

Abstract
The voltage-gated sodium channel is the site of action of more than six classes of neurotoxins and drugs that alter its function by interaction with distinct, allosterically coupled receptor sites. Batrachotoxin (BTX) is a steroidal alkaloid that binds to neurotoxin receptor site 2 and causes persistent activation. BTX binding is inhibited allosterically by local anesthetics. We have investigated the interaction of BTX with amino acid residues I1760, F1764, and Y1771, which form part of local anesthetic receptor site in transmembrane segment IVS6 of type IIA sodium channels. Alanine substitution for F1764 (mutant F1764A) reduces tritiated BTX-A-20-α-benzoate binding affinity, causing a 60-fold increase in Kd. Alanine substitution for I1760, which is adjacent to F1764 in the predicted IVS6 transmembrane alpha helix, causes only a 4-fold increase in Kd. In contrast, mutant Y1771A shows no change in BTX binding affinity. For wild-type and mutant Y1771A, BTX shifted the voltage for half-maximal activation ≈40 mV in the hyperpolarizing direction and increased the percentage of noninactivating sodium current to ≈60%. In contrast, these BTX effects were eliminated completely for the F1764A mutant and were reduced substantially for mutant I1760A. Our data suggest that the BTX receptor site shares overlapping but nonidentical molecular determinants with the local anesthetic receptor site in transmembrane segment IVS6 as well as having unique molecular determinants in transmembrane segment IS6, as demonstrated in previous work. Evidently, BTX conforms to a domain–interface allosteric model of ligand binding and action, as previously proposed for calcium agonist and antagonist drugs acting on l-type calcium channels.
Voltage-gated sodium channels are responsible for the rapid depolarization during the initial phase of the action potential in most excitable cells (1). The brain sodium channel consists of a pore-forming α subunit of ≈260 kDa associated with a 36-kDa β1 subunit and a 33-kDa β2 subunit (reviewed in ref. 2). The primary structure of the α subunit is organized in four homologous domains (I-IV) that contain six transmembrane segments (S1-S6) and an additional membrane-associated segment between S5 and S6 and are connected by large intracellular loops (3, 4).
Potent neurotoxins specifically bind to more than six distinct but allosterically coupled receptor sites on the voltage-gated sodium channel and modify its function with lethal consequences (2, 5–7). The sodium channel is also the site of action for local anesthetics and certain anticonvulsant and antiarrhythmic drugs that are used therapeutically to decrease sodium channel activity in syndromes of hyperexcitability, such as cardiac arrhythmia and epileptic seizure (1, 8, 9). Although these sodium channel binding sites are distinct, allosteric interactions among the sites modulate drug and toxin binding (10–15). Neurotoxin receptor site 2 binds neurotoxins that cause persistent activation of sodium channels, including batrachotoxin (BTX), veratridine, aconitine, and grayanotoxin (10, 12, 16). BTX is a steroidal alkaloid from the skin secretions of South American frogs of the genus Phyllobates (16). Binding of BTX shifts the voltage dependence of sodium channel activation in the negative direction, blocks inactivation, and alters ion selectivity (17–20). Its binding to site 2 is nearly irreversible and is highly dependent on the state of the channel with a strong preference for open sodium channels. The effects of BTX on activation and inactivation suggest that its binding site may contain amino acids that are crucial for both the activation and inactivation gating mechanisms, and its effects on ion selectivity suggest that the toxin may bind to a site near the channel pore. Binding of toxins at neurotoxin receptor site 2 is enhanced allosterically by the binding of α-scorpion toxins to neurotoxin receptor site 3 (10), brevetoxins to neurotoxin receptor site 5 (11, 14), and pyrethroid insecticides binding at an additional receptor site (13, 14, 21). Toxin binding at site 2 is inhibited allosterically by binding of tetrodotoxin at neurotoxin receptor site 1 (15) and by local anesthetics and related anticonvulsant and antiarrhythmic drugs (22–26). Local anesthetic block of BTX-modified sodium channels requires higher concentrations of drug than block of unmodified channels but does not displace BTX from its receptor (18, 27, 28).
In previous work from this laboratory, the S6 segment of domain I was identified as one molecular component of the BTX receptor site of brain sodium channels by photoaffinity labeling with a photoreactive toxin derivative and antibody mapping of the labeled peptides (29). Subsequent site-directed mutagenesis of the skeletal muscle sodium channel showed that replacement of I433, N434, or L437 within segment IS6 with the positively charged residue lysine abolishes BTX effects (30), suggesting that these amino acid residues in segment IS6 are components of the BTX receptor site. Photolabeling experiments identify only amino acids that are associated directly with the photoreactive group on the ligand, so additional critical interactions may not be detected. We hypothesized that the strong allosteric interaction between local anesthetics and BTX may imply an additional binding region for BTX in close physical proximity to the local anesthetic binding site. Previous work has identified amino acid residues in the S6 segment of domain IV as important molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant binding (31–33). Therefore, we have investigated these amino acids as possible components of the BTX receptor site.
EXPERIMENTAL PROCEDURES
Materials.
