

Abstract
Antisense oligonucleotides can regulate gene expression in living cells. As such, they regulate cell function and division, and can modulate cellular responses to internal and external stresses and stimuli. Although encouraging results from preclinical and clinical studies have been obtained and significant progress has been made in developing these agents as drugs, they are not yet recognized as effective therapeutics. Several major hurdles remain to be overcome, including problems with efficacy, off-target effects, delivery and side effects. The lessons learned from antisense drug development can help in the development of other oligonucleotide-based therapeutics such as CpG oligonucleotides, RNAi and miRNA.
Keywords: Antisense, RNAi, Drug targeting, cancer, chemotherapy
Historical perspective
The development of the antisense approach started in the late 1970s after the discovery that the expression of a specific gene product could be inhibited using a short complementary DNA sequence [1]. Since then, the antisense strategy has enjoyed exponential gains in interest and has been the subject of more than 16,000 publications. Between the appearance of the first publication about the antisense strategy in 1978 and the beginning of intensive antisense research (commencing in the early 1990s) very few studies were published. The increase in antisense research was largely a result of improvements in the methods used for DNA sequencing and synthesizing oligonucleotides [2, 3]. Other major milestones in the development of antisense strategies include numerous discoveries about antisense chemistry. The most notable discovery was the addition of a phosphorothioate backbone to the oligonucleotides, leading to a significant increase in their stability without major changes in their ability to hybridize with their target mRNA [3]. Other chemical modifications, including the development of DNA/RNA mixed-backbone oligonucleotides [4] and other oligonucleotides barely resembling DNA (such as peptide nucleic acid (PNA) and locked nucleic acid (LNA) structures) [3], have been made to increase the efficacy, stability and patentability of the antisense molecules.
One antisense oligonucleotide, Vitravene™ (Fomivirsen), was approved in 1998 for use against cytomegalovirus-induced retinitis by local injection [5]. However, although many antisense oligonucleotides targeting several genes that are important to severe human diseases such as cancer and infectious diseases have been in clinical trials for many years; some of them even in multiple clinical Phase III trials, none of these systemically administered antisense oligonucleotides have won marketing approval from regulatory bodies such as the FDA. Unfortunately, interest and investment in this area appears to be declining.
Proposed mechanisms of action for antisense oligonucleotides
Although antisense oligonucleotides have long been recognized as a naturally occurring gene regulation approach, the precise mechanisms of action for antisense molecules remain unclear. This is one of the major criticisms that many antisense researchers face. Two major mechanisms are widely accepted, namely physical blockage and RNase H activation [3, 6]. Other mechanisms observed for the antisense oligonucleotides include interference with mRNA processing and transport, and formation of a triplex directly with DNA [7, 8]. Other types of antisense oligonucleotides with different chemical modifications [for example: morpholino, peptide nucleic acid (PNA) or locked nucleic acid (LNA) modified oligonucleotides] are capable of acting by other mechanisms, but primarily rely on translation arrest [9, 10]. The mechanism by which the oligonucleotides exert their effects largely depends on their structure and chemistry.
Other mechanisms of action
While in vitro studies suggested that modifying the oligonucleotides would lead to differences in stability, uptake and activity, there were many other sequence-specific and structure-specific non-antisense effects in vivo that were not anticipated. Many of the structure-specific (but sequence-independent) effects were due to the modifications made to the oligonucleotides. Because of their negative charge, the phosphorothioate oligonucleotides have a tendency to bind serum proteins. As a result of this, these oligonucleotides were found to affect coagulation because they bind thrombin [11]. In addition to coagulopathies, the phosphorothioate modified oligonucleotides have a tendency to accumulate in the liver, and their administration leads to elevated transaminases, indicative of liver damage [12]. Although the effects are relatively modest, modified oligonucleotides have also been shown to stimulate the immune system [13]. Despite these unforeseen effects, the oligonucleotides are relatively safe, and have been administered at doses of up to 15 mg/kg to non-human primates [14], and Oblimersen (Genasense) has been given to patients for up to six cycles of 7 days at a 3 mg/kg/d dose with no severe adverse effects [15].
While the structure-specific effects were relatively moderate, a major unexpected sequence-dependent side effect of certain antisense oligonucleotides was intense immune system stimulation. In the mid–late 1990s it was discovered that certain motifs within the nucleotide sequences, containing either unmethylated CG motifs or GGGG motifs, were capable of stimulating an immune response – including stimulation of B cells and dendritic cells, and increased secretion of inflammatory cytokines [16, 17]. Although somewhat detrimental to the development of antisense oligonucleotides due to their unforeseen activity, oligonucleotides containing an immunostimulatory sequence have since been developed specifically to stimulate the immune system. The CpG oligonucleotides (also called immunostimulatory or immunomodulatory oligonucleotides) are currently being used in clinical trials for cancer, asthma and allergies, and as vaccine adjuvants [18, 19]. These unexpected and non-targeted effects of the antisense oligonucleotides have been partially responsible for their lack of success in the clinic, and explain why only one oligonucleotide has gained FDA approval.
Table 1 gives examples of first-generation antisense oligonucleotides under clinical development. The severity of the side effects of first-generation antisense ODNs is mostly dependent on the presence of certain sequence motifs, such as CpG dinucleotides [44]. Almost all of the oligoncleotides contains at least one CpG motif. The safety and efficacy of several second-generation mixed-backbone antisense ODNs, without CpG motifs, are presently being evaluated in clinical trials [45, 46].
Table 1.
