline chemotherapy | IL-2 | T cells (×106)/kg | completed Rx | immune response | OS (years) | Follow Up |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| New Dx with metastasis | |||||||||||||||
| 3 | 16 | M | AR | 1998 | NA | Bladder | bladder, lymph nodes, bone marrow | chemo, SCT, XRT | CR | mod | 124.4 | y | + | 9.2 | NED* |
| 13 | 33 | M | ESFT | 2000 | NA | L femur | L femur with pulmonary mets | chemo, XRT | CR | low | 207.2 | y | − | 7.2 | NED* |
| 29 | 11 | F | ESFT | 2002 | NA | R fibula | R fibula & pulmonary | chemo, XRT | CR | none | 99.6 | y | − | 5.3 | NED* |
| Recurrence (>1yr after frontline therapy) | |||||||||||||||
| 22 | 18 | F | AR | 1998 | 2000 | R neck/sinus | breast | chemo, SX | CR | low | 153.3 | y | − | 7.3 | NED* |
| 6 | 39 | M | ESFT | 1983 | 1998 | 10th L rib | R diaphragm | chemo, SX | PR | mod | 108.9 | y | + | 9.7 | recurred; currently NED × one year |
| 10 | 3 | F | AR | 1995 | 1998 | R orbit | R pre-auricular LN | SX, chemo, SCT | PD | mod | 249.6 | n | + | 9.4 | disease progression during immunotherapy, currently NED for > 5 years |
| 11 | 28 | M | ESFT | 1980 | 1998 | L tibia | pulmonary mets | chemo | CR | mod | 74.0 | y | + | 9.4 | recurred; currently NED for > 3 years |
| 5 | 16 | F | ESFT | 1995 | 1998 | R thigh | kidney | chemo | CR | mod | 203.0 | y | + | 9.1 | NED* |
| 16 | 21 | M | ESFT | 1991 | 2001 | Neck | neck | chemo | CR | low | 141.7 | y | − | 6.6 | NED* |
| 20 | 6 | F | AR | 1997 | 2000 | L ankle | s/p resection of local recurrence | chemo | CR | low | 244.5 | y | − | 7.8 | NED* |
| 27 | 26 | F | ESFT | 1994 | 2002 | R arm | R clavicle/L pleural mass with rib/pulmonary | XRT, chemo | PR | none | 197.2 | y | − | 5.1 | recurred; currently NED for > 3 years |
Abbreviation: dx, diagnosis; NA, not applicable; LN, lymph node; SX, surgery; SCT, stem cell transplant; XRT, radiation treatment; CR, complete response; PR, partial response; PD, progression of disease; IL-2, interleukin 2; mod, moderate; OS, overall survival; AWD, alive with disease; NED, no evidence of disease.
NED since initiating immunotherapy
Discussion
Lymphop enia-induced alterations in T cell homeostasis provide a fertile milieu for inducing immune responses(33), especially when adoptively transferred T cells are incorporated into the regimen(15, 34). Using this paradigm(30, 35), we administered tumor vaccines with autologous T cells to patients with high-risk pediatric sarcomas following cytoreductive chemotherapy. Based upon evidence that chromosomal translocations can provide targets for T cell immune recognition(36–39) and preliminary results in ESFT and AR xenografts(40), the tumor vaccines comprised DCs pulsed with translocation breakpoint peptides. In the first two cohorts, the immune restoration regimen included rhIL-2. However, because we observed rhIL-2 induced expansion of CD4+CD25+ regulatory T cells (25), the third cohort did not receive rhIL-2. Only the third cohort received indinavir, an HIV protease inhibitor that has shown immunomodulatory effects in vitro(41). The sequential modifications of the immunotherapy administered during the conduct of this trial resulted in each immunotherapy cohort being comprised of limited numbers of patients receiving different regimens, a design which makes it difficult to parse the contribution of varying elements within the immunotherapy to the final outcome. One feature common to all patients initiating immunotherapy however was the receipt of a sizable dose of autologous T cells during the period of lymphopenia that followed standard cytoreductive therapy. Approximately 1 × 108 T cells/kg were administered, a dose estimated to represent 2% of the total body T cell pool(42), and which has been associated with anti-leukemic effects when administered as donor lymphocyte infusions following allogeneic transplantation(43).