Tritiated BTX-A-20-α-benzoate ([3H]BTX-B) was from Dupont/NEN (specific activity, 39.9 Ci/mmol). The synthetic pyrethroid derivative RU 51049 was a generous gift from Denis Guedin (Roussel Uclaf, Romainville, France) and was dissolved in glycerol formal (Sigma) to make a 10 mM stock solution. [3H]brevetoxin 3 (PbTx-3, specific activity, 14.9 Ci/mmol), PbTx-3, and PbTx-1 were generous gifts from Daniel Baden (University of Miami). Both PbTx-1 and PbTx-3 were dissolved in ethanol to make 1 mM stock solutions. BTX (0.5 mM stock solution in ethanol) was a generous gift of John Daly (National Institutes of Health, Bethesda, MD). Veratridine (200 mM in glycerol formal) was obtained from Sigma. All other chemicals were reagent grade from commercial sources.
Molecular Biology.
Site-directed mutagenesis for rIIA sodium channel cDNAs in the pVA2580 vector has been described (34). To construct cDNAs for mammalian cell expression, blunt-ended 1,719-bp fragments encoding F1764A and Y1771A were excised by using EcoRV and were subcloned into pCDM8 containing the remainder of the sodium channel. Clones were transformed into MC1061 cells (Invitrogen), were screened by PCR for orientation, and were sequenced by using the ABI Prism dye terminator cycle sequencing kit (Perkin–Elmer Applied Biosystems). The mutation I1760A was amplified by PCR in a 600-bp cDNA fragment from a BstEII site to a silent XhoI site inserted at L1776 and was subcloned into the rIIA sodium channel cDNA in pCDM8 containing the silent XhoI site. cDNA clones were sequenced through the entire PCR fragment.
Cell Culture, Transfection, and Membrane Preparation.
tsA-201 cells, which are human embryonic kidney cells (HEK293) that have been transfected stably with simian virus 40 large tumor antigen (Robert Dubridge, Cell Genesis, Foster City, CA), were maintained at 37°C in 10% CO2 in DMEM/F12 medium (GIBCO/BRL/Life Technologies) supplemented with 10% fetal bovine serum (Gemini Biological Products, Calabasas, CA), 20 μg/ml penicillin, and 10 μg/ml streptomycin (Gemini Biological Products). cDNAs for the sodium channel α and β1 subunits were transfected transiently by calcium phosphate precipitation as described (35). The channel subunits were added in a 1:0.1 α:β1 molar ratio that previously was determined to be the optimal concentration for α subunit expression. After 36 hours, cells were washed twice with PBS and were harvested by using a cell scraper in a homogenization buffer containing 100 mM Tris (pH 7.4), 2 mM EDTA, and 500 mM choline chloride supplemented with 34 ng/ml phenylmethanesulfonyl fluoride and 1 μM benzamidine, 1 μM pepstatin A, 1 μg/ml aprotinin, and 1 μg/ml leupeptin. Cells were homogenized for 2 × 15 s with a polytron homogenizer and 10 times with a dounce homogenizer at 0°C. The homogenates were centrifuged at 200 × g for 5 min to remove the cellular debris, and the supernatant was centrifuged at 45,000 × g for 30 min to pellet the membrane fraction. Membranes were rehomogenized in binding buffer (50 mM Hepes, pH 7.5/130 mM choline Cl/5.4 mM KCl/0.5 mM MgSO4/5.5 mM glucose/0.01% Emulphor) and were stored at −80°C.
In Vitro Binding.
Binding reactions were carried out in 1 ml of binding buffer containing ≈500 μg of total protein (1/10 of a confluent 150-mm cell culture plate) in borosilicate glass tubes. For the BTX binding assays, 20 μl each of RU51049 to a final concentration of 10 μM and PbTx-1 to a final concentration of 100 nM were added to each tube. [3H]BTX-B was added at final concentrations ranging from 0.1 to 6 nM. Nonspecific binding was determined by the addition of veratridine to 10 μM and was subtracted from the total binding to determine specific binding. The reactions were incubated at 32°C for 4 h, were washed three times with wash buffer (163 mM choline Cl/5 mM Hepes, pH 7.5/1.8 mM CaCl2/0.8 mM MgSO4) containing 0.1 mg/ml BSA, and bound ligand was collected by vacuum filtration through GF-C glass fiber filters that were equilibrated in wash buffer containing 0.3% polyethyleneimine. PbTx binding experiments were conducted by using concentrations from 0.5 to 10 nM [3H]PbTx-3, and nonspecific binding was measured in the presence of 5 μM PbTx-3. The reactions were incubated for 3 h at 4°C, were washed with wash buffer containing 0.01% Triton X-100 and 0.2 mg/ml lysozyme, and were filtered though GF-C filters that had been equilibrated in wash buffer containing Triton X-100 and lysozyme. Kd and maximum binding were determined by fitting to a nonlinear one-site binding curve (prism, GraphPad, San Diego).