Examples of antisense oligonucleotides in clinical trials
| Drug | Developer | Chemistry | Target | Disease | Highest Clinical Phase | Verdict | References |
|---|---|---|---|---|---|---|---|
| GEM 231 | Hybridon (Idera) | MBO | PKA R1α subunit | Cancer | II | Still in trials | 20 |
| Affinitak (Aprinocarsen, ISIS 3521/LY900003) | ISIS/Lilly | PS | PKC-α | Cancer | III | NSCLC- low efficacy, toxicity (epistaxis, thrombosis) | 21, 22 |
| Aezea (OL(1)p53/EL-625) | Eleos Pharma | PS | p53 | Cancer (AML) | II | Still in trials | 23 |
| Alicaforsen (ISIS 2302) | ISIS/Atlantic Healthcare | PS | ICAM-1 | Crohn’s Disease, IBD | Phase II | Still in trials | 24, 25 |
| ISIS 104838 | ISIS/Orasense | PS | TNF-α | Rheumatoid arthritis | II | Completed, awaiting publication | 24, 25 |
| GEM 91 (Trecorvirsen)/92 | Hybridon | PS/MBO | Gag | HIV | Phase II/I | Low efficacy, decreased platelets | 26 |
| Vitravene (Fomivirsen) | ISIS/Novartis | PS | CMV IE | CMV retinitis | Marketed | FDA approved | 25 |
| ISIS 2503 | ISIS | PS | H-Ras | Cancer | Phase II | Still in trials | 24 |
| EZN-2968 | Enzon Pharmaceuticals | LNA | Hif-1α | Cancer | I | Still in trials | 24, 27 |
| G4460/LR 3001 | Inex/Genta | PS | c-Myb | Cancer | II | Orphan drug for CML | 24, 28 |
| LErafAON | NeoPharm | PS | c-Raf | Cancer | I/II | Being redesigned due to toxicity | 29 |
| ISIS 5132 | ISIS | PS | c-Raf | Cancer | II | Still in trials | 25 |
| Genasense (Oblimersen/G3139) | Genta | PS | Bcl-2 | Cancer | III/NDA; I/II | Still in trials | 24, 28 |
| SPC2996 | Santaris Pharma | LNA | Bcl-2 | CLL (chronic lymphocytic leukemia) | I/II | Still in trials | 24, 30 |
| OGX-427 | ISIS/OncoGene X | MBO | Hsp27 | Cancer | Phase I | Phase I planned to begin in 2007 | 24, 25 |
| LY2181308 | Lilly | MOE gapmer | Survivin | Cancer | Phase I/II | Still in trials | 25 |
| LY2275796 | Lilly | MOE | eIF-4E | Cancer | Phase I | Still in trials | 25 |
| OGX-011 | ISIS/OncoGene X | MBO | Clusterin | Cancer | Phase II | Still in trials | 24, 25 |
| Veglin | VasGene Therapeutics | PS | VEGF | Cancer | Phase I/II | Still in trials | 31 |
| AP12009 | Antisense Pharma | PS | TGF-βII | Cancer | Phase II | Still in trials | 32 |
| GTI-2501 | Lorus Therapeutics | PS | Ribonucleotide Reductase R1 | Cancer | Phase II | Still in trials | 33 |
| GTI-2040 | Lorus Therapeutics | PS | Ribonucleotide Reductase R2 | Cancer | Phase II | Still in trials | 24, 33 |
| AEG 35156 | Aegera Pharma | MBO | XIAP | Cancer | Phase I/II | Still in trials | 24, 34 |
| MG 98 | MethylGene/MGI Pharma/British Biotech | MBO | DNA methyltransferase | Cancer | Phase II | No longer in trials | 24, 35 |
| EPI-2010 (RASON) | Epigenesis/Genta | PS | Adenosine A1 receptor | Asthma | Phase II | No longer in trials due to low efficacy | 36, 37 |
| ISIS 301012 | ISIS | MOE | Apo-B100 | Cardiovascular disease (High cholesterol) | Phase II | Still in trials | 25 |
| ISIS 113715 | ISIS | MOE | PTP-1B | Diabetes | Phase II | Still in trials | 25 |
| ISIS 14803 | ISIS/Merck | PS | HCV IRES | HCV | Phase I | No longer in development | 38 |
| ISIS 325568 | ISIS/Ortho-McNeil | MOE | Glucagon receptor | Type II diabetes | Phase I/II | Still in trials | 24, 25 |
| AVI-4065 | AVI Biopharma | Phosphoroamidate-Morpholino (PMO) | HCV | HCV | Phase II | Still in trials | 24, 39 |
| AVI-5126 | AVI Biopharma | PMO | CABG | Phase I | Still in trials | 39 | |
| AVI-6001 | AVI Biopharma | PMO | Influenza/avian flu | Phase I | Still in trials | 39 | |
| AVI-6002 | AVI Biopharma | PMO | Ebola virus | Phase I | Still in trials | 39 | |
| Resten-NG/MP | AVI Biopharma | PMO | c-Myc | Restenosis | Phase II | Still in trials | 39 |
| AVI-4557 | AVI Biopharma | PMO | Cyp 3A4 | Cancer/metabolism | Phase Ib | No longer in trials | 39 |
| AVI-4658 | AVI Biopharma | PMO | Dystrophin (exon-skipping) | Duchenne’s Muscular Dystrophy | Phase I/II | Still in trials | 24, 39 |
| ATL1102 | ISIS/Antisense Therapeutics Ltd | MOE | VLA-4 (CD49d) | Multiple Sclerosis | Phase II | Still in trials | 25, 40 |
| Not yet named | University of Pittsburgh | Phosphorothioate | CD40, CD80, CD86 in dendritic cells | Diabetes type I | Phase I | Still in trials | 24 |
| VRX496 | VIRxSYS corporation | Not published | CD4+ T cells with a lentiviral vector expressing an anti-env oligo | HIV | Phase II | Still in trials | 24, 41 |
| ASM8-003 | Topigen Pharmaceuticals | Phosphorothioate | CCR3 and common beta chain of IL-3/5 and GM-CSF receptors | Allergies/asthma | Phase I/II | Still in trials | 24, 42 |
| iCo-007 | iCo Therapeutics Inc | Phosphorothioate | c-Raf | Diabetic retinopathy | Phase I | Still in trials | 24, 43 |
Antisense therapy: hype versus reality
When the antisense strategy was first introduced, it was recognized that it could represent a specific, systemic gene silencing strategy. Successful development of such a strategy could allow an almost endless variety of human diseases to be treated, provided that a particular gene had been identified and characterized for the disease. Once the synthetic chemistry of the oligonucleotides had been simplified, making the oligonucleotides more readily available, the strategy gained favor as the new ‘hot’ technology. Antisense oligonucleotides have been used for a variety of purposes, including target validation, gene function studies and as experimental therapy for different diseases. The antisense oligonucleotides have been used most frequently for cancer-related targets including oncogenes, signaling molecules and mutant tumor suppressor genes [47–49], although pathogen-associated and other disease-related gene products such as ICAM-1 and TNF-α (for Crohn’s disease and Rheumatoid Arthritis, respectively) have also been targeted [50, 51].
Hundreds of antisense oligonucleotides have been examined in preclinical studies (generally in rodents) and preliminary clinical trials, and many have shown significant activity, both against the target gene product and against the disease [6, 52, 53]. However, the overall results of clinical trials of antisense oligonucleotides have been mixed. There have been clinical trials of more than thirty antisense oligonucleotides (Table 1), with the vast majority being developed for cancer therapy. While many of the oligonucleotides did not make it past Phase I trials, several have been in more advanced trials, even up to filing an NDA (new drug application with the US FDA). Box 1 contains case studies of two of these oligonucleotides; Vitravene and Oblimersen.