With regard to feasibility, the results demonstrate that immune cells can be harvested prior to cytoreductive therapy without undue treatment delays and monocytes collected from this patient population reliably generated dendritic cells meeting release criteria from all patients. Importantly however, only 30 of 52 patients apheresed following initial enrollment eventually initiated immunotherapy, due to progressive disease which rendered them unable to meet eligibility criteria (n=11) for immunotherapy, patient choice (n=10) or death due to complications of therapy (n=1). While progressive disease was not itself an exclusion criteria for receipt of immunotherapy, it is important to note that only 2 patients with progressive disease during cytoreductive therapy initiated immunotherapy. Thus, new approaches for inducing an effective remission remain critical for improving outcomes for sizable numbers of this patient populations since the period of minimal residual neoplastic disease that is required to accomplish this therapy was not achievable in a sizable fraction of this patient population.
Nonetheless, an “intent-to-treat” analysis revealed overall 5-yr survival rate for the 52 patients apheresed of 31%, which compares favorably with rates reported in the literature. For the 30 patients who initiated immunotherapy, OS 5-year was 43%, however, selection factors no doubt contribute to this favorable survival rate since patients with rapidly progressive disease did not initiate immunotherapy. Moreover, the requirement for a prolonged chemotherapy free interval as part of the eligibility criteria for enrollment with recurrent disease selects for patients with more indolent disease as compared to other studies of recurrent disease in the same histologies. Indeed, some patients enrolled following “late” tumor recurrences experienced exceptionally indolent disease courses, both prior to and following immunotherapy (Table 3, patients #6 and #11). This is illustrated graphically in figures 3D/3E, which demonstrates improved OS for recurrent patients as compared to newly diagnosed metastatic patients, despite the fact that EFS were not significantly different. When evaluating only the newly diagnosed metastatic subset, where this selection factor did not come into play, the 5-year OS for the entire cohort was 23%.
Similar to findings in some other studies of immunotherapy(44), survival did not correlate with immune response to tumor peptide vaccination. This could indicate that the immunotherapy administered here did not alter the natural history of disease in these patients and that the favorable survival in the immunotherapy recipients relates to selection factors alone. Alternatively, the peripheral blood may not be optimal for measuring immune reactivity since activated immune cells traffic to tissues in order to effect antitumor immune responses. Finally, it is also possible that adoptive transfer of autologous T cells as performed here contributed to control of micrometastatic disease independent of vaccine responses. We previously published that patients with ESFT have circulating T cells which recognize and lyse autologous tumors(45), and in animal models of sarcoma, adoptive transfer of T cells during a period of minimal residual disease prevented the development of overt metastatic disease(46). Similarly, improved immune reconstitution measured by the lymphocyte count on Day 15 following autologous stem cell transplantation correlates with improved survival in several cancers(47) and younger ESFT and rhabdomyosarcoma patients, who experience more rapid immune reconstitution(30), consistently have better outcomes than older individuals(7, 48). Thus, it remains possible that more effective immune reconstitution, rendered via young age or immunotherapy, diminishes the rate of metastatic recurrence.