Electrophysiological Studies.
tsA-201 cells were cotransfected with a cDNA encoding the human CD8 marker protein (EBO-pCD-leu2; American Type Culture Collection) in addition to sodium channel α and β1 subunits by using the calcium phosphate precipitation and antibody-labeling method as described (36). Successfully transfected cells then were identified with magnetic polystyrene microspheres coated with anti-CD8 antibody (ref. 37; Dynabeads M-450 CD8, Dynal, Great Neck, NY). Cells were maintained as described above.
Whole-cell sodium currents were recorded from tsA-201 cells expressing wild-type (WT) or mutant sodium channels (38, 39). Patch pipettes were pulled from 75-μl micropipette glass (VWR Scientific) and were fire polished before use. The external recording solution consisted of (in mM): 140 NaCl, 10 Hepes, 1 MgCl2, 0.4 CdCl2, 25 CsCl, 5 BaCl2 (pH 7.3 with NaOH; 300–305 microosmoles/liter. The internal recording solution consisted of (in mM): 188.9 N-methyl d-glucamine, 40 Hepes, 4 MgCl2, 1 NaCl, 0.1 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate acid, 25 phosphocreatine, 2–4 Na2ATP, 0.2 Na3GTP, 0.1 leupeptin (pH 7.2 with H2S04; 270–275 microosmoles/liter. BTX was prepared as a concentrated (0.5 mM) stock in ethanol and was diluted in internal solution before recording. BTX was applied intracellularly at a concentration of 10 μM in the recording pipette.
Electrode resistances were typically 3–6 megaohms in the bath. Recordings were obtained by using an Axon Instruments 1C patch clamp (Axon Instruments, Foster City, CA). Voltage pulses were delivered, and currents were recorded by using a personal computer running basic-fastlab software to control a data acquisition interface (Indec Systems, Santa Cruz, CA). Series resistance compensation (70–80%) was used routinely. Data were collected by using standard voltage step protocols. Conductance (g) was calculated as g(V) = I(V − Vrev), where I was the current, V was the test pulse voltage, and Vrev was the measured reversal potential. Conductance–voltage curves were fit with a single Boltzmann function of the form:
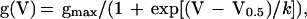 |
where V0.5 was the half activation voltage, k was a slope factor, and gmax was the maximum conductance. Least-squares curve fitting and statistical analysis were done by using sigma plot (Jandel, San Rafael, CA) and igor pro (WaveMetrics, Lake Oswego, OR). Statistics are presented as means ± SEM.
RESULTS AND DISCUSSION
High-Affinity Binding of [3H]BTX-B to Sodium Channels in Transfected Cells.
Measurement of BTX-B binding to sodium channels in vitro is difficult because of high levels of nonspecific binding and relatively low affinity of the hydrophobic ligand. As shown (14, 29), [3H]BTX-B binding affinity is enhanced greatly by the addition of the brevetoxin PbTx-1 and the pyrethroid RU51049 to the binding mixture through the allosteric interactions among the receptor sites for these three ligands. These toxins all enhance the probability that the sodium channel is in the open state and may enhance [3H]BTX-B binding by shifting sodium channels persistently to the high affinity open state. In the presence of these toxins, [3H]BTX-B binding is highly specific (>90%). Sodium channel α subunits were expressed transiently alone or in combination with the β1 subunit in tsA-201 cells, and membrane fractions were prepared and analyzed for specific BTX binding. As shown in Fig. 1, [3H]BTX-B binds with high affinity to the α subunit of the voltage-gated sodium channel. Coexpression of the β1 subunit increases the total number of BTX binding sites without changing binding affinity. This is consistent with previous results showing that coexpression of β1 subunits increases the number of saxitoxin binding sites but does not affect the affinity of the channel for that toxin (40). The [3H]BTX-B binding affinity of expressed sodium channels in the presence of PbTx-1 and RU51049 is 0.84 ± 0.03 nM (n = 5). This agrees with previous results (14) for reconstituted sodium channels from rat brain, suggesting that β2 subunits, which are present in reconstituted channels but not in the transfected cells, do not play a major role in BTX binding. These data also show that the sodium channel α subunit alone is sufficient for high-affinity BTX binding. However, to maximize the number of sodium channels, we cotransfected the α and β1 subunits in all subsequent experiments.
Figure 1.

Binding of BTX to transiently expressed sodium channels. [3H]BTX-B binding was measured in the presence of 100 nM PbTx-1 and 10 μM RU51049. Scatchard analysis was conducted on membrane preparations of tsA-201 cells transfected with the type IIA α subunits only (closed triangles) or α + β1 subunits (closed squares). Kd was 0.84 nM for both, and maximum binding was 46 fmol/mg total protein (α alone) and 94 fmol/mg protein (α + β1). This experiment was repeated three times with similar results. B/F, (Bound [3H]BTX-B)/(Free [3H]BTX-B).
Effect of Mutations in the Local Anesthetic Receptor Site on BTX and PbTx Affinity.