Box.1. Case study: Vitravene and Oblimersen
Drug Name: Genasense™ (Oblimersen sodium/G3139)
Target: Bcl-2
Rationale: Blc-2 is an oncogene that has been implicated in a variety of human cancers (54)
Disease target(s): Solid tumors and hematological cancers
Pre-clinical forecast: Positive: G3139 demonstrated significant decreases in Bcl-2, led to anti-tumor effects and was well-tolerated in animals (55, 56)
Clinical trials: Some positive suggesting that Genasense was well-tolerated and could exert anti-cancer effects, and that it may be especially useful for purging bone marrow of lymphoma patients. There were also some negative trial results, indicating that the agent was not effective and that it had toxic side-effects (55–57).
FDA experience: An NDA filed in 2004 for melanoma was rejected, but is being appealed.
Future forecast: Trials are still ongoing, and even if the appeal is unsuccessful, it is still likely that another NDA will be filed (for a different cancer type).
Drug Name: Vitravene™ (Fomivirsen/ISIS 2922)
Target: HIV-associated cytomegalovirus retinitis (CMV-Intermediate Early gene (IE55))
Rationale: Inhibiting the proliferation of CMV can preserve the sight of patients
Disease target(s): CMV
Pre-clinical forecast: Potent anti-viral effects with no retinal toxicity at up to 5 uM (rabbits, pigs, primates) and an acceptable pharmacokinetic profile (58–60).
Clinical trials: Positive
FDA experience: Vitravene was approved by the FDA in 1998
Future forecast: Although effective, the applications of Vitravene are limited because of its target and its route of administration (intravitreal). Moreover, it is possible that some CMV mutants may be innately resistant (or others may develop resistance) to the antisense oligonucleotide due to different IE sequences (61).
Clinical trials of antisense oligonucleotides
Many oligonucleotides were designed to decrease the expression of oncoproteins such as Bcl-2, c-Raf, H-Ras, c-Myc, c-Myb and XIAP [6, 62, 63]. Others have focused on cell signaling molecules implicated in cancer initiation or progression, including the tumor suppressor p53 (mutant), VEGF, IGF-1R, TGF-BII, PKA, and PKCα [6, 62, 63]. Still, other cancer-related molecules have been targeted including survivin, clusterin, ribonucleotide reductase, and DNA methyltransferase [6, 62, 63].
The results of the studies with these agents have varied widely, although numerous antisense oligonucleotides are still being evaluated in human clinical trials. Those that are no longer in development were discontinued for various reasons, including toxicity (Affinitak/Aprinocarsen) and low efficacy (GEM 91, LErafAON). Others have been shelved temporarily until a systemic antisense agent gains FDA approval (facilitating their application process). The failure of some of the oligonucleotides could have been due to the selection of patients, study endpoints or due to the sequence or chemistry of the oligonucleotides.
Several antisense oligonucleotides have a promising future, including GEM231 and AVI4658. These two oligonucleotides are of particular interest because their target-specific and therapeutic effects can be monitored easily by a blood test or biopsy, respectively [64, 65], facilitating clinical trials. As noted in Box 1, Genasense has been under investigation in the clinic for many years, and may also still have a bright future. Its target (Bcl-2) has been implicated in numerous cancers, and the agent has shown promise in several clinical trials [57]. It is possible that this drug could eventually gain approval by the FDA. However, there are a few issues, including its ‘second generation’ chemistry and the presence of a CpG motif that may be thwarting its success. Santaris Pharma has designed an anti-Bcl-2 antisense oligonucleotide with LNA modifications, which may be superior to Genasense in terms of decreased side effects [30].
Improving clinical trials of antisense oligonucleotides
Other improvements made to new trials are changes to the methods and endpoints of the clinical trials themselves. Following demonstration of activity in animal models, most early clinical trials of antisense oligonucleotides focused solely on the toxicity of the compounds, and did not evaluate the expression of the target in patients. In agreement with the push for novel agents to fail early, many Phase I studies are now examining toxicity and target expression. There is also now a push to be able to examine the target either directly (e.g. in biopsy specimens before/after treatment) or via biomarkers. Certain targets have serum biomarkers that can be easily measured to determine the effects of the antisense oligonucleotide (see Box 1 [65, 66]). While biomarkers do not exist (or are as yet unknown) for many targets, validation of the effects of the antisense oligonucleotide in tissue can be accomplished by methods commonly used by pathologists and molecular biologists. For example, immunohistochemistry can be used to assess the expression and localization of most oncogenes or signaling molecules. For those that are more difficult to detect due to compartmentalization or low expression, cell preparations can be made to separate/concentrate the specific organelles or compartments, then Western blotting and RT-PCR can be done to evaluate expression of the target.
Because many clinical trials failed in Phase II/III due to a lack of observed efficacy, monitoring expression of the target during Phase I trials would allow investigators to determine whether the oligonucleotide was ineffective because it was not reaching the target tissue in human patients (which could be a result of stability or delivery issues) or whether the inhibition of the target was not causing the desired effect (e.g. due to overlapping, complementary, or mutated pathways), thus preventing the continued administration of an ineffective agent.
Perhaps the most important improvement that is being made to antisense oligonucleotide trials is that they are now being combined with other agents. Given the heterogeneous nature of tumors, it is preferable to have more than one target for therapy. This helps to prevent the escape of cancer cells that have inherent mutations or develop new mutations to a single targeted pathway. Moreover, numerous preclinical studies suggest that targeting signaling molecules or oncogenes (e.g. MDM2) with antisense oligonucleotides can sensitize tumors to treatment with chemotherapeutic agents or radiation [67, 68]. It is possible that the oligonucleotides that produced underwhelming results in clinical studies as single agents might produce better anticancer effects if given in combination with another agent. It may be of interest to examine this possibility, at least at the preclinical level, before an antisense drug is considered ineffective.
It also bears mentioning that much is being learned about carcinogenesis and cancer progression. Targeting a single oncogene may inhibit growth of cells or even induce apoptosis, but most advanced tumors have numerous issues that have to be addressed in order to effectively eradicate the tumor. Targeting multiple oncogenes or pathways may be required to make any headway in tumor destruction. In addition, there are other emerging theories that discount targeting of oncogenes completely. For example, it has been postulated that chromosomal damage underlies most tumor initiation, progression and drug resistance [69]. In this case, therapies that repair or replace defective DNA would be required for optimal tumor destruction and long-term patient survival, while targeting oncogenes would be a temporary fix, at best. Nevertheless, as has been evidenced by successful treatment of numerous patients with agents targeting oncogenes (Herceptin®, Gleevec®, Tarceva®, etc), oncogenes still appear to represent valid targets for patient therapy, and can produce some long-lasting anti-cancer effects.