Patients on this study experienced profound lymphopenia following cytoreductive therapy(25), but essentially all patients generated brisk immune responses to influenza during immunotherapy whereas the DC vaccines were relatively ineffective. Several factors likely contribute to this finding. First, both the breakpoint region and the E7 peptide represented neoantigens for most patients, which provide a more stringent test of immune competence than the generation of responses to a recall antigen which influenza represents. Second, while the breakpoint peptides used as immunogens in this trial bind to some HLA molecules(39), they appear poorly immunogenic overall(32). Third, the DCs administered in this study may not be optimal for inducing primary immune responses. Immature DCs, as administered to the first cohort, are now known to be suboptimal for inducing immune responses(49), and the CD40L-matured DCs used in cohorts 2 and 3 produced relatively low levels of IL-12 (data not shown) a critical immunostimulatory cytokine. Further, intravenous infusion of DCs, as administered to cohort 1, appears less effective than other routes of administration(18). Fourth, while lymphopenia provides opportunities for generating immune responses, it also presents inherent barriers, including expansion of CD4+CD25+ regulatory cells capable of broad immune suppression(25) and which contribute to the suppression of antitumor immunity. CD4+CD25+ Tregs were preferentially expanded by rhIL2 therapy in cohorts 1 and 2 of this study, and significant increases in the frequency of these cells also occurred in response to lymphopenia alone in cohort 3. Although indinavir was incorporated into cohort 3 based upon preclinical data suggesting that it could augment immune reconstitution in patients not infected with HIV, we did not observe any beneficial impact of indinavir in this study with regard to overall immune reconstitution and/or vaccine responsiveness.
In summary, consolidative immunotherapy, as delivered here, incorporates scientific principles to provide a framework for administering immune based therapy in chemoresponsive cancer. It appears feasible for a sizable fraction of patients with high-risk pediatric sarcomas who experience a good clinical response to frontline cytoreductive therapy, and patients receiving an autologous lymphocyte infusion demonstrate sufficient immunocompetence to generate vaccine induced immune responses to influenza. However, improvements in the regimen are needed to induce strong and sustained immune responses to tumor antigens in a majority of patients. Modifications under study include alternative approaches for inducing DC maturation(50) and the use of more immunogenic antigens. While the final determination of the effectiveness of immunotherapies such as this will require analysis of survival outcomes in randomized studies using an intention-to-treat analysis, pilot studies such as this that carefully assess immune endpoints are essential for optimizing immunotherapy for cancer(50).
Acknowledgments
This manuscript is dedicated to the memory of Charles S. Carter whose innovation and dedication to the research and development of cellular immunotherapy played a pivotal role in the completion of this and many other cellular therapy trials conducted throughout the world. The investigators would also like to thank the patients and the families of patients who consented to receive investigational therapy on this study as well as the staff of the Pediatric Oncology Branch and the NIH Clinical Center for exemplary care of these patients. This work was supported by the Intramural Research Program of the US National Institutes of Health, National Cancer Institute, Center for Cancer Research. We would also like to thank Drs. Seth Steinberg, and Terry Fry for their careful review of the manuscript.
References