To examine the interactions of BTX with the local anesthetic receptor site, we measured BTX binding to WT sodium channels and to channels having mutations in the local anesthetic receptor site. Based on our previous studies (31), we examined mutants with substitutions of alanine for I1760, F1764, or Y1771 in transmembrane segment IVS6 that substantially reduce local anesthetic block. Binding of [3H]BTX-B to membranes from cells expressing mutant Y1771A yielded a Kd that was identical to that for WT sodium channels (Fig. 2 A and B). In contrast, the Kd values for binding to mutants I1760A and F1764A were 4- and 60-fold greater than WT, respectively. These results indicate that F1764 is essential for BTX binding and that I1760 also plays an important role in the interaction.
Figure 2.

BTX and brevetoxin binding to sodium channels with point mutations in the local anesthetic receptor site. (A) Scatchard analysis of binding of [3H]BTX-B in the presence of 10 μM RU51049 and 100 nM PbTx-1 for WT (closed squares), I1760A (closed triangles), F1764A (open squares), and Y1771A (open circles). Nonspecific binding was determined by the addition of excess unlabeled veratridine. The data were normalized with respect to maximum binding. (B) Averages of Kd values for BTX binding determined from three experiments. (C) Scatchard analysis of binding of [3H]PbTx-3. Nonspecific binding was determined in the presence of excess unlabeled PbTx-3. (D) Averages of Kd values for PbTx binding determined from three experiments. %B/F, Percent maximum bound (3H]BTX-B)/(Free [3H]BTX-B).
To determine whether effects of these mutations were specific to local anesthetics and BTX, we examined binding of PbTx, which is not thought to interact allosterically with local anesthetic binding. As illustrated in Fig. 2 C and D, we found that mutations I1760A, F1764A, and Y1771A had no effect on PbTx-3 binding affinity. This result shows that the effects of mutations of these amino acids are specific to binding of BTX and local anesthetics and are not global effects that disrupt all toxin binding to sodium channels.
Interaction of Pyrethroids with BTX on Local Anesthetic-resistant Sodium Channels.
We next examined whether allosteric modulation of BTX binding by the pyrethroid RU51049 also was affected by these three mutations. All experiments were normalized for the total number of binding sites relative to the WT sodium channel as measured by [3H]PbTx-3 binding. Little or no specific binding was observed for BTX alone or for BTX plus PbTx-1. RU51049 significantly increased the specific binding of BTX to each of these sodium channel mutants (Fig. 3), indicating that allosteric interactions between pyrethroids and BTX were retained in the mutants. PbTx-1 caused a further increase in binding of [3H]BTX-B to RU51049-modified WT sodium channels as well as to I1760A and Y1771A mutant channels, indicating that PbTx-1 bound to and modulated RU51049-bound sodium channels in these experiments. In contrast, PbTx did not enhance binding of [3H]BTX-B to F1764A channels with or without RU51049, suggesting that this mutation disrupted the allosteric interaction between the PbTx and BTX binding sites in addition to reducing the affinity for BTX binding.
Figure 3.

Allosteric interaction of neurotoxins with BTX binding to sodium channels with point mutations in the local anesthetic receptor site. BTX binding at a concentration of 5 nM is shown for WT (striped bars), I1760A (open bars), F1764A (stippled bars), and Y1771A (closed bars). The means ± SEM of three replicates are shown. The data were corrected for the Bmax for PbTx binding in parallel experiments to account for differences in the total number of binding sites in each sample.
Effects of BTX on Sodium Channel Function.
BTX drastically alters the functional properties of voltage-gated sodium channels (18, 20). It shifts the voltage dependence of activation in the hyperpolarizing direction by as much as 30 mV and inhibits both fast and slow inactivation, causing the channel to remain open persistently. This effect is highly state-dependent, with BTX binding selectively to channels in the open state. WT and mutant sodium channel cDNAs were cotransfected with the β1 subunit into tsA-201 cells and were studied by whole-cell voltage-clamp recording. Depolarization of transfected cells expressing WT sodium channels gave rise to a rapidly activating and inactivating inward current (Fig. 4A). Including 10 μM BTX in the recording pipette had little effect on WT sodium channels in cells that were not stimulated strongly (Fig. 4A, Control). This is expected because BTX binds preferentially to the open state. Subsequent application of a 5-Hz train of 2,000 depolarizing pulses to 0 mV resulted in BTX modification of a substantial fraction of the WT sodium channels, as indicated by incomplete inactivation of the sodium current (Fig. 4A, BTX). The mean fraction of noninactivating current increased from 6.7 to 60.3% for WT channels after BTX modification (Fig. 5A).
Figure 4.

Effects of BTX on the functional properties of WT channels. (A) Representative current traces elicited by a 50-ms test pulse to 0 mV from a holding potential of −120 mV before and after BTX modification. After formation of a high resistance seal and establishment of the whole-cell recording configuration with a recording pipette containing BTX, a brief voltage pulse protocol was applied to confirm the quality of the voltage clamp, and then the cell was stimulated by depolarization to 0 mV. The current elicited by this first stimulation is labeled “Control”. After ≈10 min of whole-cell dialysis, a series of 2,000 pulses to 0 mV for 50 ms each was applied, and then the sodium current was recorded in a second test pulse (trace labeled “BTX”). (B) Peak current–voltage relationship for type IIA sodium channels before (closed circles) and after (open circles) BTX modification. (C) Conductance–voltage activation curves generated before and after BTX modification. Note that the data after BTX modification are fitted with the sum of two Boltzmann relationships. Data in all panels that were obtained after BTX modification have been normalized to the maximum current observed before modification.