Other nucleic-acid-based therapeutics: can lessons from antisense therapy help?
Although only Vitravene has been approved by the FDA its approval opened the door for other oligonucleotide-based therapies, including ribozymes, RNA interference, aptamers and other types of gene therapy to enter the clinic. These strategies, while working by different mechanisms, are all based upon the principle that administration, or plasmid-driven expression, of exogenous DNA or RNA can be used to regulate the type and extent of expression of targeted gene products. Figure 1 shows the timeline for the development of these different strategies by denoting the number of PubMed publications focusing on them each year.
Figure 1. Ups and downs of RNA/DNA-based therapeutics and gene therapy.
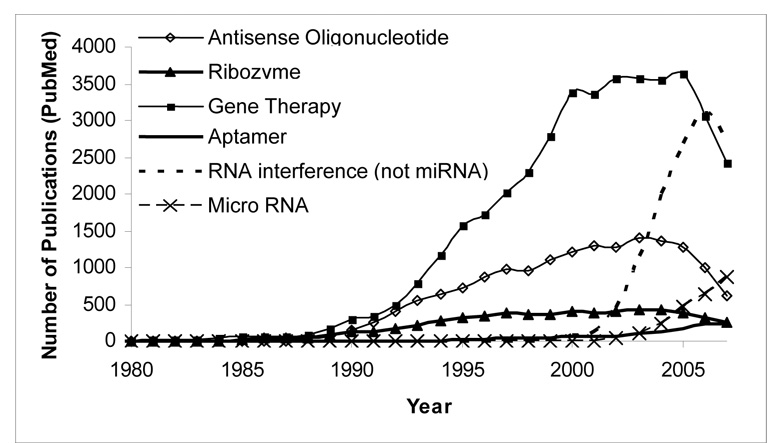
The number of publications are based on PUBMED citations
Ribozymes
Ribozymes are based upon catalytic RNA originally found in the protozoan tetrahymena [70]. While the naturally occurring ribozymes are self-splicing, modifications have yielded catalytic oligonucleotides that can cleave a targeted RNA sequence or revise the mRNA to generate correct sequences that can be translated into normal proteins [71]. Much like antisense oligonucleotides, ribozymes can be targeted to a variety of molecules, and have been developed as experimental therapeutics for cancer, infectious diseases and other human diseases, such as sickle cell anemia [48, 72, 73]. Although ribozymes can be manipulated somewhat, their catalytic nature is highly dependent upon their structure, decreasing the ability to modulate their chemistry to improve pharmacokinetics, efficacy or toxicity. There have been several ribozymes evaluated in clinical trials, and two ribozymes, one for HIV (OZ1) and another for cancer (Angiozyme) were recently examined in clinical trials in the USA [24]. No ribozymes have been approved by the FDA.
Aptamers
While aptamers, like antisense oligonucleotides and ribozymes, are short stretches of RNA or DNA, they work by a distinct mechanism of action. Unlike the other nucleotide-based strategies, complementarity is not important for the activity of aptamers; their tertiary and quaternary structures determine their binding [74]. Aptamers have specific 3-dimensional structures that can form complexes with target proteins and inhibit their activity [74]. For this reason, aptamers can be considered ‘chemical antibodies’. Although they can bind proteins like antibodies, aptamers are not immunogenic, even at doses up to 1000 times the therapeutic dose in primates [75]. Combining the properties of nucleotide-based therapies and antibodies, aptamers can be used to target extracellular and cytoplasmic proteins. Moreover, aptamers are amenable to all of the modifications that can be made to other nucleotide therapeutics, although care must be taken to preserve the structure, particularly the binding region [76].
With the recent improvements in HTS, it is possible to screen libraries of aptamers using Systematic Evolution of Ligands by Exponential Enrichment (SELEX) to determine the best aptamer for a particular target rapidly [77]. Aptamers can theoretically be targeted to almost any molecule and, like other nucleotide-based therapies, have been examined in preclinical models of cancer and other diseases [74, 78]. Several aptamers have been in recent clinical trials (i.e. REG1 targeting factor IXa and EYE001 targeting VEGF) and Macugen™ (Pegatinib), which targets VEGF, was approved by the FDA for use against wet macular degeneration in 2004.
RNA Interference
Although it was discovered only a decade ago [79], RNA interference (RNAi) has since become a standard for various types of laboratory research, and its pioneers were awarded the 2006 Nobel Prize in Medicine for their work. Like the antisense and ribozyme strategies, RNAi relies on complementarity between the RNA and its target mRNA to bring about destruction of the target. In vivo, long stretches of dsRNA can interact with the DICER endoribonuclease to be cleaved into short (21–23 nt) dsRNA with 3’ overhangs [80]. Then, the endogenous or synthetic short stretches of dsRNA enter the multinuclease-containing RNA-induced silencing complex (RISC) and these enzymes lead to specific cleavage of complementary targets [80]. While short (<23nt) segments of RNA are generally considered optimal for gene silencing [81], it has also been shown that longer (<30nt) sequences can lead to efficient, and perhaps even more potent, gene silencing. Rose et al demonstrated that a 27-mer siRNA was able to achieve 100-fold stronger gene silencing than a similar shorter sequence (although processing of the blunt 27-mer led to production of various siRNA of 19–23nt) [82]. Although longer sequences lead to generation of an interferon response by cells, it is not clear whether this response will be stronger for a 27-mer than a 21-mer sequence, thus 27-mers should still be considered for future applications.
At present, there are several different types of commonly used RNAi: short-interfering RNA (siRNA), short-hairpin RNA (shRNA) and micro RNA (miRNA), all of which can inhibit expression of the target gene product. The siRNA and shRNA (generally 20–22 nt in length, but they can be up to 30nt) were designed to overcome issues with immune system stimulation and complete translation arrest observed when longer RNA sequences were used for RNAi, and to optimize the silencing effects [81]. Despite the recent discovery of RNAi, several siRNA molecules have already been evaluated in human clinical trials [83]. These include a siRNA targeting IL-10 for treatment of preeclampsia, VEGF and VEGFR-1 for macular degeneration, and BCR-ABL for CML [24]. Other RNAi molecules are likely to be in clinical trials soon.
Another type of RNAi, miRNA, is a naturally occurring mammalian post-transcriptional gene regulatory system [84]. It has been suggested that miRNA-mediated regulation of gene products may account for various disease states, including cancer [85, 86]. A miRNA expressed by the Kaposi’s sarcoma virus was recently shown to mimic an endogenous human miRNA (miR-155) that regulates cell growth, promoting B cell transformation [87]. Thus, naturally occurring endogenous and exogenous miRNAs represent targets for therapy [88]. Similarly, it will probably be possible to use miRNAs like siRNA or antisense oligonucleotides to target endogenous gene regulation.
