

Abstract
Mitochondria are dynamic organelles that frequently move, divide, and fuse with one another to maintain their architecture and functions. However, the signaling mechanisms involved in these processes are still not well characterized. In this study, we analyze mitochondrial dynamics and morphology in neurons. Using time-lapse imaging, we find that Ca2+ influx through voltage-dependent Ca2+ channels (VDCCs) causes a rapid halt in mitochondrial movement and induces mitochondrial fission. VDCC-associated Ca2+ signaling stimulates phosphorylation of dynamin-related protein 1 (Drp1) at serine 600 via activation of Ca2+/calmodulin-dependent protein kinase Iα (CaMKIα). In neurons and HeLa cells, phosphorylation of Drp1 at serine 600 is associated with an increase in Drp1 translocation to mitochondria, whereas in vitro, phosphorylation of Drp1 results in an increase in its affinity for Fis1. CaMKIα is a widely expressed protein kinase, suggesting that Ca2+ is likely to be functionally important in the control of mitochondrial dynamics through regulation of Drp1 phosphorylation in neurons and other cell types.
Introduction
Mitochondria play an essential function in cells through the production of energy and the ability to regulate intracellular Ca2+. As such, they are involved in a variety of cellular processes, including survival, proliferation, and apoptosis (Kroemer and Reed, 2000; Shaw and Nunnari, 2002; Fannjiang et al., 2004; Oakes and Korsmeyer, 2004; Szabadkai et al., 2004; Youle and Karbowski, 2005; Karbowski et al., 2006). Mitochondria are also dynamic organelles and move through the cell with frequent fission and fusion processes that influence their morphology (Bereiter-Hahn and Voth, 1994; Rube and van der Bliek, 2004; Chan, 2006).
Recent studies have shed light on the molecular mechanisms involved in mitochondrial fission and fusion. In mammalian cells, mitochondrial fusion is regulated by Fzo1, OPA1, and Mgm1 (Bossy-Wetzel et al., 2003; Lee et al., 2004). In contrast, dynamin-related protein 1 (Drp1), Fis1, and possibly other proteins such as MTP18 play an important role in mitochondrial fission (Stojanovski et al., 2004; Tondera et al., 2005; Yu et al., 2005). A critical role is played by the Drp1, which translocates to mitochondria and forms a complex with Fis1 on the mitochondrial surface. There, Drp1's GTPase activity is coupled to outer membrane scission (Yoon et al., 2001; Barsoum et al., 2006). Mitochondrial fission is likely to be similar to the process involved in plasma membrane endocytosis mediated by the GTPase activity of dynamin, such as in synaptic vesicle endocytosis (Takei et al., 1995). Recent studies suggest that posttranslational modifications including phosphorylation, ubiquitination, and SUMOylation are likely involved in the control of mitochondrial fission (Cerveny et al., 2007; Detmer and Chan, 2007). However, it is still unclear how extracellular stimuli might modulate intracellular signaling processes to control mitochondrial dynamics and morphology.
In neurons, mitochondrial trafficking to pre- and postsynaptic sites is likely to play an important role in control of basal synaptic transmission and plasticity (Tang and Zucker, 1997; Li et al., 2004). In dendrites, mitochondrial movement and morphology are regulated by synaptic activity, whereas in axons, mitochondrial movement responds to changes in axonal outgrowth (Chada and Hollenbeck, 2003; Ruthel and Hollenbeck, 2003; Li et al., 2004). Mitochondrial movement and morphology are also regulated by nitric oxide, and alterations in mitochondrial morphology may play a role in apoptosis (Barsoum et al., 2006; Wasiak et al., 2007). Although mitochondrial movement and fission/fusion play critical functional roles in neurons, it is unclear how these changes in mitochondrial morphology and movement are coupled to alterations in synaptic activity.
In the present study, we find that intracellular signaling associated with activation of voltage-dependent Ca2+ channels (VDCCs) regulates mitochondrial dynamics and morphology in neurons. Ca2+ signaling via VDCCs stimulates mitochondrial fragmentation, and this involves activation of Ca2+/calmodulin-dependent protein kinase I (CaMKI) and phosphorylation of Drp1 at serine 600 (Ser600). These results identify an important mechanism involved in Drp1-regulated mitochondrial dynamics and highlight a key role for Ca2+ in the control of mitochondrial dynamics.
Results
Effects of high K+ on mitochondrial dynamics and morphology in neurons
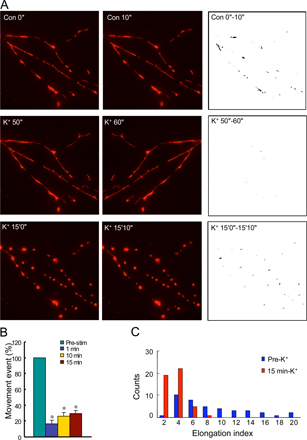
Cultured hippocampal neurons were transfected with pDsRed2-Mito to label mitochondria. The movement and morphology of mitochondria were monitored using live-cell imaging. As previously demonstrated (Ligon and Steward, 2000), mitochondria were found in neuronal axons and dendrites, exhibiting an elongated shape and dynamic movement. Treatment with 45 mM K+ for 15 min had a marked effect on mitochondrial shape and movement. Elongated mitochondria became much shorter and rounder in morphology (Fig. 1 A and Fig. S1, A and C, available at http://www.jcb.org/cgi/content/full/jcb.200802164/DC1). In time-lapse imaging, 45-mM K+ stimulation was found to trigger a rapid halt to movement (mitochondrial movement was reduced to 16.5% of control at 1 min, recovering to 26.3% at 10 min and 29.6% at 15 min; Fig. S1, A and B). High K+ treatment also caused mitochondria to form ringlike structures that were the sites of fission events giving rise to smaller, round mitochondrial structures (Fig. 1 B).
Figure 1.

Effects of high K+ on mitochondrial dynamics and morphology in neurons. (A) Neurons (10 DIV) were transfected with pDsRed2-Mito to label mitochondria. Neurons (at 11 DIV) were then treated with 45 mM K+ for 15 min, and mitochondrial fluorescence was analyzed by time-lapse imaging. Arrows show ringlike mitochondria formation in dendrites. Bar, 10 μm. (B) Higher magnification in dendrites show details of ringlike mitochondrial formation induced by treatment with 45 mM K+. Arrows show mitochondrial fission. Time in minutes after application of high K+ is indicated at the bottom of each panel. Bar, 1 μm. (C) Ultrastructure of mitochondria analyzed by electron microscopy. Panels i and ii show examples of mitochondria from control neurons. The remaining panels (iii–vii) show examples of mitochondria in neurons treated with 45 mM K+ for 15 min. (ii) Mitochondria in dendrites from control neurons were rod shaped, and their cristae were clear. (iii–vii) Mitochondria from neurons treated with 45 mM K+ formed clusters that exhibited less electron-dense matrices and contained less cristae. (v–vii) 45-mM K+ treatment caused mitochondrial fission. Arrows in v–vii show connections between dividing mitochondria. The dividing mitochondria shared continuous outer membrane with separate inner membranes (arrowhead in vi). Bars: (i and iii)1 μm; (ii and iv–vii) 200 nm.
Mitochondrial morphology was also assessed by electron microscopy. Normal mitochondria had a rodlike shape with clear cristae (Fig. 1 C, i and ii). After high K+ treatment, mitochondria appeared empty with less electron-dense material and less cristae (Fig. 1 C, iii–vii). Mitochondria undergoing division were frequently observed. In some cases, the dividing mitochondria were found to be connected by a narrow necklike membrane (Fig. 1 C, v–vii).