Figure 5.

Effects of BTX on the functional properties of mutant sodium channels. (A) Bar graph summarizing the means and SEM values for the increase in the fraction of noninactivating sodium current after BTX modification of WT and mutant sodium channels in response to 2,000 5-Hz depolarizing pulses (Inset). An asterisk indicates statistical significance (P < 0.05, Student’s t test) for n = 7 WT, 10 I1760A, 7 F1764, or 6 Y1771A cells. (B–D) Representative current traces elicited by a 50-ms test pulse to 0 mV from a holding potential of −120 mV before and after BTX modification of the indicated mutant channel. (E) Voltage for half-maximal activation of BTX-modified WT and mutant sodium channels. These values were calculated from a single Boltzmann fit to conductance–voltage curves for the noninactivating component of sodium current at the end of 50-ms test pulses after BTX modification.
BTX also causes a hyperpolarizing shift in the voltage dependence of activation of modified sodium channels, as observed in current–voltage (I/V) relationships (Fig. 4B). Because of the more negative voltage dependence of BTX-modified sodium channels, two voltage-dependent components were apparent in activation curves in the presence of the toxin, one with voltage dependence similar to unmodified channels and one with the shifted voltage dependence of BTX-modified channels (Fig. 4C). The noninactivating sodium current at the end of the depolarizing test pulses resulted primarily from BTX-modified sodium channels. The BTX-modified channels had half-maximal activation at −59.9 ± 4.3 mV compared with −19.5 ± 3.1 mV for control WT sodium channels, a 40-mV negative shift.
Effects of Mutations in the Local Anesthetic Receptor Site on BTX Modification of Sodium Channel Function.
Mutant sodium channels were expressed and tested by using similar protocols as for WT, and the effects on persistent sodium currents are illustrated in Fig. 5. As expected based on the binding data presented above, mutation Y1771A had no effect on BTX modification of the channel. The increase in noninactivating sodium current (Fig. 5 A and D) and the negative shift in the voltage dependence of activation (−36 mV) in the presence of BTX were all similar to WT. In contrast, alanine substitution at F1764 abolished the effects of BTX. Even after 2,000 repetitive pulses, no change in noninactivating sodium current was noted (Fig. 5 A and B). Thus, although substitution of alanine for F1764 and Y1771 gave rise to mutant sodium channels with similar biophysical characteristics, including a significant noninactivating sodium current, BTX effects are normal for Y1771A but are abolished for F1764A.
Substitution of alanine for I1760 resulted in a mutant sodium channel with intermediate sensitivity to BTX (Fig. 5 A, C, and E). For this mutant, the fraction of noninactivating sodium current after BTX modification increased to only 35.9% (Fig. 5A), significantly less than that observed for WT channels. In addition, the voltage dependence of activation of BTX-modified channels was significantly less shifted than WT (shift = −31.8 ± 2.8 mV). Thus, using the same protocol, BTX is a less effective modifier of mutant I1760A than of WT channels.
The reduced affinity and weaker effects of BTX on mutant I1760A might result from an increase in the rate of dissociation of bound BTX compared with WT. We tested this hypothesis by inducing BTX modification with 2,000 depolarizing pulses and then holding BTX-modified WT or I1760A channels at −120 mV with brief, 5-ms depolarizations to 0 mV once every 30 s to test the degree of channel modification. Because BTX modification favors the open state, closing and deactivating the BTX-modified sodium channels at −120 mV was expected to favor BTX dissociation if it could escape from its binding site. However, this protocol did not greatly diminish the level of BTX modification of WT sodium channels, as assessed from the comparable level of noninactivating sodium current in the 1st and 29th pulses (Fig. 6 A and C). Thus, BTX binding to WT channels is slowly reversible under these conditions. In contrast, when the same protocol is applied to I1760A, the noninactivating sodium current caused by BTX modification was reversed consistently (Fig. 6 B and C). These data indicate that reversal of BTX effect is enhanced ≈10-fold in I1760A, providing independent support for the reduced affinity for BTX binding to mutants I1760A observed in biochemical experiments. In addition, the electrophysiological experiments demonstrate that F1764 is required for the functional effects of BTX and that I1760 is required for the slow reversal of BTX effects on sodium channel function. Because the electrophysiological experiments were carried out in the absence of the allosteric modulators PbTx and RU51049, they also confirm that the effects of these mutations are directly on BTX binding and action.
Figure 6.