In fact, a recent study demonstrated that an array of miRNAs (although not a single miRNA sequence) could be used to inhibit the BCR-ABL oncogene implicated in leukemia [89]. It is also possible to use antisense oligonucleotides to knockdown expression of miRNAs, inhibiting their function in vivo. One research group demonstrated that the use of a PNA (peptide nucleic acid) or LNA/2-O-methyl oligonucleotide could decrease expression of miRNA-122 in human and rodent cell lines [90]. These findings suggest that miRNA can be successfully targeted or can be used to regulate gene expression. A related type of RNAi, piRNA/rasiRNA (Piwi-associated interfering RNA/repeat-associated siRNA), has also been discovered. The piRNAs are only starting to be characterized, but appear to regulate gene silencing, cell differentiation and gametogenesis, and could also represent targets for therapy [91, 92]. While no miRNAs or piRNAs have been examined in clinical trials (as targets or for therapy) it is likely that, given the rapid advances made in RNAi, they will be in the near future.
Gene Therapy
Although all of these approaches can loosely be considered gene therapy, the topic often refers to strategies meant to increase expression of normal (wildtype) gene products. Chief among these strategies are those using viral vectors (adenoviruses, adeno-associated viruses, retroviruses, pox viruses, etc) to deliver the gene of interest, although other strategies, such as nanoparticles, are also being used. Gene therapy can theoretically be used for any gene with a known sequence, and has been examined extensively for cancer therapy. Gene therapy is especially attractive for genetic disorders such as Muscular Dystrophy, Cystic Fibrosis and SCID (severe combined immunodeficiency); because they are well-characterized, the mutations responsible for most cases of the disease are known, and because there are no existing treatments that are sufficiently effective. There are currently more than 650 ongoing gene therapy clinical trials [24] but, despite the number of clinical trials, no gene therapy strategies that introduce genetic material to replace or supply missing or faulty genes have been approved by the FDA.
Thus, while antisense oligonucleotides were among the first nucleotide-based therapies to be investigated, a variety of other strategies exist. The lessons learned from antisense oligonucleotides, the problems encountered in their clinical development and the forecast for their future success will have a major impact on the future development of the other related strategies. Likewise, the findings from trials of the nucleotide-based agents representing other strategies are likely to influence the future development of antisense oligonucleotides.
Concluding remarks: can the antisense oligonucleotide strategy be salvaged?
In the past three decades, many lessons have been learned in the development of antisense oligonulceotides as a novel class of therapeutics. Table 2 briefly summarizes the major obstacles encountered during the various steps in their development, some of which still need to be addressed. It might be argued that these obstacles also apply to other agents, such as small molecule inhibitors (eg: Gleevec® targeting the bcr-abl translocation). The development of Gleevec® is a success story for rational drug design; a single target was selected, an agent that inhibited it was developed, tested and transitioned without major issues to the clinic. However, comparing the clinical failures of the various antisense agents tested with a single small molecule skews the perception of the success of the antisense strategy. Numerous small molecule inhibitors of various targets have been designed, and most have failed in the clinic. Thus, it is not surprising that of the few antisense oligonucleotides that have been examined in clinical trials, only one has gained FDA approval. Moreover, the early trials of antisense strategies were not properly designed to test the agents, and it is possible that better-designed trials might have revealed activity that was not apparent using the existing methods.
Table 2.
Lessons learned during antisense drug development
| Stage/Goals | Approaches and Requirements |
|---|---|
| 1. Design of Oligonucleotides | Watson-Crick pairing rules do not guarantee specificity |
| Off-target effects may not be avoidable | |
| Purity is an important factor | |
| SAR may not be apparent | |
| 2. Target Validation | Specific down-regulation of target genes may not be sufficient |
| Off-target effects may not be fully identified | |
| Feedback regulation may minimize the efficacy of down-regulation | |
| Compensation by other pathways may offset the antisense effects | |
| 3. In vitro/in vivo Activity | The mRNA level may not reflect gene expression |
| mRNA level may not correlate with protein level | |
| The mRNA or protein level may not correlate with activity | |
| An in vitro/in vivo relationship may be lacking | |
| 4. Pharmacology | Pharmacokinetics and pharmacodynamics are largely lacking |
| Studies of oligonucleotide metabolism are lacking | |
| In vitro and in vivo uptake can be completely different | |
| Approaches to study off-target effects are lacking | |
| Drug delivery approaches are lacking | |
| Drug-drug interaction studies are lacking | |
| 5. Toxicology | The safety of oligos has not been confirmed in humans |
| Toxicokinetic profiles of oligonuleotides are lacking | |
| Species differences are unknown | |
| Studies of drug-drug interactions in relation to toxicity are lacking | |
| Studies of mechanisms of toxicity are lacking | |
| 6. Clinical trials | Pharmacokinetic studies are limited |
| Biomarkers for monitoring drug and responses are lacking | |
| Pharmacodynamic studies are extremely limited | |
| Pharmacogenomic approaches are not well developed or applied | |
| Phase III trials are largely limited | |
| 7. Chemical Synthesis | Oligo synthesis on a large-scale and GMP-quality is limited, expensive Impurity is still an issue |
| 8. Regulatory Issues | Understanding of the novel class of therapeutics is limited |
| Novel clinical trial approaches are lacking | |
| Standards for combination therapy for safety and efficacy are not well established |
Similarly, investigators have been writing off gene therapy as a whole, including antisense oligonucleotide-based strategies, as ineffective, dangerous and too difficult to develop. While this might be the case for individual agents, it may be possible to overcome the reputation that this has earned the class as a whole. A number of improvements have been, and are still being, incorporated into the antisense agents themselves, as well as to the clinical trials examining them. In addition to the improvements already mentioned (e.g. changes in the chemistry and structures of the oligonucleotides, elimination of sequences causing side effects, better clinical study endpoints and combination therapy) there are also improvements being made to the selection of targets. Compared with the initial studies of antisense molecules, there are now more known targets, the targets are better characterized and it is possible to select better sequences to decrease expression of the target. Moreover, although these improvements have been demonstrated to improve the preclinical efficacy and safety of the antisense compounds, the original “second generation” compounds with phosphorothioate modified structures have demonstrated their activity and efficacy both in preclinical and in clinical studies (eg: Genasense). Thus, while the traditional antisense molecules were the forerunners of the more “evolved” molecules, they are not buried in the past, and many traditional antisense molecules are still undergoing clinical evaluation. Moreover, the discovery and design of novel strategies such as RNAi do not preclude the utility of the antisense oligonucleotides. For example, it has been shown that antisense oligonucleotides (that exert their functions via RNase H) are as effective as siRNA [93], and in fact, well-designed antisense molecules may be more effective, at least in vitro, than siRNA targeting the same molecule [94].