VDCC-associated Ca2+ signaling is required for the effects of high K+ on mitochondrial dynamics
To investigate the signaling mechanisms involved in the effects of high K+ on mitochondria, we first measured intracellular Ca2+ level after high K+ treatment. As expected, high K+ stimulation triggered a rapid increase in intracellular Ca2+. The increased Ca2+ was sustained during the 20-min treatment with high K+ and then rapidly recovered to a normal level after washout of K+ (Fig. 2 A). The changes in mitochondrial movement and morphology were observed in response to various concentrations of K+ from 10 to 100 mM (not depicted) but, importantly, were reversible after 15-min treatment with high K+ and subsequent washout (Fig. 2 B). Furthermore, we found that there was little detectable neuronal death 24 h after high K+ treatment (15 min) and subsequent washout (Fig. 2 C). In contrast, as expected, treatment with glutamate (500 μM for 15 min) resulted in obvious cell death when examined 24 h later.
Figure 2.

High K+ induces elevation in intracellular Ca2+ and reversible changes in mitochondrial morphology. (A) Intracellular Ca2+ level was measured in hippocampal neurons (12 DIV) in response to treatment with 45 mM K+. Graphs show normalized ratios of F340/F380 in four different neurons (thin lines) and the mean value (bold line). (B) Neurons transfected with pDsRed2-Mito were analyzed before addition of 45 mM K+ and 15 min after treatment with high K+. Arrows show ringlike mitochondria formations. After 15 min, 45 mM K+ was removed by exchange of media, and mitochondria were imaged after 3 h. The same result was obtained in three separate experiments (n = 3). Bars, 10 μm. (C) Neurons (12 DIV) were untreated or treated for 15 min with 500 μM glutamate or 45 mM K+. After the 15-min treatment, the media was replaced by conditioned medium from 10-d-old neuronal cultures. 24 h later, cell death was measured by Hoechst 33258 and MAP2 staining. Data were obtained from at least five visual fields under a 20× objective from at least three independent experiments for each group. Error bars indicate SEM in each group. Bars, 40 μm.
To investigate the identity of Ca2+ channels involved in the regulation of mitochondrial dynamics, we examined the effects of a series of VDCC inhibitors (Fig. 3). All of the VDCC blockers (N, P/Q, and L type) attenuated the high K+–induced changes in mitochondrial movement (Fig. 3 B). However, only the N-type VDCC blocker, P/Q-type VDCC blocker (conotoxin-MVIIC or a mix of conotoxin-GVIA and agatoxin-IVA), and P/Q-type blocker agatoxin IVA prevented high K+–induced changes in mitochondrial morphology (Fig. 3, A and B). These results suggest that either L-, N-, or P/Q-type VDCC may be involved in K+-induced mitochondrial dynamics but that coactivation of N and P/Q types plays the most important role.
Figure 3.

VDCC-associated Ca2+ signaling is involved in the effects of high K+ on mitochondria. (A) Neurons (10 DIV) transfected with pDsRed2-Mito were analyzed by time-lapse imaging. Neurons (at 11 DIV) were then preincubated with 10 μM nifedipine (L-type blocker), 1 μM Cono-MVIIC (N and P/Q blocker), 10 μM UO126 (MAPK inhibitor), 20 μM KN93 (CaMK inhibitor), 20 μM KN92 (control for KN93), and 1 μM FK506 and were incubated in 45 mM K+ for 15 min. Bars, 10 μm. (B) Mitochondrial length was measured through analysis of the elongation index (calculated by the square of longest length divided by the area). The elongation index after 15 min of high K+ stimulation was normalized to the value before stimulation. Mitochondrial movement was measured by time-lapse fluorescence microscopy. The movement events after high K+ stimulation were normalized to the value before stimulation. Neurons were either untreated (No sti), incubated with 45 mM K+ for 15 min (K+), or preincubated with various pharmacological agents before incubation with 45 mM K+ as in A and also including 1 μM Cono-GVIA (N-type blocker), 0.1 μM Aga-IVA (P/Q-type blocker), or Cono-GVIA plus Aga-IVA. The results shown are from five experiments and represent the mean ± SEM (n = 5) in each group. Values were analyzed using one-way ANOVA followed by post-Tukey test. *, P < 0.05 versus K+ stimulation.
CaMKIα mediates VDCC-dependent mitochondrial changes
We next investigated the role of intracellular Ca2+-dependent signaling pathways that might be involved in control of mitochondrial dynamics. FK506 and cyclosporin, which act through inhibition of the Ca2+-dependent protein phosphatase, calcineurin, had no effect on high K+–induced changes in mitochondrial shape and movement (Fig. 3, A and B). U0126, a blocker of extracellular signal-regulated kinase signaling, also had no effect on K+-induced changes (Fig. 3, A and B). However, KN93, an antagonist of CaMKs, almost completely blocked the K+-induced changes in mitochondrial shape and movement (Fig. 3, A and B). KN92, a negative control for KN93, had no effect. The inhibitory effect of KN93 was not caused by an effect on Ca2+ influx because KN93 (or KN92) had no effect on high K+–induced elevation in intracellular Ca2+ (unpublished data). KN93 is a general inhibitor of CaMKs, including CaMKI, CaMKII, and CaMKIV, but recent studies in neurons have highlighted that CaMKI may be an important target for KN93 (Schmitt et al., 2005). Therefore, we assessed the possible contribution of CaMKIα, the most abundant CaMKI isoform (Hook and Means, 2001), in the regulation of mitochondrial dynamics and morphology. Expression of a dominant-negative CaMKIα (CaMKIα-DN) blocked the effect of high K+ on mitochondrial length and movement (Fig. 4, A and C). In addition, expression of constitutively active CaMKIα (CaMKIα-CA) alone resulted in inhibition of mitochondrial movement and caused mitochondria to become shorter in length (Fig. 4, B and D).
Figure 4.

CaMKIα mediates VDCC-dependent mitochondrial changes after high K+. (A) Neurons (10 DIV) were cotransfected with pDsRed2-Mito and GFP (negative control; top) or GFP–CaMKIα-DN. Neurons (at 11 DIV) were treated with 45 mM K+ for 15 min. GFP fluorescence is shown in the left panels, and mitochondrial fluorescence is shown before (Pre-K+) and after high K+ treatment in the middle and right panels. Bars, 20 μm. (B) Neurons were transfected with GFP–CaMKIα-CA and pDsRed2-Mito. GFP fluorescence is shown in the left panel, and mitochondrial fluorescence is shown in the right panel. Bar, 20 μm. (C) Mitochondrial movement or length was measured for neurons that were either untreated (no stimulation) or treated with 45 mM K+ as shown in A. The movement events after high K+ stimulation were normalized to the value obtained before stimulation. The elongation index after 15 min of high K+ stimulation was also normalized to the value obtained before stimulation. Data were obtained from >150 mitochondria in three to five independent experiments for each condition. Error bars indicate SEM in each group. *, P < 0.001. (D) Mitochondrial movement or elongation index was measured for neurons that were either untreated (Control; transfected with GFP) or after transfection with GFP–CaMKI-CA as in B. Mitochondrial movement was measured by time-lapse fluorescence microscopy. Mitochondrial length was measured through analysis of the elongation index. Data were obtained from at least 350 mitochondria in 7–10 neurons for each condition. Error bars indicate SEM in each group. *, P < 0.001.
Ser600 of Drp1 is phosphorylated by CaMKIα
We next investigated whether proteins involved in mitochondrial fission might be phosphorylated by CaMKIα. Drp1 is an important protein involved in the scission of mitochondrial membranes (Frank et al., 2001; Smirnova et al., 2001; Yoon et al., 2003). Based on the consensus sequence of CaMKI (Lee et al., 1994; Matsushita and Nairn, 1998), there is a potential evolutionary conserved phosphorylation site at Ser600 in Drp1-3 (Drp1 isoform 3 as well as other isoforms; Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200802164/DC1). In vitro assays indicated that recombinant Drp1 was phosphorylated by CaMKIα but was not phosphorylated by CaMKII (Fig. 5 A). Mutation of Ser600 (to alanine) resulted in a loss of phosphorylation by CaMKIα (Fig. 5 B). Phosphorylation of Ser600 was further confirmed by a phosphoantibody that specifically recognized phospho-Ser600 (Fig. 5 C). We then examined the phosphorylation of Drp1 by CaMKIα in intact cells. In HeLa cells, high K+ treatment increased phosphorylation of Drp1 at Ser600, and the phosphorylation was prevented after down-regulation of CaMKIα levels using RNAi (control studies indicated that CaMKIα was down-regulated by >80% using human CaMKIα RNAi; Fig. 5 D and Fig. S2 B). In primary cultured neurons, high K+ treatment also increased phosphorylation of Drp1 at Ser600, and this was prevented by preincubation with KN93 (Fig. 5, E and F). Together, these results strongly suggest that Drp1 is a physiological substrate for CaMKIα and that phosphorylation can be selectively stimulated through activation of VDCCs.