- 1.Arndt CA, Crist WM. Common musculoskeletal tumors of childhood and adolescence. N Engl J Med. 1999;341:342–352. doi: 10.1056/NEJM199907293410507. [DOI] [PubMed] [Google Scholar]
- 2.Stevens MC, Rey A, Bouvet N, et al. Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: third study of the International Society of Paediatric Oncology--SIOP Malignant Mesenchymal Tumor 89. J Clin Oncol. 2005;23:2618–2628. doi: 10.1200/JCO.2005.08.130. [DOI] [PubMed] [Google Scholar]
- 3.Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348:694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]
- 4.Oeffinger KC, Mertens AC, Sklar CA, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355:1572–1582. doi: 10.1056/NEJMsa060185. [DOI] [PubMed] [Google Scholar]
- 5.Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing's tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing's Sarcoma Study Group. J Clin Oncol. 2000;18:3108–3114. doi: 10.1200/JCO.2000.18.17.3108. [DOI] [PubMed] [Google Scholar]
- 6.Meyers PA, Krailo MD, Ladanyi M, et al. High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing's sarcoma does not improve prognosis. J Clin Oncol. 2001;19:2812–2820. doi: 10.1200/JCO.2001.19.11.2812. [DOI] [PubMed] [Google Scholar]
- 7.Burdach S, Jurgens H. High-dose chemoradiotherapy (HDC) in the Ewing family of tumors (EFT) Crit Rev Oncol Hematol. 2002;41:169–189. doi: 10.1016/s1040-8428(01)00154-8. [DOI] [PubMed] [Google Scholar]
- 8.Koscielniak E, Klingebiel TH, Peters C, et al. Do patients with metastatic and recurrent rhabdomyosarcoma benefit from high-dose therapy with hematopoietic rescue? Report of the German/Austrian Pediatric Bone Marrow Transplantation Group. Bone Marrow Transplant. 1997;19:227–231. doi: 10.1038/sj.bmt.1700628. [DOI] [PubMed] [Google Scholar]
- 9.Meyers PA. High-dose therapy with autologous stem cell rescue for pediatric sarcomas. Curr Opin Oncol. 2004;16:120–125. doi: 10.1097/00001622-200403000-00006. [DOI] [PubMed] [Google Scholar]
- 10.Breneman JC, Lyden E, Pappo AS, et al. Prognostic factors and clinical outcomes in children and adolescents with metastatic rhabdomyosarcoma--a report from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 2003;21:78–84. doi: 10.1200/JCO.2003.06.129. [DOI] [PubMed] [Google Scholar]
- 11.Raney RB, Jr, Crist WM, Maurer HM, Foulkes MA. Prognosis of children with soft tissue sarcoma who relapse after achieving a complete response. A report from the Intergroup Rhabdomyosarcoma Study I. Cancer. 1983;52:44–50. doi: 10.1002/1097-0142(19830701)52:1<44::aid-cncr2820520110>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 12.Vassal G, Couanet D, Stockdale E, et al. Phase II trial of irinotecan in children with relapsed or refractory rhabdomyosarcoma: a joint study of the French Society of Pediatric Oncology and the United Kingdom Children's Cancer Study Group. J Clin Oncol. 2007;25:356–361. doi: 10.1200/JCO.2006.06.1960. [DOI] [PubMed] [Google Scholar]
- 13.Wagner LM, McAllister N, Goldsby RE, et al. Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatr Blood Cancer. 2007;48:132–139. doi: 10.1002/pbc.20697. [DOI] [PubMed] [Google Scholar]
- 14.Saylors RL, 3rd, Stine KC, Sullivan J, et al. Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. J Clin Oncol. 2001;19:3463–3469. doi: 10.1200/JCO.2001.19.15.3463. [DOI] [PubMed] [Google Scholar]
- 15.Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dazzi F, Szydlo RM, Goldman JM. Donor lymphocyte infusions for relapse of chronic myeloid leukemia after allogeneic stem cell transplant: where we now stand. Exp Hematol. 1999;27:1477–1486. doi: 10.1016/s0301-472x(99)00096-x. [DOI] [PubMed] [Google Scholar]