In conclusion, the antisense strategy is far from dead and, although the results of clinical trials have been unimpressive, it appears that the strategy still has a viable future. As more information is uncovered about the human genome, its regulation and the response of human cells to exogenous nucleotides, better agents can be designed. Moreover, the improvements made to the strategy should soon start to yield more-favorable results in clinical trials.
Acknowledgements
The work was supported in part by NIH/NCI grants R01 CA112029 and R01 CA121211. E.R. Rayburn was supported in part by the USA Department of Defense Prostate Cancer Research Program (grant number W81XWH-06-1-0063) and an NIH T32 (CA075930) “Cancer gene therapy training program” Training Grant/Postdoctoral Appointment‥
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
TEASER
Various problems have been encountered and resolved during the development of antisense oligonucleotides, and the resolution of the remaining problems may facilitate the development of both antisense and other strategies.
References
- 1.Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. U.S.A. 1978;75:280. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geiser T. Large-scale economic synthesis of antisense phosphorothioate analogues of DNA for preclinical investigations. Ann. N.Y. Acad. Sci. 1990;616:173. doi: 10.1111/j.1749-6632.1990.tb17838.x. [DOI] [PubMed] [Google Scholar]
- 3.Kurreck J. Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem. 2003;270:1628. doi: 10.1046/j.1432-1033.2003.03555.x. [DOI] [PubMed] [Google Scholar]
- 4.Kandimalla ER, et al. Mixed backbone antisense oligonucleotides: design, biochemical and biological properties of oligonucleotides containing 2′-5′-ribo- and 3′-5′-deoxyribonucleotide segments. Nucleic Acids Res. 1997;25:370. doi: 10.1093/nar/25.2.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crooke ST. Vitravene--another piece in the mosaic. Antisense Nucleic Acid Drug Dev. 1998;8:vii. doi: 10.1089/oli.1.1998.8.vii. [DOI] [PubMed] [Google Scholar]
- 6.Rayburn ER, et al. Antisense-based cancer therapeutics: are we there yet? Expert Opin. Emerg. Drugs. 2006;11:337. doi: 10.1517/14728214.11.2.337. [DOI] [PubMed] [Google Scholar]
- 7.Crooke ST. Molecular mechanisms of action of antisense drugs. Biochim. Biophys. Acta. 1999;1489:31. doi: 10.1016/s0167-4781(99)00148-7. [DOI] [PubMed] [Google Scholar]
- 8.Hanvey JC, et al. DNA triple-helix formation at physiologic pH and temperature. Antisense Res. Dev. 1991;1:307. doi: 10.1089/ard.1991.1.307. [DOI] [PubMed] [Google Scholar]
- 9.Boiziau C, et al. Mechanisms of the inhibition of reverse transcription by antisense oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 1992;89:768. doi: 10.1073/pnas.89.2.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokota T, et al. Potential of oligonucleotide-mediated exon-skipping therapy for Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2007;7:831. doi: 10.1517/14712598.7.6.831. [DOI] [PubMed] [Google Scholar]
- 11.Henry SP, et al. Inhibition of coagulation by a phosphorothioate oligonucleotide. Antisense Nucleic Acid Drug Dev. 1997;7:503. doi: 10.1089/oli.1.1997.7.503. [DOI] [PubMed] [Google Scholar]
- 12.Agrawal S, et al. Toxicologic effects of an oligodeoxynucleotide phosphorothioate and its analogs following intravenous administration in rats. Antisense Nucleic Acid Drug Dev. 1997;7:575. doi: 10.1089/oli.1.1997.7.575. [DOI] [PubMed] [Google Scholar]
- 13.Liang H, et al. Activation of human B cells by phosphorothioate oligodeoxynucleotides. J. Clin. Invest. 1996;98:1119. doi: 10.1172/JCI118894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webb MS, et al. Toxicity and toxicokinetics of a phosphorothioate oligonucleotide against the c-myc oncogene in cynomolgus monkeys. Antisense Nucleic Acid Drug Dev. 2001;11:155. doi: 10.1089/108729001300338681. [DOI] [PubMed] [Google Scholar]
- 15.O'Brien S, et al. Randomized phase III trial of fludarabine plus cyclophosphamide with or without oblimersen sodium (Bcl-2 antisense) in patients with relapsed or refractory chronic lymphocytic leukemia. J. Clin. Oncol. 2007;25:1114. doi: 10.1200/JCO.2006.07.1191. [DOI] [PubMed] [Google Scholar]
- 16.Krieg AM, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 17.Prasad V, et al. Oligonucleotides tethered to a short polyguanylic acid stretch are targeted to macrophages: enhanced antiviral activity of a vesicular stomatitis virus-specific antisense oligonucleotide. Antimicrob. Agents Chemother. 1999;43:2689. doi: 10.1128/aac.43.11.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat. Rev. Drug Discov. 2006;5:471. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 19.Wang H, et al. Synthetic oligodeoxynucleotides containing deoxycytidyl-deoxyguanosine dinucleotides (CpG ODNs) and modified analogs as novel anticancer therapeutics. Curr. Pharm. Des. 2005;11:2889. doi: 10.2174/1381612054546707. [DOI] [PubMed] [Google Scholar]
- 20.Goel S, et al. A phase I safety and dose escalation trial of docetaxel combined with GEM231, a second generation antisense oligonucleotide targeting protein kinase A R1alpha in patients with advanced solid cancers. Invest. New Drugs. 2006;24:125. doi: 10.1007/s10637-006-2378-x. [DOI] [PubMed] [Google Scholar]
- 21.Paz-Ares L, et al. Phase III study of gemcitabine and cisplatin with or without aprinocarsen, a protein kinase C-alpha antisense oligonucleotide, in patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 2006;24:1428. doi: 10.1200/JCO.2005.04.3299. [DOI] [PubMed] [Google Scholar]
- 22.Kelland L. Discontinued drugs in 2005: oncology drugs. Expert Opin. Investig. Drugs. 2006;15:1309. doi: 10.1517/13543784.15.11.1309. [DOI] [PubMed] [Google Scholar]
- 23. http://eleosinc.com/ (viewed 3/11/08)