Figure 5.

CaMKIα phosphorylates recombinant Drp1 and endogenous Drp1 in cells. (A) Recombinant Drp1 was incubated with either CaMKIα or CaMKII as indicated in the presence of [32P]ATP. Proteins were separated by SDS-PAGE and analyzed by autoradiography and Coomassie staining. (B) Wild-type Drp1 or mutant Drp1 in which Ser600 was replaced by alanine was incubated with CaMKIα and [32P]ATP, and proteins were separated by SDS-PAGE and analyzed by autoradiography. (C) Recombinant Drp1 was incubated with CaMKIα in the absence or presence of ATP. Proteins were separated by SDS-PAGE and analyzed by immunoblotting using a phosphospecific antibody selective for phospho-Ser600. (D) HeLa cells were transfected without (control siRNA) or with siRNA selective for CaMKIα. HeLa cells were incubated without (−) or with 45 mM K+ for 15 min (+). Proteins were separated by SDS-PAGE and analyzed by immunoblotting with phospho-Ser600 or Drp1 antibodies. (E) Neurons were incubated for the indicated times with 45 mM K+ in the absence or presence of KN93 (20 μM added 30 min before high K+). Proteins were separated by SDS-PAGE and analyzed by immunoblotting with phospho-Ser600 or Drp1 antibodies as indicated. (F) Neurons were untreated (control) or treated with 45 mM K+ for 15 min. Proteins were separated by SDS-PAGE and analyzed by immunoblotting with phospho-Ser600 or total Drp1 antibodies as indicated (bottom). Bar graphs show cumulative data from three independent experiments for each condition. Error bars indicate SEM in each group. *, P < 0.05.
Drp1-S600 phosphorylation is required for the effects of high K+ on mitochondrial dynamics
We next examined the functional consequences of Drp1 phosphorylation on mitochondrial morphology. These studies were performed both in HeLa cells and in cultured neurons. In HeLa cells, we down-regulated Drp1 expression using RNAi and substituted expression with wild-type Drp1 or mutant Drp1 in which Ser600 was changed to alanine (S600A) to mimic the dephospho-state or to aspartic acid (S600D) to mimic the phospho-state (Fig. 6). The RNAi treatment reduced expression of endogenous Drp1 significantly, and the level of expression of the wild-type and mutant RNAi-resistant forms of YFP-Drp1 were slightly higher than that of the normal levels of the endogenous protein (Fig. 6 A). High K+ treatment of HeLa cells also resulted in a rapid appearance of fragmented mitochondria in control cells or in cells in which endogenous Drp1 expression was reduced, although wild-type RNAi-resistant YFP-Drp1 or the YFP-Drp1-S600D mutant was expressed (Fig. 6, B and C). In contrast, expression of the RNAi-resistant YFP-Drp1-S600A resulted in attenuation of the effect of high K+ treatment on mitochondrial fragmentation.
Figure 6.

Drp1-S600 phosphorylation is required for the effects of high K+ on mitochondrial dynamics. (A) HeLa cells were first transfected with control siRNA (siCont; Invitrogen) and siRNA for Drp1 (siDrp1) before transfection with the Drp1 plasmids. 1 d later, HeLa cells were transfected with GFP, YFP-Drp1R, YFP-Drp1R-S600A, or YFP-Drp1R-S600D together with pDsRed2-Mito. Proteins were separated by SDS-PAGE and analyzed by immunoblotting using Drp1 and actin antibodies. (B) HeLa cells were treated without (No sti) or with 20 mM K+ (high K+), and mitochondrial fluorescence was analyzed by time-lapse imaging. Mitochondrial length was measured through analysis of the elongation index. Bars, 5 μm. (C) The elongation index was normalized to the value before stimulation. Values shown are the mean ± SEM (n = 5) in each group. Values were analyzed using one-way ANOVA followed by post-Tukey test. *, P < 0.01. (D) Neurons (10 DIV) were cotransfected with pDsRed2-Mito and GFP, YFP-Drp1, or YFP-Drp1-S600A as indicated. Neurons (at 11 DIV) were then treated without (No sti) or with 45 mM K+ for 15 min, and mitochondrial fluorescence was analyzed by time-lapse imaging. Bars, 10 μm. (E) Mitochondrial length was measured through analysis of the elongation index. Values shown are the mean ± SEM (n = 5) in each group. Values were analyzed using one-way ANOVA followed by post-Tukey test. *, P < 0.05; **, P < 0.005.
We also transfected neurons with pDsRed2-Mito and GFP, YFP-Drp1, or YFP-Drp1-S600A and examined mitochondrial morphology during K+ stimulation. In these experiments, endogenous expression of Drp1 was not down-regulated with RNAi. As expected, high K+ treatment of neurons transfected with GFP or wild-type YFP-Drp1 resulted in the rapid appearance of fragmented mitochondria throughout dendrites (Fig. 6, D and E). In contrast, expression of YFP-Drp1-S600A significantly inhibited the effects of high K+ treatment on mitochondrial morphology.
Drp1's mitochondrial localization is regulated by CaMKIα-dependent phosphorylation
In mammalian cells, Drp1 is believed to translocate to the outer mitochondrial membrane, where it interacts with Fis1 and perhaps other proteins (Yoon et al., 2001). We transfected neurons with YFP-Drp1 and examined the localization of the protein (Fig. 7). Under control conditions, wild-type YFP-Drp1 was found evenly distributed throughout the cytoplasm (Fig. 7 A). High K+ treatment resulted in the rapid appearance of punctate YFP-Drp1 staining throughout the cytoplasm (Fig. 7 A; quantitative analysis of exogenous Drp1 foci in neurons shown in Fig. 7 B). Preincubation with KN93 attenuated the appearance of the punctate YFP-Drp1 stain in response to high K+ treatment (Fig. 7, A and B). In addition, high K+ had no significant effect on the cytoplasmic localization of the mutant YFP-Drp1-S600A (Fig. 7, A and B). We also performed biochemical fractionation assays to examine the distribution of endogenous Drp1 after high K+ stimulation. Phosphorylated Drp1 rapidly translocated to the mitochondrial fraction after high K+ treatment, and this was blocked by pretreatment with KN93 (Fig. 7, C and D).
Figure 7.

CaMKIα-dependent phosphorylation regulates Drp1 intracellular distribution in neurons. (A) Neurons (10 DIV) were transfected with YFP-Drp1 or YFP-Drp1-S600A. Neurons (at 11 DIV) were untreated (left) or treated with (right) 45 mM K+ or preincubated with 20 μM KN93 for 30 min before high K+ stimulation. Bars, 10 μm. (B) Quantitative analysis of exogenous Drp1 foci. Data were obtained from at least five independent experiments for each condition shown in A. Control is YFP-Drp1 without stimulation. Error bars indicate SD in each group. *, P < 0.05. (C) Neurons were treated with 45 mM K+ in the absence or presence of 20 μM KN93 for various times as indicated. Proteins in mitochondrial fractions were separated by SDS-PAGE and analyzed by immunoblotting using Drp1 T-Drp1, P-Drp1, or Tom40 (for quantification of mitochondrial membrane) antibodies. (D) Quantitative analysis of Drp1 translocation. Drp1 levels were normalized to Tom40 levels at the time points indicated in Fig. 7 C. Cumulative results from three independent experiments for each condition are shown. Error bars indicate the mean ± SEM in each group.