- 17.Mu LJ, Kyte JA, Kvalheim G, et al. Immunotherapy with allotumour mRNA-transfected dendritic cells in androgen-resistant prostate cancer patients. Br J Cancer. 2005;93:749–756. doi: 10.1038/sj.bjc.6602761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thurner B, Haendle I, Roder C, et al. Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med. 1999;190:1669–1678. doi: 10.1084/jem.190.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Small EJ, Schellhammer PF, Higano CS, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–3094. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greenberg PD. Adoptive T cell therapy of tumors: mechanisms operative in the recognition and elimination of tumor cells. Adv Immunol. 1991;49:281–355. doi: 10.1016/s0065-2776(08)60778-6. [DOI] [PubMed] [Google Scholar]
- 22.Sondak VK, Wagner PD, Shu S, Chang AE. Suppressive effects of visceral tumor on the generation of antitumor T cells for adoptive immunotherapy. Arch Surg. 1991;126:442–446. doi: 10.1001/archsurg.1991.01410280040005. [DOI] [PubMed] [Google Scholar]
- 23.Collins RH, Jr, Shpilberg O, Drobyski WR, et al. Donor leukocyte infusions in 140 patients with relapsed malignancy after allogeneic bone marrow transplantation. J Clin Oncol. 1997;15:433–444. doi: 10.1200/JCO.1997.15.2.433. [DOI] [PubMed] [Google Scholar]
- 24.Dagher R, Long LM, Read EJ, et al. Pilot trial of tumor-specific peptide vaccination and continuous infusion interleukin-2 in patients with recurrent Ewing sarcoma and alveolar rhabdomyosarcoma: an inter-institute NIH study. Med Pediatr Oncol. 2002;38:158–164. doi: 10.1002/mpo.1303. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Chua KS, Guimond M, et al. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat Med. 2005;11:1238–1243. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 26.Dagher R, Pham TA, Sorbara L, et al. Molecular confirmation of Ewing sarcoma. J Pediatr Hematol Oncol. 2001;23:221–224. doi: 10.1097/00043426-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 27.Czerniecki BJ, Carter C, Rivoltini L, et al. Calcium ionophore-treated peripheral blood monocytes and dendritic cells rapidly display characteristics of activated dendritic cells. J Immunol. 1997;159:3823–3837. [PubMed] [Google Scholar]
- 28.Merino ME, Navid F, Christensen BL, et al. Immunomagnetic purging of Ewing's sarcoma from blood and bone marrow: quantitation by real-time polymerase chain reaction. J Clin Oncol. 2001;19:3649–3659. doi: 10.1200/JCO.2001.19.16.3649. [DOI] [PubMed] [Google Scholar]
- 29.Wong EC, Maher VE, Hines K, et al. Development of a clinical-scale method for generation of dendritic cells from PBMC for use in cancer immunotherapy. Cytotherapy. 2001;3:19–29. doi: 10.1080/146532401753156377. [DOI] [PubMed] [Google Scholar]
- 30.Mackall CL, Fleisher TA, Brown MR, et al. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N Engl J Med. 1995;332:143–149. doi: 10.1056/NEJM199501193320303. [DOI] [PubMed] [Google Scholar]
- 31.Mackall CL. T-cell immunodeficiency following cytotoxic antineoplastic therapy: a review. Oncologist. 1999;4:370–378. [PubMed] [Google Scholar]
- 32.Rodeberg DA, Nuss RA, Heppelmann CJ, Celis E. Lack of effective T-lymphocyte response to the PAX3/FKHR translocation area in alveolar rhabdomyosarcoma. Cancer Immunol Immunother. 2005;54:526–534. doi: 10.1007/s00262-004-0625-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asavaroengchai W, Kotera Y, Mule JJ. Tumor lysate-pulsed dendritic cells can elicit an effective antitumor immune response during early lymphoid recovery. Proc Natl Acad Sci U S A. 2002;99:931–936. doi: 10.1073/pnas.022634999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mackall CL, Bare CV, Granger LA, Sharrow SO, Titus JA, Gress RE. Thymic-independent T cell regeneration occurs via antigen-driven expansion of peripheral T cells resulting in a repertoire that is limited in diversity and prone to skewing. J Immunol. 1996;156:4609–4616. [PubMed] [Google Scholar]