- 24. www.clinicaltrials.gov (viewed 3/11/08)
- 25. http://www.isispharm.com (viewed 3/11/08)
- 26.Ivanova G, et al. Anti-HIV activity of steric block oligonucleotides. Ann. N.Y. Acad. Sci. 2006;1082:103. doi: 10.1196/annals.1348.033. [DOI] [PubMed] [Google Scholar]
- 27. http://www.enzon.com (viewed 3/11/08)
- 28. http://www.genta.com/home/ (viewed 3/11/08)
- 29.Dritschilo A, et al. Phase I study of liposome-encapsulated c-raf antisense oligodeoxyribonucleotide infusion in combination with radiation therapy in patients with advanced malignancies. Clin. Cancer Res. 2006;12:1251. doi: 10.1158/1078-0432.CCR-05-1260. [DOI] [PubMed] [Google Scholar]
- 30. http://www.santaris.com/ (viewed 3/11/08)
- 31. http://www.vasgene.com/ (viewed 3/11/08)
- 32. http://www.antisense-pharma.com (viewed 3/11/08)
- 33. http://www.lorusthera.com (viewed 3/11/08)
- 34. http://www.aegera.com (viewed 3/11/08)
- 35. http://www.methylgene.com (viewed 3/11/08)
- 36.Ball HA, et al. Sense and antisense: therapeutic potential of oligonucleotides and interference RNA in asthma and allergic disorders. Clin. Rev. Allergy Immunol. 2004;27:207. doi: 10.1385/CRIAI:27:3:207. [DOI] [PubMed] [Google Scholar]
- 37. http://www.epigene.com (viewed 3/11/08)
- 38.Georgopapadakou N. Discontinued drugs in 2005: anti-infectives. Expert Opin. Investig. Drugs. 2007;16:1. doi: 10.1517/13543784.16.1.1. [DOI] [PubMed] [Google Scholar]
- 39. http://www.avibio.com (viewed 3/11/08)
- 40. http://www.antisense.com.au/_home.asp (viewed 3/11/08)
- 41. http://www.virxsys.com/ (viewed 3/11/08)
- 42. www.topigen.com (viewed 3/11/08)
- 43. http://www.icotherapeutics.com/ (viewed 3/11/08)
- 44.Agrawal S, Kandimalla ER. Antisense and/or immunostimulatory oligonucleotide therapeutics. Curr. Cancer Drug Targets. 2001;1:197. doi: 10.2174/1568009013334160. [DOI] [PubMed] [Google Scholar]
- 45.Goel S, et al. A safety study of a mixed-backbone oligonucleotide (GEM231) targeting the type I regulatory subunit alpha of protein kinase A using a continuous infusion schedule in patients with refractory solid tumors. Clin. Cancer Res. 2003;9:4069. [PubMed] [Google Scholar]
- 46.Mani S, et al. Clinical studies in patients with solid tumors using a second-generation antisense oligonucleotide (GEM 231) targeted against protein kinase A type I. Ann. N.Y. Acad. Sci. 2003;1002:252. doi: 10.1196/annals.1281.028. [DOI] [PubMed] [Google Scholar]
- 47.Chan JH, et al. Antisense oligonucleotides: from design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006;33:533. doi: 10.1111/j.1440-1681.2006.04403.x. [DOI] [PubMed] [Google Scholar]
- 48.Rayburn E, et al. RNA Silencing Technologies in Drug Discovery and Target Validation. Lett. Drug Des. and Disc. 2005;2:1. [Google Scholar]
- 49.Dean NM, Bennett CF. Antisense oligonucleotide-based therapeutics for cancer. Oncogene. 2003;22:9087. doi: 10.1038/sj.onc.1207231. [DOI] [PubMed] [Google Scholar]
- 50.Yacyshyn B, et al. A randomized, double-masked, placebo-controlled study of alicaforsen, an antisense inhibitor of intercellular adhesion molecule 1, for the treatment of subjects with active Crohn's disease. Clin. Gastroenterol. Hepatol. 2007;5:215. doi: 10.1016/j.cgh.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 51.Sewell KL, et al. Phase I trial of ISIS 104838, a 2′-methoxyethyl modified antisense oligonucleotide targeting tumor necrosis factor-alpha. J. Pharmacol. Exp. Ther. 2002;303:1334. doi: 10.1124/jpet.102.036749. [DOI] [PubMed] [Google Scholar]
- 52.Leonetti C, Zupi G. Targeting different signaling pathways with antisense oligonucleotides combination for cancer therapy. Curr. Pharm. Des. 2007;13:463. doi: 10.2174/138161207780162917. [DOI] [PubMed] [Google Scholar]
- 53.Redell MS, Tweardy DJ. Targeting transcription factors for cancer therapy. Curr. Pharm. Des. 2005;11:2873. doi: 10.2174/1381612054546699. [DOI] [PubMed] [Google Scholar]
- 54.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klasa RJ, et al. Oblimersen Bcl-2 antisense: facilitating apoptosis in anticancer treatment. Antisense Nucleic Acid Drug Dev. 2002;12:193. doi: 10.1089/108729002760220798. [DOI] [PubMed] [Google Scholar]
- 56.Tolcher AW. Targeting Bcl-2 protein expression in solid tumors and hematologic malignancies with antisense oligonucleotides. Clin. Adv. Hematol. Oncol. 2005;3:635. [PubMed] [Google Scholar]
- 57.Kim R, et al. Antisense and nonantisense effects of antisense Bcl-2 on multiple roles of Bcl-2 as a chemosensitizer in cancer therapy. Cancer Gene Ther. 2007;14:1. doi: 10.1038/sj.cgt.7700986. [DOI] [PubMed] [Google Scholar]
- 58.Flores-Aguilar M, et al. Evaluation of retinal toxicity and efficacy of anti-cytomegalovirus and anti-herpes simplex virus antiviral phosphorothioate oligonucleotides ISIS 2922 and ISIS 4015. J. Infect. Dis. 1997;175:1308. doi: 10.1086/516461. [DOI] [PubMed] [Google Scholar]
- 59.Perry CM, Balfour JA. Fomivirsen. Drugs. 1999;57:375. doi: 10.2165/00003495-199957030-00010. discussion 381. [DOI] [PubMed] [Google Scholar]
- 60.Leeds JM, et al. Pharmacokinetics of a potential human cytomegalovirus therapeutic, a phosphorothioate oligonucleotide, after intravitreal injection in the rabbit. Drug Metab. Dispos. 1997;25:921. [PubMed] [Google Scholar]
- 61.Mulamba GB, et al. Human cytomegalovirus mutant with sequence-dependent resistance to the phosphorothioate oligonucleotide fomivirsen (ISIS 2922) Antimicrob. Agents Chemother. 1998;42:971. doi: 10.1128/aac.42.4.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de Nigris F, et al. Targeting c-Myc, Ras and IGF cascade to treat cancer and vascular disorders. Cell Cycle. 2006;5:1621. doi: 10.4161/cc.5.15.3138. [DOI] [PubMed] [Google Scholar]
- 63.Crooke ST. Potential roles of antisense technology in cancer chemotherapy. Oncogene. 2000;19:6651. doi: 10.1038/sj.onc.1204093. [DOI] [PubMed] [Google Scholar]