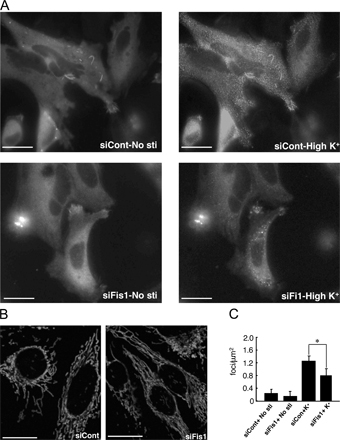
Similar studies were performed in HeLa cells in which endogenous Drp1 was down-regulated with RNAi and replaced by expression of either wild-type or mutant YFP-Drp1 (Fig. 8). High K+ treatment also induced the rapid appearance of punctate YFP-Drp1 in HeLa cells (Fig. 8, A and B). This was accompanied by fragmentation of mitochondria (Fig. 6, A and B). Similar to neurons, high K+ had no significant effect on the cytoplasmic localization of YFP-Drp1-S600A. However, somewhat unexpectedly, the YFP-Drp1-S600D protein was found diffusely distributed in the cytosol under basal conditions, and high K+ treatment also resulted in the rapid appearance of punctate staining of this phosphomimetic mutant protein.
Figure 8.

High K+ treatment regulates Drp1 dynamics in HeLa cells. (A) HeLa cells were transfected with control siRNA (siCont) and siRNA for Drp1 (siDrp1) before the transfection with GFP, YFP-Drp1R, or YFP-Drp1R-S600A and mito-DsRed as described above. HeLa cells were then either untreated or incubated with 20 mM K+ for 15 min. (B) Quantitative analysis of exogenous Drp1 foci. Data were obtained from the results from at least five independent experiments for each condition shown in A. Values were analyzed using one-way ANOVA followed by post-Turkey test. Control is siDrp1 without stimulation. Error bars indicate SD in each group. *, P < 0.01 versus siDrp1+YFP-Drp1R+No stimulation. Bars, 10 μm.
In mammalian cells, Drp1 has been shown to regulate mitochondrial fission through an interaction with Fis1 (Yoon et al., 2003). Therefore, we examined in vitro whether CaMKIα-dependent phosphorylation of Drp1 could facilitate the interaction of Drp1 with hFis1. In the absence of prior phosphorylation by CaMKIα, a small amount of Drp1-6His was pulled down by GST-hFis1. Phosphorylation of wild-type Drp1 by CaMKIα resulted in a substantial increase in the amount of Drp1-6His pulled down by GST-hFis1 (Fig. 9 A). The amount of Drp1 bound to GST-Fis1 increased in proportion to the level of phosphorylation of Drp1 (Fig. 9 B). In contrast to wild-type Drp1, there was little measurable binding found between Drp1-S600A and GST-Fis1 (Fig. 9 C). Finally, after down-regulation of Fis1 with siRNA in HeLa cells, the effect of high K+ on the punctate distribution of YFP-Drp1 was reduced (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200802164/DC1).
Figure 9.

Phosphorylation regulates Drp1 and hFis1 binding in vitro. (A) Recombinant Drp1-6His was preincubated in the absence or presence of CaMKIα (KI) and ATP and mixed with GST-hFis1 as indicated. Proteins were recovered using glutathione-Sepharose and analyzed by immunoblotting using an anti-6His antibody to detect Drp1 (top; bottom shows Drp1 input). Quantitation of the results is shown in the bar graph. The results were otained in three independent experiments. Error bars indicate SEM in each group. (B) Drp1-6His was maximally phosphorylated by CaMKI, and the sample was mixed in different ratios with nonphosphorylated Drp1-6His (values are expressed as percent P-Drp1/Drp1; see x axis). GST pull-down assays were performed as in A. The data represent means from three independent experiments. (C) The binding of mutant Drp1-S600A-6His to GST-hFis1 was compared with unphosphorylated and phosphorylated wild-type Drp1-6His. Drp1 was detected by immunoblotting as described above. Similar results were obtained from two independent experiments.
Discussion
In this study, we have demonstrated that Ca2+-dependent signaling has profound effects on mitochondrial dynamics. The regulation of mitochondrial dynamics in response to high K+ stimulation involved activation of N-type VDCCs, P/Q-type VDCCs, and, to a lesser degree, L-type VDCCs and subsequent Ca2+ entry into cells. In response to VDCC activation, mitochondria exhibit significant morphological and dynamic changes. Mitochondria very rapidly stopped moving and formed ringlike structures that accompanied the division process. An elongated mitochondrial morphology was rapidly recovered, and neuronal viability was not impaired after washout of high K+ from the medium. These results were obtained from studies using live-cell imaging with mitochondrial-targeted fluorescent protein as well as by electron microscopy. The electron micrographs showed that the high K+ treatment resulted in mitochondria that appeared hollow with a less electron-dense matrix and less cristae. The ringlike formation and hollow mitochondrial changes during fission are also consistent with results from studies in which hFis1 was overexpressed in HeLa cells (Yoon et al., 2003).
The effect of Ca2+ on mitochondrial movement and morphology required the activity of CaMKI. KN93, a CaMK inhibitor known to inhibit CaMKI in neurons, almost completely blocked the effects of high K+ on mitochondrial dynamics. More specifically, expression of CaMKIα-DN blocked the effect of high K+ on mitochondrial movement and morphology, although CaMKIα-CA mimicked the effects of high K+. Moreover, our experiments indicated that phosphorylation of Drp1 by CaMKIα was likely to play an important role in the effect of high K+. Our results indicated that Ca2+ influx via VDCCs resulted in activation of CaMKIα and the phosphorylation of Drp1 at Ser600. Ser600 is included within a consensus CaMKI phosphorylation site in the C-terminal GTPase effector domain (GED; Fig. S2 A). In contrast to dynamin, Drp1 does not have a pleckstrin homology domain, and the GED is necessary and sufficient for interaction with mitochondria (Pitts et al., 2004). The GED is conserved in all Drp1 isoforms, and all are likely to be regulated by CaMKIα-dependent phosphorylation.
The recruitment of Drp1 to the cytoplasmic side of the outer mitochondrial membrane is dependent on Fis1 (Yoon et al., 2003). Fis1 is a small 17-kD protein that interacts with the outer mitochondrial membrane via its C terminus (Mozdy et al., 2000) and interacts with Drp1 via two N-terminal tetratricopeptide repeat motifs (Yu et al., 2005). The expression level of Fis1 appears to be the rate-limiting factor in mitochondrial fission through its ability to interact with Drp1 and recruit it in an active conformation to allow scission of the mitochondrial membrane. Our results showed that in intact cells, phosphorylation of Drp1 by CaMKIα was necessary for translocation of the protein to mitochondria. In addition, expression of a (dephospho) mutant Drp1 in which Ser600 was changed to alanine was able to attenuate the effect of high K+ on mitochondrial morphology. Moreover, in vitro, prior phosphorylation of Drp1 by CaMKIα significantly increased the binding of Drp1 and Fis1. Finally, reduction of Fis1 expression was able to attenuate the effect of high K+ on Drp1 translocation. Together, these results suggest that activation of CaMKIα and phosphorylation of Drp1 are necessary for the increased mitochondrial fission observed after exposure of cells to high K+. Interestingly, a mutant Drp1 in which Ser600 was changed to aspartate to mimic the phosphorylated state behaved like the wild-type protein under basal and high K+ conditions. This result indicates that the S600D mutation does not mimic phosphorylation by CaMKI and also suggests that CaMKI likely regulates the function of other proteins involved in Drp1 translocation to mitochondria.