- 35.Hakim FT, Memon SA, Cepeda R, et al. Age-dependent incidence, time course, and consequences of thymic renewal in adults. J Clin Invest. 2005;115:930–939. doi: 10.1172/JCI22492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Worley BS, van den Broeke LT, Goletz TJ, et al. Antigenicity of fusion proteins from sarcoma-associated chromosomal translocations. Cancer Res. 2001;61:6868–6875. [PubMed] [Google Scholar]
- 37.Yotnda P, Firat H, Garcia-Pons F, et al. Cytotoxic T cell response against the chimeric p210 BCR-ABL protein in patients with chronic myelogenous leukemia. J Clin Invest. 1998;101:2290–2296. doi: 10.1172/JCI488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yotnda P, Garcia F, Peuchmaur M, et al. Cytotoxic T cell response against the chimeric ETV6-AML1 protein in childhood acute lymphoblastic leukemia. J Clin Invest. 1998;102:455–462. doi: 10.1172/JCI3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van den Broeke LT, Pendleton CD, Mackall C, Helman LJ, Berzofsky JA. Identification and epitope enhancement of a PAX-FKHR fusion protein breakpoint epitope in alveolar rhabdomyosarcoma cells created by a tumorigenic chromosomal translocation inducing CTL capable of lysing human tumors. Cancer Res. 2006;66:1818–1823. doi: 10.1158/0008-5472.CAN-05-2549. [DOI] [PubMed] [Google Scholar]
- 40.Goletz TJ, Zhan S, Pendleton CD, Helman LJ, Berzofsky JA. Cytotoxic T cell responses against the EWS/Fli-1 Ewing's sarcoma fusion protein and the PAX3/FKHR alveolar rhabdomyosarcoma fusion protein; Proceedings of the American Association for Cancer Research; 1995. [Google Scholar]
- 41.Sloand EM, Kumar PN, Kim S, Chaudhuri A, Weichold FF, Young NS. Human immunodeficiency virus type 1 protease inhibitor modulates activation of peripheral blood CD4(+) T cells and decreases their susceptibility to apoptosis in vitro and in vivo. Blood. 1999;94:1021–1027. [PubMed] [Google Scholar]
- 42.Westermann J, Pabst R. Lymphocyte subsets in the blood: a diagnostic window on the lymphoid system? Immunol Today. 1990;11:406–410. doi: 10.1016/0167-5699(90)90160-b. [DOI] [PubMed] [Google Scholar]
- 43.Porter D, Levine JE. Graft-versus-host disease and graft-versus-leukemia after donor leukocyte infusion. Semin Hematol. 2006;43:53–61. doi: 10.1053/j.seminhematol.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 44.Banchereau J, Ueno H, Dhodapkar M, et al. Immune and clinical outcomes in patients with stage IV melanoma vaccinated with peptide-pulsed dendritic cells derived from CD34+ progenitors and activated with type I interferon. J Immunother (1997) 2005;28:505–516. doi: 10.1097/01.cji.0000171292.79663.cb. [DOI] [PubMed] [Google Scholar]
- 45.Zhang H, Merchant MS, Chua KS, et al. Tumor expression of 4-1BB ligand sustains tumor lytic T cells. Cancer Biol Ther. 2003;2:579–586. doi: 10.4161/cbt.2.5.545. [DOI] [PubMed] [Google Scholar]
- 46.Merchant MS, Melchionda F, Sinha M, Khanna C, Helman L, Mackall CL. Immune reconstitution prevents metastatic recurrence of murine osteosarcoma. Cancer Immunol Immunother. 2006 doi: 10.1007/s00262-006-0257-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Porrata LF, Markovic SN. Timely reconstitution of immune competence affects clinical outcome following autologous stem cell transplantation. Clin Exp Med. 2004;4:78–85. doi: 10.1007/s10238-004-0041-4. [DOI] [PubMed] [Google Scholar]
- 48.Blakely ML, Andrassy RJ, Raney RB, et al. Prognostic factors and surgical treatment guidelines for children with rhabdomyosarcoma of the perineum or anus: a report of Intergroup Rhabdomyosarcoma Studies I through IV, 1972 through 1997. J Pediatr Surg. 2003;38:347–353. doi: 10.1053/jpsu.2003.50106. [DOI] [PubMed] [Google Scholar]
- 49.Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193:233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Czerniecki BJ, Koski GK, Koldovsky U, et al. Targeting HER-2/neu in early breast cancer development using dendritic cells with staged interleukin-12 burst secretion. Cancer Res. 2007;67:1842–1852. doi: 10.1158/0008-5472.CAN-06-4038. [DOI] [PubMed] [Google Scholar]