- 64.Aartsma-Rus A, van Ommen GJ. Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA. 2007;13:1609. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang H, et al. Extracellular activity of cyclic AMP-dependent protein kinase as a biomarker for human cancer detection: distribution characteristics in a normal population and cancer patients. Cancer Epidemiol. Biomarkers Prev. 2007;16:789. doi: 10.1158/1055-9965.EPI-06-0367. [DOI] [PubMed] [Google Scholar]
- 66.Nesterova M, et al. Autoantibody biomarker opens a new gateway for cancer diagnosis. Biochim. Biophys. Acta. 2006;1762:398. doi: 10.1016/j.bbadis.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 67.Wang H, et al. Chemosensitization and radiosensitization of human cancer by antisense anti-MDM2 oligonucleotides: in vitro and in vivo activities and mechanisms. Ann. N.Y. Acad. Sci. 2003;1002:217. doi: 10.1196/annals.1281.025. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Z, et al. Radiosensitization by antisense anti-MDM2 mixed-backbone oligonucleotide in in vitro and in vivo human cancer models. Clin. Cancer Res. 2004;10:1263. doi: 10.1158/1078-0432.ccr-0245-03. [DOI] [PubMed] [Google Scholar]
- 69.Duesberg P, et al. Aneuploidy and cancer: from correlation to causation. Contrib. Microbiol. 2006;13:16. doi: 10.1159/000092963. [DOI] [PubMed] [Google Scholar]
- 70.Inoue T, et al. Intermolecular exon ligation of the rRNA precursor of Tetrahymena: oligonucleotides can function as 5′ exons. Cell. 1985;43:431. doi: 10.1016/0092-8674(85)90173-4. [DOI] [PubMed] [Google Scholar]
- 71.Sullenger BA, Cech TR. Ribozyme-mediated repair of defective mRNA by targeted, trans-splicing. Nature. 1994;371:619. doi: 10.1038/371619a0. [DOI] [PubMed] [Google Scholar]
- 72.Grassi G, et al. Therapeutic potential of hammerhead ribozymes in the treatment of hyper-proliferative diseases. Curr. Pharm. Biotechnol. 2004;5:369. doi: 10.2174/1389201043376760. [DOI] [PubMed] [Google Scholar]
- 73.Bartolome J, et al. Ribozymes as antiviral agents. Minerva Med. 2004;95:11. [PubMed] [Google Scholar]
- 74.Ireson CR, Kelland LR. Discovery and development of anticancer aptamers. Mol. Cancer Ther. 2006;5:2957. doi: 10.1158/1535-7163.MCT-06-0172. [DOI] [PubMed] [Google Scholar]
- 75.Que-Gewirth NS, Sullenger BA. Gene therapy progress and prospects: RNA aptamers. Gene Ther. 2007;14:283. doi: 10.1038/sj.gt.3302900. [DOI] [PubMed] [Google Scholar]
- 76.Wilson C, Keefe AD. Building oligonucleotide therapeutics using non-natural chemistries. Curr. Opin. Chem. Biol. 2006;10:607. doi: 10.1016/j.cbpa.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 77.Djordjevic M. SELEX experiments: new prospects, applications and data analysis in inferring regulatory pathways. Biomol. Eng. 2007;24:179. doi: 10.1016/j.bioeng.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 78.Dyke CK, et al. First-in-human experience of an antidote-controlled anticoagulant using RNA aptamer technology: a phase 1a pharmacodynamic evaluation of a drug-antidote pair for the controlled regulation of factor IXa activity. Circulation. 2006;114:2490. doi: 10.1161/CIRCULATIONAHA.106.668434. [DOI] [PubMed] [Google Scholar]
- 79.Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 80.Bernstein E, et al. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 81.Elbashir SM, et al. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rose SD, et al. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 2005;33:4140. doi: 10.1093/nar/gki732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Fougerolles A, et al. Interfering with disease: a progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007;6:443. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001;107:823. doi: 10.1016/s0092-8674(01)00616-x. [DOI] [PubMed] [Google Scholar]
- 85.Georges M, et al. Polymorphic miRNA-mediated gene regulation: contribution to phenotypic variation and disease. Curr. Opin. Genet. Dev. 2007;17:166. doi: 10.1016/j.gde.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 86.Jeyaseelan K, et al. MicroRNAs as therapeutic targets in human diseases. Expert Opin. Ther. Targets. 2007;11:1119. doi: 10.1517/14728222.11.8.1119. [DOI] [PubMed] [Google Scholar]
- 87.Gottwein E, et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450:1096. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Esau CC, Monia BP. Therapeutic potential for microRNAs. Adv. Drug Deliv. Rev. 2007;59:101. doi: 10.1016/j.addr.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 89.McLaughlin J, et al. Sustained suppression of Bcr-Abl-driven lymphoid leukemia by microRNA mimics. Proc. Natl. Acad. Sci. U.S.A. 2007;104:20501. doi: 10.1073/pnas.0710532105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fabani MM, Gait MJ. miR-122 targeting with LNA/2′-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA peptide conjugates. RNA. 2008;14:336. doi: 10.1261/rna.844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zamore PD. RNA silencing: genomic defence with a slice of pi. Nature. 2007;446:864. doi: 10.1038/446864a. [DOI] [PubMed] [Google Scholar]
- 92.Taubert H, et al. Expression of the stem cell self-renewal gene Hiwi and risk of tumour-related death in patients with soft-tissue sarcoma. Oncogene. 2007;26:1098. doi: 10.1038/sj.onc.1209880. [DOI] [PubMed] [Google Scholar]
- 93.Vickers TA, et al. Efficient reduction of target RNAs by small interfering RNA and RNase H-dependent antisense agents. A comparative analysis. J. Biol. Chem. 2003;28:7108. doi: 10.1074/jbc.M210326200. [DOI] [PubMed] [Google Scholar]
- 94.Miyagishi M, et al. Comparison of the suppressive effects of antisense oligonucleotides and siRNAs directed against the same targets in mammalian cells. Antisense Nucleic Acid Drug Dev. 2003;13:1. doi: 10.1089/108729003764097296. [DOI] [PubMed] [Google Scholar]