Drp1 and Fis1 were found previously to interact by fluorescence resonance energy transfer and coimmunoprecipitation experiments. The proteins can also interact in vitro (Yoon et al., 2003). However, this interaction is normally transient (Wasiak et al., 2007), requiring the use of a chemical cross-linker to observe coimmunoprecipitation or interaction in vitro (Yoon et al., 2003). The precise molecular details of the interaction of Drp1 and Fis1 remain to be established. A study in yeast suggests that a third protein termed Mdv1 is required for the interaction between Dnm1 and Fis1 (Hoppins et al., 2007). However, a recent study suggests that Saccharomyces cerevisiae Fis1 directly binds Dnm1 and not Mdv1 in vitro (Wells et al., 2007). Interestingly, this binding is negatively regulated by a short N-terminal region of S. cerevisiae Fis1. Notably, the N-terminal region of Fis1 is not well conserved between yeast and mammals. Moreover, Mdv1 is not found in mammalian cells, and human Fis1 does not rescue deletion of the yeast gene (Suzuki et al., 2003; Stojanovski et al., 2004). Together, these observations suggest that the interactions between Drp1 and Fis1 in mammalian cells may be distinct from those occurring between the equivalent proteins in yeast. Phosphorylation of Drp1 by CaMKIα may therefore represent a mechanism by which the affinity of Drp1 and Fis1 is increased in mammalian cells.
During the revision of this manuscript, several other recent studies also found that Drp1 is phosphorylated in mammalian cells. Two independent studies found that cAMP-dependent PKA phosphorylated Drp1 at the same site as CaMKI (different residue numbers reflect the use of different Drp1 isoforms; Chang and Blackstone, 2007; Cribbs and Strack, 2007). CaMKI and PKA are known to have overlapping substrate specificity and, for example, phosphorylation of a common serine residue in synapsin I has been found to be phosphorylated in response to high K+ treatment or increased cAMP levels (Nairn and Greengard, 1987). Although there were some differences between the two studies, there was a general agreement on the conclusion. Results obtained from the use of phosphosite mutants of Drp1 indicated that increased phosphorylation of Drp1 was associated with increased mitochondrial length or area, results consistent with decreased mitochondrial fission. Moreover, the study by Cribbs and Strack (2007) found that expression of a phosphomimetic form of Drp1 was associated with increased resistance to proapoptotic treatments. These studies did not find any effect of phosphorylation on Drp1 self-assembly.
The basis for the difference between our study and these two recent studies is not clear (Chang and Blackstone, 2007; Cribbs and Strack, 2007). It is notable, however, that there was general agreement that the basal level of phosphorylation of native Drp1 was very low. Based on the results from these two studies, a low level of phosphorylation of Drp1 would predict that mitochondria would be expected to be highly fragmented and cells would be vulnerable to apoptosis. However, this situation is generally the opposite of what was actually observed. In our studies, mitochondria were highly elongated, especially in neuronal dendrites, consistent with the idea that a low basal phosphorylation of Drp1 favors mitochondrial fusion and increased phosphorylation favors mitochondrial fission. Another notable feature of these two studies is that they did not find any effect of increased cAMP levels on mitochondrial morphology. This result is consistent with the idea that phosphorylation of Drp1 at Ser600, although necessary for regulation of mitochondrial morphology, is not sufficient on its own and that activation of CaMKI has additional roles in regulation of Drp1 interaction with mitochondria. It will be interesting to investigate whether Ca2+- and cAMP-dependent signaling pathways can act synergistically to regulate mitochondrial morphology.
Another recent study found that rat Drp1 is phosphorylated by Cdk1/cyclin B, leading to stimulation of mitochondrial fission during mitosis (Taguchi et al., 2007). The CaMKIα and Cdk1 phosphorylation sites are very close in the C terminus of Drp1, highlighting the important role of this region of the protein in regulation of mitochondrial fission. Phosphorylation at the C terminus, at least in the case of CaMKIα, presumably results in alterations in the conformation of Drp1 and increases affinity of Drp1 for Fis1. It will be of interest to investigate whether phosphorylation of Drp1 by Cdk1 also stimulates fission through an increase in its affinity for Fis1 and to examine the interrelationship between the phosphorylation of these sites.
Recent studies have found that mitochondria distribution and movement in dendrites is regulated by synaptic activity (Li et al., 2004; Chang and Reynolds, 2006; Sung et al., 2008). K+-dependent depolarization or titanic stimulation was associated with a redistribution of mitochondria to dendritic spines, presumably to support the high metabolic requirement at these sites during synaptic activation. Moreover, increased Drp1 activity was found to be associated with increased mitochondrial fission, redistribution of mitochondria to dendrites, density of dendritic spines, and synaptic activity (Li et al., 2004). Preferential activation of CaMKs is found with protocols that increase long-term potentiation at excitatory synapses (Xia and Storm, 2005). CaMKIα-dependent phosphorylation of Drp1 and altered mitochondrial dynamics are therefore likely to play an important role in changes in synaptic morphology and function that accompany long-term potentiation.
Our results indicated that the effects of high K+ treatment were rapid, occurring within minutes, and were reversible. Moreover, high K+ treatment had no effect on cell death. This latter finding is of interest because several previous studies have suggested that mitochondrial fission is involved in cell death (Frank et al., 2001; Fannjiang et al., 2004; Lee et al., 2004; Youle and Karbowski, 2005). In particular, Drp1 has been found to be associated with proapoptotic members of the Bcl-2 family at sites of membrane scission during apoptosis (Wasiak et al., 2007). However, recent studies have also suggested that mitochondrial fission, although necessary for certain types of cell death, is not itself sufficient to cause apoptosis (Szabadkai et al., 2004; Meuer et al., 2007). The results of the current study add support to this hypothesis. Indeed, in preliminary experiments, we found that pretreatment with high K+ in neurons could prevent release of cytochrome c from mitochondria and prevent apoptosis in response to glutamate treatment (unpublished data). CaMKIα-dependent phosphorylation of Drp1 may play an important role in this protective process.
In conclusion, these results provide important new insights into the understanding of intracellular signaling pathways that regulate mitochondrial dynamics and morphology. We found that mitochondrial dynamics and morphology are controlled by Ca2+ influx through VDCCs. In turn, VDCCs are coupled to the activation of CaMKIα, resulting in phosphorylation of Drp1. CaMKIα-dependent phosphorylation of Drp1 facilitates its interaction with Fis1 and leads to increased mitochondrial fission. In addition to the phosphorylation of Drp1, CaMKIα may also influence other aspects of Drp1 translocation to mitochondria. Mitochondrial morphology is regulated by a balance between fission and fusion. Although we cannot rule out the possibility that mitochondrial fragmentation induced by high K+ is also accompanied by inhibition of mitochondrial fusion, the results indicate that the effects of high K+ are mediated by activation of CaMKI and phosphorylation of Drp1. This suggests that the fission process is the most likely target for the effects of high K+. Although our studies focused on regulation of Drp1 in neurons, we also found that CaMKI could regulate Drp1 function in HeLa cells. Ca2+ release from the ER has also been found to promote the translocation of Drp1 from the cytoplasm to the outer mitochondrial membrane of nonneuronal cells (Breckenridge et al., 2003). CaMKI is a ubiquitously expressed protein kinase. Thus, CaMKI-dependent phosphorylation and regulation of Drp1 may be a general phenomenon found in many different types of cells.
Materials and methods
DNA construction
PDsRed2-Mito (Clontech Laboratories, Inc.) is a mammalian expression vector that encodes the fluorescent protein DsRed2 tagged with the mitochondrial targeting sequence from subunit VIII of human cytochrome c oxidase. CaMKIα-DN was generated by mutating K49 to E to disrupt catalytic activity. CaMKIα-CA was obtained by truncating the autoinhibitory and CaM-binding domains (Yokokura et al., 1995; Matsushita and Nairn, 1998). The wild-type Drp1 (isoform 3) plasmid was provided by A.M. van der Bliek (University of California, Los Angeles, Los Angeles, CA; Smirnova et al., 1998). Drp1-S600A and S600D site-directed mutagenesis was performed using a Quickchange kit (Stratagene) according to the manufacturer's instructions. To replace endogenous Drp1 with RNAi-resistant YFP-Drp1, the underlined nucleotide T was changed to C to introduce a silent mutation in the Drp1 siRNA target sequence (1417-CGTAAAAGGTTGCCTGTTACA). Wild-type Drp1 and Drp1-S600A were cloned into pGEX-6p-1 and PET 21a+ for GST-Drp1 and Drp1-6His protein purification, respectively. CaMKK-433 and CaMKI-293 were also cloned into PET 21a+ for recombinant protein purification as described previously (Matsushita and Nairn, 1998). hFis1 cDNA was cloned by RT-PCR from HeLa cells. hFis1-ΔC (without 30 amino acids at the C terminus) was cloned into the pGEX-6p-1 vector for GST–hFis1-C protein purification.