I1760 is required for irreversible binding of BTX to the sodium channel. After BTX modification, the membrane potential was held at −120 mV, and 5-ms test pulses to 0 mV were applied once every 30 s (see Inset). (A) Representative current traces from a type IIA sodium channel at 0 and 840 s after repolarization to −120 mV. Note that there is little reversal of the BTX effect after holding the channel in the closed conformation for 15 min. (B) Representative current traces from a mutant I1760A channel 0 s before and 840 s after repolarization to −120 mV. For this mutant, BTX modification is more rapidly reversible. (C) Rate of reversal of the effect of BTX at −120 mV for WT (filled circles) and mutant (open circles) sodium channels. Normalized sodium current recorded at the end of each 50-ms test pulse is plotted versus time at −120 mV.
Mechanism of BTX Effects on Sodium Channel Gating.
It is interesting that BTX, which prevents sodium channel inactivation, binds to the IVS6 region of the sodium channel that is required for fast inactivation. Combined mutation of F1764 and V1774 to alanine completely disrupts sodium channel fast inactivation, and mutation of either of those residues partially blocks inactivation (34). Thus, the effects of BTX on fast inactivation may be mediated by its interaction with segment IVS6. The peptide α-scorpion and sea anemone toxins, which also prevent fast inactivation, interact with a receptor site that includes the extracellular end of transmembrane segment IVS4 (41). These toxins are proposed to slow inactivation by preventing the outward movement of the IVS4 voltage sensor on depolarization and thereby slowing or preventing coupling of activation to inactivation (41). Segment IVS4 is thought to be packed close to transmembrane segment IVS6 in the folded structure of the α subunit (42) and to be involved in coupling activation to inactivation (41, 43). BTX binding to the amino acid residues we have identified in segment IVS6 may allow the toxin to alter the voltage-dependent movements of the adjacent IVS4 voltage sensor and thereby affect both activation and coupling of activation to inactivation. α-scorpion toxins and sea anemone toxins are strong allosteric enhancers of BTX binding and action (10, 12, 44). Their allosteric effects may result from altering the movement of the IVS4 voltage sensor in a way that is favorable for BTX binding and channel activation but unfavorable for fast inactivation.
Overlapping Receptor Sites for BTX and Local Anesthetics.
Our results demonstrate that BTX interacts with a region of the sodium channel that also binds local anesthetics. Fig. 7 illustrates a modified version of the model presented previously for association of local anesthetic drugs with the sodium channel (31), showing the interaction of BTX in this region. Amino acids I1760, F1764, and Y1771 fall on the same face of the predicted IVS6 α helix. The point on the BTX molecule that interacts with IVS6 is not yet defined. However, it can be assumed that the dimethylpyrrolidone carboxylic acid moiety does not interact with segment IVS6 because previous photoaffinity labeling experiments with a BTX derivative having a photoreactive group in this position labeled only segment IS6 (29). Similarly, mutagenesis experiments (30) also directly implicate the IS6 segment in BTX binding and action. Therefore, we propose a model in which BTX binds to the sodium channel with its dimethylpyrrolidone carboxylic acid group directed toward segment IS6 and its steroid moiety toward segment IVS6 (Fig. 7).
Figure 7.

A model for BTX binding on transmembrane segments IS6 and IVS6 of the type IIA sodium channel. This model shows the potential localization of the BTX binding site relative to the previously known location of the local anesthetic binding site. The local anesthetic is depicted as binding in front of the plane of the S6 α helices, presumably within the pore, and BTX behind the same plane and not within the pore. The dotted lines illustrate the amino acid residues that are thought to interact with bound BTX. Their precise points of interaction on the BTX molecule still are defined incompletely. The IS6 segment is illustrated in an outward position relative to the IVS6 segment based on the experiments of Chiamvimonvat et al. (47) on disulfide crosslinking of pore residues. I433, N434, and L437 are illustrated based on the experiments of Trainer et al. (29) and Wang and Wang (30). I1760, F1764, and Y1771 in segment IVS6 are illustrated based on the experiments of Ragsdale et al. (31) and the results presented here.
Because local anesthetics are thought to block sodium channels by physically occluding the pore (1) while the larger BTX molecule activates sodium channels and does not occlude the pore (18, 20), the sites of binding of these two classes of agents must be distinct. Previous experiments have shown that local anesthetics and other sodium channel blocking drugs are indirect allosteric inhibitors of BTX binding and action (22–24, 26), consistent with separate binding sites. Moreover, local anesthetics can block sodium channels activated by BTX without displacing the toxin, although higher concentrations are required than for unmodified sodium channels (27, 28). Our results show that BTX and sodium channel blocking drugs interact with adjacent and possibly overlapping sites in the IVS6 transmembrane segment. Therefore, their allosteric interaction likely takes place over a short distance within the protein structure. Their separate binding sites may involve interactions with opposite faces of the sidechains of the same amino acid residues in the IVS6 segment, allowing local anesthetics to bind in the pore and block it while BTX interacts with some of the same residues and alters channel gating.
Comparison with Calcium Channel Agonists and Antagonists.