Primary neuronal culture
The hippocampus from ∼18–19-embryonic day fetal Wistar rats was treated with 0.125% trypsin (Invitrogen) and 0.004% DNase-I (Sigma-Aldrich) at 37°C for 15 min and mechanically dissociated. Neurons were plated on poly-l-lysine (Sigma-Aldrich) and laminin (Roche)-coated glass-bottomed or plastic-bottomed 35-mm culture dishes (cell density was ∼25,000–30,000/35-mm dish for microscopy or ∼45,000–50,000/35-mm dish for Western blotting). Cells were maintained in culture medium with DME, 100 μg/ml penicillin-streptomycin (Invitrogen), and 10% fetal calf serum (Invitrogen). Cultures were maintained at 37°C in a 95% air, 5% CO2 humidified incubator. On the second day, the culture medium was replaced with medium containing DME, 2% B-27 supplement (Invitrogen), and 5% fetal calf serum. On the fourth day, the culture medium was replaced with medium containing DME, α-MEM, F-12 nutrient mixture, 2% B-27 supplement, 0.34% glucose, 25 μM 5-fluoro-deoxyuridine, 25 μM uridine, 1 mM kynurenic acid (Sigma-Aldrich), and 1% fetal calf serum. Cultures were used for experiments on days 9–12.
Transfection procedures
Transfection for neurons was performed with Lipofactamine 2000 (Invitrogen) according to the manufacturer's instructions. After 6 h of incubation, the medium containing Lipofactamine was replaced with normal culture medium. The cells were viewed 18–24 h after transfection by fluorescence microscopy. Predesigned siRNA for human CaMKIα (Thermo Fisher Scientific), human Fis1 (Thermo Fisher Scientific), and human Drp1 (Sigma-Aldrich) was introduced with DharmaFECT Transfection reagents according to the manufacturer's instructions.
Time-lapse imaging of mitochondria or Drp1
Mitochondrial imaging was performed with an inverted fluorescence microscope (Axiovert 200; Carl Zeiss, Inc.) with excitation at 545 nm using neuronal culture medium or DME (Invitrogen) with 10% FBS at 30°C. Illumination and exposure to the CCD camera (ORCA; Hamamatsu Photonics) were controlled and synchronized with AquaCosmos software (Hamamatsu Photonics). 63×/1.4 NA (Plan Apochromat; Carl Zeiss, Inc.) or 100×/1.45 NA (Plan Fluar; Carl Zeiss, Inc.) objectives were used to view the cells, and a narrow bandpass rhodamine filter was used when capturing the fluorescence images. GFP was visualized with excitation at 480 nm using an FITC filter. Time-lapse imaging was captured automatically with a selected interval. Drp1 imaging was performed with a laser confocal microscope (Fluoview 300; Olympus). 60×/1.0 NA or 40×/0.9 NA WLSM (PIauApo) objectives (Olympus) were used to view the cells. YFP or GFP was visualized using a BA 505–525 barrier filter. Mito-DsRed was viewed using a BA 565IF filter. Time-lapse imaging was also captured automatically with a selected interval. To analyze Drp1 foci, we selected random regions in neurons. The areas of these regions were also different in different cells, but their areas (micrometers squared) were automatically calculated by the software (Fluoview 300; Olympus). The background was then subtracted, and the number of Drp1 foci was counted and divided by the area to obtain foci/micrometers squared for Drp1 distribution in cells.
Analysis of mitochondrial movement and morphology
AquaCosmos software (Hamamatsu Photonics) was used to analyze mitochondrial movement and morphology. Mitochondrial movement was evaluated by analyzing the pDsRed2-Mito signal with time-lapse imaging using neuronal culture medium or DME (Invitrogen) with 10% FBS at 30°C. Pixel intensity difference was obtained by image subtraction between successive images at 10-s intervals, and the difference after subtraction of the background was calculated as the movement during the 10-s period. Image differences between six sequential images were averaged. The pixel area of the image was normalized to the total mitochondrial pixel area before subtraction. Morphology was evaluated by elongation index. Mitochondria were picked up after subtraction of background by using AquaCosmos software (Hamamatsu Photonics). The length and the area of an individual mitochondria were measured, and the elongation index was calculated by the square of the length divided by the area (i.e., when the elongation index is larger, mitochondria are longer). The elongation index after 15 min of high K+ stimulation was normalized to the value measured before stimulation to obtain the percent change in elongation index.
Intracellular Ca2+ measurement
Hippocampal cultures were incubated with Fura-2 AM (5 μM for 30 min) and washed with Hepes-buffered salt solution composed of 119 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, and 20 mM glucose in 25 mM Hepes (buffered to pH 7.4 with NaOH) at 30°C. Ca2+ measurement was performed with an epifluorescence inverted microscope (Axiovert 200; Carl Zeiss, Inc.) equipped with a 20×/NA 0.75 Fluar UV objective and Ca2+ ratio imaging system (Hamamatsu Photonics). Ratiometric measurement of Fura-2 fluorescence was obtained by sequential excitation at 340- and 380-nm lights from a xenon arc lamp. The intracellular Ca2+ change was indicated as the ratio of emitted fluorescence at 340- and 380-nm excitations.
Neuronal cell death assay
Hippocampal neuronal cultures were exposed to 500 μM glutamate or 45 mM of high K+ for15 min. After treatment, the cultures were washed and incubated with normal media taken from a batch of neurons cultured in parallel. After 24 h of incubation, the cultures were fixed with 4% PFA for 10 min. Cultures were stained with MAP2 antibody and Hoechst 33258 and analyzed by an inverted fluorescence microscope (Axiovert 200; Carl Zeiss, Inc.) using a 20×/0.75 NA Fluar objective. Neurons with condensed Hoechst 33258 staining were counted as dead cells.
Electron microscopy
Hippocampal neurons cultured on glass coverslips were washed with 0.1 M sodium cacodylate buffer, pH 7.4, and fixed with 2% glutaraldehyde and 3% PFA in 0.1 M sodium cacodylate buffer. After washing in 0.1 M sodium cacodylate buffer, the neurons were postfixed with 2% OsO4 in the same buffer, block stained in 2% aqueous uranyl acetate, dehydrated in ascending alcohols and propylene oxide, and finally embedded in epoxy resin Quetol 812 (Nisshin EM Co.) between a Teflon support and the coverglass. After Quetol 812 polymerization, the coverglass was removed by dipping the sample alternatively into liquid nitrogen and hot water. Neurons to be sectioned were selected by observing the flat-embedded resin plate under the light microscope. Areas containing the selected neurons were cut out and mounted on resin blocks for thin sectioning. 60–70-nm sections were stained with 2% aqueous uranyl acetate and lead citrate, and micrographs were taken at 75 kV in a transmission electron microscope (H-7100; Hitachi).
Purification of recombinant proteins
Purification of recombinant proteins was performed as described previously (Matsushita and Nairn, 1998). In brief, BL21 cells containing each expression plasmid were grown overnight at 37°C. The proteins were expressed in these cells after induction with 0.2 mM isopropyl1-thio-β-d-galactopyranoside. The expressed proteins were purified using a column of glutathione–Sepharose 4 Fast Flow (GE Healthcare) for GST proteins or Probond Nickel-Chelating resin (Invitrogen) for 6His-tagged proteins. Fractions were collected, and protein was analyzed by SDS-PAGE and staining with Coomassie brilliant blue. Fractions containing expressed protein were pooled and dialyzed against PBS. Protein concentrations were determined using a protein assay kit (Bio-Rad Laboratories). Aliquots were stored at −80°C.
Phosphorylation assays
The CaMK kinase assay buffer (100 μl) contained 50 mM Hepes, pH 7.5, 10 mM magnesium acetate, 1 mM EGTA, 5 mM dithiothreitol, 2.4 μM calmodulin, and 2 mM CaCl2. For CaMKII assays, 20 mM Hepes, pH 7.4, 10 mM MgCl2, and 1 mM dithiothreitol were used. The final concentrations of kinases or proteins were 500 ng/ml CaMKKα-433-6His, 1 μg/ml CaMKIα-293-6His, and 10 μg/ml CaMKII, GST-Drp1, or GST-Drp1-S600A. Reactions were initiated by the addition of γ-[32P]ATP (100 μM; 2–5 × 102 cpm/pmol) and incubated at 30°C for 30 min. Reactions were terminated by the addition of 100 μl SDS sample buffer (1% SDS, 60 mM Tris-HCl, pH 6.8, 5% [vol/vol] glycerol, and 0.2 M β-mercaptoethanol) and boiling at 100°C for 5 min. Samples were analyzed by SDS-PAGE (8% polyacrylamide) and autoradiography. For samples analyzed by immunoblotting, γ-[32P]ATP was replaced by nonradioactive ATP.
Drp1 phosphospecific antibody
A peptide corresponding to residues 594–608 of Drp1-3 (PVARKLS(p)AREQRDCE) was chemically phosphorylated at residue Ser600, and a rabbit polyclonal antibody was generated by Genosys (Sigma-Aldrich) essentially as described previously (Tomizawa et al., 2003).
Immunoblotting
After treatment of cultured neurons (9–12 days in vitro [DIV]), dishes were washed once with ice-cold PBS, and adherent cells were scraped with lysis buffer A (20 mM Tris, pH 7.4, 150 mM NaCl, 10 mM sodium orthovanadate, 20 mM sodium fluoride, 0.25 M sucrose, 1 mM dithiothreitol, 500 nM okadaic acid, and 0.5% Tween 20). Cell lysates were sonicated in 4× sample buffer, boiled for 5 min, and stored at −20°C until used. Proteins were analyzed by SDS-PAGE and immunoblotting on nitrocellulose membranes. Total Drp1 was probed with Drp1 monoclonal antibody (1:1,000 dilution; BD Biosciences), and phosphorylated Drp1 was probed with phospho-Ser600 antibody (1:1,000), both at 4°C overnight. HRP-conjugated secondary antibody was used at 1:2,000, and blotting signals were detected with standard protocols.
GST pull-down experiments
10 μg/ml of recombinant 6His-tagged Drp1 was phosphorylated by CaMKIα as described above using nonradioactive ATP. After incubation for 60 min at 30°C, 10 μg/ml GST-hFis1 was added followed by GST–Sepharose beads in the presence of PBS, pH 7.4, 1% BSA, and 0.1% Tween 20. Samples were mixed by gentle shaking at 4°C for at least 2 h and washed four times in washing buffer (20 mM Tris-HCl, pH 7.4, 500 mM NaCl, and 1% Tween 20). The GST beads were collected, proteins were eluted with sample buffer, and samples were analyzed by SDS-PAGE and immunoblotting. Drp1 bound to GST-hFis1 was detected with anti-6His monoclonal antibody (mouse IgG1, 1:5,000; BD Biosciences). For binding kinetics analysis, phosphorylated Drp1-6His was prepared as described above. The phosphorylated Drp1 was then mixed in different ratios with nonphosphorylated Drp1, and the percentage of Drp1-P relative to the total Drp1 was calculated. Drp1-6His bound to GST-Fis1 was detected by immunoblotting analysis.
Preparation of mitochondria-enriched fractions
Plated neurons were washed once with ice-cold PBS and scraped in lysis buffer A (20 mM Tris, pH 7.4, 150 mM NaCl, 10 mM sodium orthovanadate, 20 mM sodium fluoride, 0.25 M sucrose, 1 mM dithiothreitol, 500 nM okadaic acid, Complete 1 tablet/50 ml lysis buffer A, and 0.5% Tween 20). Cells were lysed by passing them 10 times through a 28-G 1/2 needle and centrifuged at 1,300 g for 15 min to remove nuclei and unbroken cells. The supernatant was collected and centrifuged again for 15 min at 5,000 g to pellet the mitochondria-enriched fraction. The mitochondrial pellets were diluted with 4× sample buffer, boiled for 6 min at 100°C, and stored at −30°C. The mitochondria membrane protein Tom40 was blotted (rabbit polyclonal antibody; Santa Cruz Biotechnology, Inc.) and used as an endogenous control.
Statistical analysis
The significance between the experimental groups was assessed by using one-way analysis of variance (ANOVA) followed by post-Tukey test. For single comparison, we performed an unpaired Student's t test. Data are shown as the mean ± SEM, and P < 0.05 was considered significant.
Online supplemental material
Fig. S1 shows that high K+ treatment alters mitochondrial movement in neurons. Fig. S2 shows a conserved phosphorylation CaMKI/PKA consensus motif in Drp1 proteins and down-regulation of CaMKIα level expression using RNAi. Fig. S3 shows that Fis1 regulates K+-induced Drp1 intracellular distribution in HeLa cells. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200802164/DC1.
Supplementary Material
Acknowledgments
We are grateful to A.M. van der Bliek for Drp1 plasmids. We thank T. Ogawa for technical assistance.
This work was supported in part by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan; by grants from Preventure-Japan Science and Technology Agency; and by the National Institutes of Health (A.C. Nairn).
Abbreviations used in this paper: ANOVA, analysis of variance; CaMK, Ca2+/calmodulin-dependent protein kinase; CaMKIα-CA, constitutively active CaMKIα; CaMKIα-DN, dominant-negative CaMKIα; DIV, days in vitro; Drp1, dynamin-related protein 1; GED, GTPase effector domain; VDCC, voltage-dependent Ca2+ channel.