Recent experiments that have mapped the molecular determinants for binding of calcium channel blockers to l-type calcium channels have reached analogous conclusions to those presented here in several respects. Phenylalkylamines, which are intracellular pore-blocking drugs like local anesthetics, bind to a receptor site that includes amino acid residues in analogous positions in the IVS6 segment of the α1 subunit of l-type calcium channels to I1760, F1764, and Y1771 in sodium channels (45, 46). Dihydropyridines, which can be allosteric activators or inhibitors of calcium channels, bind to an adjacent and partly overlapping set of amino acid residues in segment IVS6 (45, 46). Both of these classes of drugs also bind to amino acid residues in transmembrane segment IIIS6 of calcium channels (45, 46). The negative allosteric interactions observed between these two classes of calcium channel ligands evidently take place over very short distances in the IIIS6 and IVS6 segments. As for sodium channels, it is suggested that single amino acid residues can participate in the binding sites for both phenylalkylamines and dihydropyridines, with different faces of their sidechains interacting with the two bound ligands. Evidently, there are fundamental structural and functional analogies among the receptor sites for these diverse pharmacological agents on sodium and calcium channels.
A Domain–Interface Model for Drug and Toxin Action.
Our finding that the receptor site for BTX contains structural elements from both domains I and IV extends the domain–interface model (45, 46) of calcium channel drug binding and action to sodium channels. Just as calcium channel agonists and antagonists bind at the interface of the IIIS6 and IVS6 segments of the calcium channel α1 subunit, our present results show that BTX activates sodium channels by binding to a site formed by the IS6 and IVS6 segments of the sodium channel α subunit. As previously found for ligands acting at the subunit interfaces of allosteric enzymes, our results show that drugs and toxins that modify the gating properties of sodium and calcium channels through allosteric interactions all bind at interfaces between homologous domains. Favorable energetic interactions of drugs and toxins bound at these sites may alter domain–domain interactions in channel gating and thereby mediate the effects of the bound ligands. Additional pharmacological sites are likely to be found at the interfaces between other domains of the ion channel proteins.
Acknowledgments
We thank Dr. Bertil Hille (University of Washington) for critical comments on the manuscript, Dr. Dan Baden (University of Miami) for the generous gift of the brevetoxin, Dr. John Daly (National Institutes of Health) for the generous gift of unlabeled BTX, and Dr. Denis Guedin (Roussel Uclaf, Romainville, France) for the generous gift of RU51049. This work was supported by National Institutes of Health Research Grant NS15751 to W.A.C., a predoctoral National Research Service Award from the National Institutes of Health (Training Grant T32 GM07270) to N.J.L., and an individual postdoctoral National Research Service Award from the National Institutes of Health to A.R.C.
ABBREVIATIONS
- BTX
batrachotoxin
- [3H]BTX-B
tritiated BTX-A-20-α-benzoate
- PbTx
brevetoxin from Gymnodinium breve (previously designated Ptychodiscus brevis)
- WT
wild-type
References
- 1.Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer; 1992. p. 607. [Google Scholar]
- 2.Catterall, W. A. (1992) Physiol. Rev. 72, Suppl. 4, S15–S48. [DOI] [PubMed]
- 3.Numa S. Harvey Lect. 1989;83:121–165. [PubMed] [Google Scholar]
- 4.Catterall W A. Annu Rev Biochem. 1995;65:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- 5.Catterall W A. Annu Rev Pharmacol Toxicol. 1980;20:15–43. doi: 10.1146/annurev.pa.20.040180.000311. [DOI] [PubMed] [Google Scholar]
- 6.Strichartz G, Rando T, Wang G K. Annu Rev Neurosci. 1987;10:237–267. doi: 10.1146/annurev.ne.10.030187.001321. [DOI] [PubMed] [Google Scholar]
- 7.Gordon D. In: Toxins and Signal Transduction. Gutman Y, Lazarowici P, editors. London: Harwood; 1997. pp. 119–149. [Google Scholar]
- 8.Butterworth J F, IV, Strichartz G R. Anesthesiology. 1990;72:711–734. doi: 10.1097/00000542-199004000-00022. [DOI] [PubMed] [Google Scholar]
- 9.Catterall W A. Trends Pharmacol Sci. 1987;8:57–65. doi: 10.1016/0165-6147(92)90079-l. [DOI] [PubMed] [Google Scholar]
- 10.Catterall W A. J Biol Chem. 1977;252:8669–8676. [PubMed] [Google Scholar]
- 11.Catterall W A, Risk M. Mol Pharmacol. 1981;19:345–348. [PubMed] [Google Scholar]