References
- Barsoum, M.J., H. Yuan, A.A. Gerencser, G. Liot, Y. Kushnareva, S. Graber, I. Kovacs, W.D. Lee, J. Waggoner, J. Cui, et al. 2006. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 25:3900–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereiter-Hahn, J., and M. Voth. 1994. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc. Res. Tech. 27:198–219. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel, E., M.J. Barsoum, A. Godzik, R. Schwarzenbacher, and S.A. Lipton. 2003. Mitochondrial fission in apoptosis, neurodegeneratin and aging. Curr. Opin. Cell Biol. 15:706–716. [DOI] [PubMed] [Google Scholar]
- Breckenridge, D.G., M. Stojanovic, R.C. Marcellus, and G.C. Shore. 2003. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 160:1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerveny, K.L., Y. Tamura, Z. Zhang, R.E. Jensen, and H. Sesaki. 2007. Regulation of mitochondrial fusion and division. Trends Cell Biol. 17:563–569. [DOI] [PubMed] [Google Scholar]
- Chada, S.R., and P.J. Hollenbeck. 2003. Mitochondrial movement and positioning in axons: the role of growth factor signaling. J. Exp. Biol. 206:1985–1992. [DOI] [PubMed] [Google Scholar]
- Chan, D.C. 2006. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 22:79–99. [DOI] [PubMed] [Google Scholar]
- Chang, C.R., and C. Blackstone. 2007. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282:21583–21587. [DOI] [PubMed] [Google Scholar]
- Chang, D.T., and I.J. Reynolds. 2006. Differences in mitochondrial movement and morphology in young and mature primary cortical neurons in culture. Neuroscience. 141:727–736. [DOI] [PubMed] [Google Scholar]
- Cribbs, J.T., and S. Strack. 2007. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8:939–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmer, S.A., and D.C. Chan. 2007. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 8:870–879. [DOI] [PubMed] [Google Scholar]
- Fannjiang, Y., W.C. Cheng, S.J. Lee, B. Qi, J. Pevsner, J.M. McCaffery, R.B. Hill, G. Basanez, and J.M. Hardwick. 2004. Mitochondrial fission proteins regulate programmed cell death in yeast. Genes Dev. 18:2785–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, S., B. Gaume, E.S. Bergmann-Leitner, W.W. Leitner, E.G. Robert, F. Catez, C.L. Smith, and R.J. Youle. 2001. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell. 1:515–525. [DOI] [PubMed] [Google Scholar]
- Hook, S.S., and A.R. Means. 2001. Ca(2+)/CaM-dependent kinases: from activation to function. Annu. Rev. Pharmacol. Toxicol. 41:471–505. [DOI] [PubMed] [Google Scholar]
- Hoppins, S., L. Lackner, and J. Nunnari. 2007. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 76:751–780. [DOI] [PubMed] [Google Scholar]
- Karbowski, M., K.L. Norris, M.M. Cleland, S.Y. Jeong, and R.J. Youle. 2006. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 443:658–662. [DOI] [PubMed] [Google Scholar]
- Kroemer, G., and J.C. Reed. 2000. Mitochondrial control of cell death. Nat. Med. 6:513–519. [DOI] [PubMed] [Google Scholar]
- Lee, J.C., Y.G. Kwon, D.S. Lawrence, and A.M. Edelman. 1994. A requirement of hydrophobic and basic amino acid residues for substrate recognition by Ca2+/calmodulin-dependent protein kinase Ia. Proc. Natl. Acad. Sci. USA. 91:6413–6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, Y.J., S.Y. Jeong, M. Karbowski, C.L. Smith, and R.J. Youle. 2004. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell. 15:5001–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., K. Okamoto, Y. Hayashi, and M. Sheng. 2004. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 119:873–887. [DOI] [PubMed] [Google Scholar]
- Ligon, L.A., and O. Steward. 2000. Movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 427:340–350. [DOI] [PubMed] [Google Scholar]
- Matsushita, M., and A.C. Nairn. 1998. Characterization of the mechanism of regulation of Ca2+/calmodulin-dependent protein kinase I by calmodulin and by Ca2+/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 273:21473–21481. [DOI] [PubMed] [Google Scholar]
- Meuer, K., I.E. Suppanz, P. Lingor, V. Planchamp, B. Goricke, L. Fichtner, G.H. Braus, G.P. Dietz, S. Jakobs, M. Bahr, and J.H. Weishaupt. 2007. Cyclin-dependent kinase 5 is an upstream regulator of mitochondrial fission during neuronal apoptosis. Cell Death Differ. 14:651–661. [DOI] [PubMed] [Google Scholar]
- Mozdy, A.D., J.M. McCaffery, and J.M. Shaw. 2000. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J. Cell Biol. 151:367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairn, A.C., and P. Greengard. 1987. Purification and characterization of Ca2+/calmodulin-dependent protein kinase I from bovine brain. J. Biol. Chem. 262:7273–7281. [PubMed] [Google Scholar]
- Oakes, S.A., and S.J. Korsmeyer. 2004. Untangling the web: mitochondrial fission and apoptosis. Dev. Cell. 7:460–462. [DOI] [PubMed] [Google Scholar]
- Pitts, K.R., M.A. McNiven, and Y. Yoon. 2004. Mitochondria-specific function of the dynamin family protein DLP1 is mediated by its C-terminal domains. J. Biol. Chem. 279:50286–50294. [DOI] [PubMed] [Google Scholar]
- Rube, D.A., and A.M. van der Bliek. 2004. Mitochondrial morphology is dynamic and varied. Mol. Cell. Biochem. 256-257:331–339. [DOI] [PubMed] [Google Scholar]
- Ruthel, G., and P.J. Hollenbeck. 2003. Response of mitochondrial traffic to axon determination and differential branch growth. J. Neurosci. 23:8618–8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, J.M., E.S. Guire, T. Saneyoshi, and T.R. Soderling. 2005. Calmodulin-dependent kinase kinase/calmodulin kinase I activity gates extracellular-regulated kinase-dependent long-term potentiation. J. Neurosci. 25:1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, J.M., and J. Nunnari. 2002. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. 12:178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova, E., D.L. Shurland, S.N. Ryazantsev, and A.M. van der Bliek. 1998. A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 143:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova, E., L. Griparic, D.L. Shurland, and A.M. van der Bliek. 2001. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell. 12:2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovski, D., O.S. Koutsopoulos, K. Okamoto, and M.T. Ryan. 2004. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J. Cell Sci. 117:1201–1210. [DOI] [PubMed] [Google Scholar]
- Sung, J.Y., O. Engmann, M.A. Teylan, A.C. Nairn, P. Greengard, and Y. Kim. 2008. WAVE1 controls neuronal activity-induced mitochondrial distribution in dendritic spines. Proc. Natl. Acad. Sci. USA. 105:3112–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M., S.Y. Jeong, M. Karbowski, R.J. Youle, and N. Tjandra. 2003. The solution structure of human mitochondria fission protein Fis1 reveals a novel TPR-like helix bundle. J. Mol. Biol. 334:445–458. [DOI] [PubMed] [Google Scholar]
- Szabadkai, G., A.M. Simoni, M. Chami, M.R. Wieckowski, R.J. Youle, and R. Rizzuto. 2004. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell. 16:59–68. [DOI] [PubMed] [Google Scholar]
- Taguchi, N., N. Ishihara, A. Jofuku, T. Oka, and K. Mihara. 2007. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282:11521–11529. [DOI] [PubMed] [Google Scholar]
- Takei, K., P.S. McPherson, S.L. Schmid, and P. De Camilli. 1995. Tubular membrane invaginations coated by dynamin rings are induced by GTP-γ S in nerve terminals. Nature. 374:186–190. [DOI] [PubMed] [Google Scholar]
- Tang, Y., and R. Zucker. 1997. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 18:483–491. [DOI] [PubMed] [Google Scholar]
- Tomizawa, K., S. Sunada, Y.F. Lu, Y. Oda, M. Kinuta, T. Ohshima, T. Saito, F.Y. Wei, M. Matsushita, S.T. Li, et al. 2003. Cophosphorylation of amphiphysin I and dynamin I by Cdk5 regulates clathrin-mediated endocytosis of synaptic vesicles. J. Cell Biol. 163:813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondera, D., F. Czauderna, K. Paulick, R. Schwarzer, J. Kaufmann, and A. Santel. 2005. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J. Cell Sci. 118:3049–3059. [DOI] [PubMed] [Google Scholar]
- Wasiak, S., R. Zunino, and H.M. McBride. 2007. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 177:439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells, R.C., L.K. Picton, S.C. Williams, F.J. Tan, and R.B. Hill. 2007. Direct binding of the dynamin-like GTPase, Dnm1, to mitochondrial dynamics protein Fis1 is negatively regulated by the Fis1 N-terminal arm. J. Biol. Chem. 282:33769–33775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, Z., and D.R. Storm. 2005. The role of calmodulin as a signal integrator for synaptic plasticity. Nat. Rev. Neurosci. 6:267–276. [DOI] [PubMed] [Google Scholar]
- Yokokura, H., M.R. Picciotto, A.C. Nairn, and H. Hidaka. 1995. The regulatory region of calcium/calmodulin-dependent protein kinase I contains closely associated autoinhibitory and calmodulin-binding domains. J. Biol. Chem. 270:23851–23859. [DOI] [PubMed] [Google Scholar]
- Yoon, Y., K.R. Pitts, and M.A. McNiven. 2001. Mammalian dynamin-like protein DLP1 tubulates membranes. Mol. Biol. Cell. 12:2894–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, Y., E.W. Krueger, B.J. Oswald, and M.A. McNiven. 2003. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 23:5409–5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle, R.J., and M. Karbowski. 2005. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 6:657–663. [DOI] [PubMed] [Google Scholar]
- Yu, T., R.J. Fox, L.S. Burwell, and Y. Yoon. 2005. Regulation of mitochondrial fission and apoptosis by the mitochondrial outer membrane protein hFis1. J. Cell Sci. 118:4141–4151. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}