- 12.Catterall W A, Morrow C S, Daly J W, Brown G B. J Biol Chem. 1981;256:8922–8927. [PubMed] [Google Scholar]
- 13.Lombet A, Mourre C, Lazdunski M. Brain Res. 1988;459:44–53. doi: 10.1016/0006-8993(88)90284-3. [DOI] [PubMed] [Google Scholar]
- 14.Trainer V L, Moreau E, Guedin D, Baden D G, Catterall W A. J Biol Chem. 1993;268:17114–17119. [PubMed] [Google Scholar]
- 15.Brown G B. J Neurosci. 1986;6:2065–2070. doi: 10.1523/JNEUROSCI.06-07-02064.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albuquerque E X, Daly J W, Witcop B. Science. 1971;172:995–1002. doi: 10.1126/science.172.3987.995. [DOI] [PubMed] [Google Scholar]
- 17.Khodorov B I. Membr Transp Processes. 1978;2:153–174. [Google Scholar]
- 18.Khodorov B I. Prog Biophys Mol Biol. 1985;45:57–148. doi: 10.1016/0079-6107(85)90005-7. [DOI] [PubMed] [Google Scholar]
- 19.Khodorov B I, Revenko S V. Neuroscience. 1979;4:1315–1330. doi: 10.1016/0306-4522(79)90159-3. [DOI] [PubMed] [Google Scholar]
- 20.Wang G K, Wang S-Y. Pflügers Arch. 1994;427:309–316. doi: 10.1007/BF00374539. [DOI] [PubMed] [Google Scholar]
- 21.Brown G B, Gaupp J E, Olsen R W. Mol Pharmacol. 1988;34:54–59. [PubMed] [Google Scholar]
- 22.Catterall W A. Mol Pharmacol. 1981;20:356–362. [PubMed] [Google Scholar]
- 23.Willow M, Catterall W A. Mol Pharmacol. 1982;22:627–635. [PubMed] [Google Scholar]
- 24.Creveling C R, McNeal E T, Daly J W, Brown G B. Mol Pharmacol. 1983;23:350–358. [PubMed] [Google Scholar]
- 25.Postma S W, Catterall W A. Mol Pharmacol. 1984;25:219–227. [PubMed] [Google Scholar]
- 26.Sheldon R S, Duff H J, Thakore E, Hill R J. J Pharmacol Exp Ther. 1994;268:187–194. [PubMed] [Google Scholar]
- 27.Wasserstrom J A, Liberty K, Kelly J, Santucci P, Myers M. Biophys J. 1993;65:386–395. doi: 10.1016/S0006-3495(93)81046-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zamponi G W, Doyle D D, French R J. Biophys J. 1993;65:80–90. doi: 10.1016/S0006-3495(93)81042-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trainer V L, Brown G B, Catterall W A. J Biol Chem. 1996;271:11261–11267. doi: 10.1074/jbc.271.19.11261. [DOI] [PubMed] [Google Scholar]
- 30.Wang S Y, Wang G K. Proc Natl Acad Sci USA. 1998;95:2653–2658. doi: 10.1073/pnas.95.5.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ragsdale D S, McPhee J C, Scheuer T, Catterall W A. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 32.Ragsdale D R, McPhee J C, Scheuer T, Catterall W A. Proc Natl Acad Sci USA. 1996;93:9270–9275. doi: 10.1073/pnas.93.17.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qu Y, Rogers J, Tanada T, Scheuer T, Catterall W A. Proc Natl Acad Sci USA. 1995;270:25696–25701. [Google Scholar]
- 34.McPhee J C, Ragsdale D S, Scheuer T, Catterall W A. Proc Natl Acad Sci USA. 1994;91:12346–12350. doi: 10.1073/pnas.91.25.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jordan M, Schallhorn A, Wurm F M. Nucleic Acids Res. 1996;24:596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Margolskee R F, McHendry-Rinde B, Horn R. BioTechniques. 1993;15:906–911. [PubMed] [Google Scholar]
- 37.Jurman M E, Boland L M, Liu Y, Yellen G. BioTechniques. 1994;17:876–881. [PubMed] [Google Scholar]
- 38.Hamill O P, Marty A, Neher E, Sakmann B, Sigworth F J. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 39.Cantrell A R, Scheuer T, Catterall W A. J Neurosci. 1997;17:7330–7338. doi: 10.1523/JNEUROSCI.17-19-07330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Isom L L, Scheuer T, Brownstein A B, Ragsdale D S, Murphy B J, Catterall W A. J Biol Chem. 1994;270:3306–3312. doi: 10.1074/jbc.270.7.3306. [DOI] [PubMed] [Google Scholar]
- 41.Rogers J C, Qu Y, Tanada T N, Scheuer T, Catterall W A. J Biol Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- 42.Fozzard H A, Hanck D A. Physiol Rev. 1996;76:887–926. doi: 10.1152/physrev.1996.76.3.887. [DOI] [PubMed] [Google Scholar]
- 43.Chen L Q, Santarelli V, Horn R, Kallen R G. J Gen Physiol. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Catterall W A, Beress L. J Biol Chem. 1978;253:7393–7396. [PubMed] [Google Scholar]
- 45.Catterall W A, Striessnig J. Trends Pharmacol Sci. 1992;13:256–262. doi: 10.1016/0165-6147(92)90079-l. [DOI] [PubMed] [Google Scholar]
- 46.Hockerman G H, Peterson B Z, Johnson B D, Catterall W A. Annu Rev Pharmacol Toxicol. 1997;37:361–396. doi: 10.1146/annurev.pharmtox.37.1.361. [DOI] [PubMed] [Google Scholar]
- 47.Chiamvimonvat N, Pérez-García M T, Ranjan R, Marban E, Tomaselli G F. Neuron. 1996;16:1037–1047. doi: 10.1016/s0896-6273(00)80127-0. [DOI] [PubMed] [Google Scholar]