

Abstract
Spatial regulation is an important feature of signal specificity elicited by cytoplasmic tyrosine kinases of the Src family (SRC family protein tyrosine kinases [SFK]). Cholesterol-enriched membrane domains, such as caveolae, regulate association of SFK with the platelet-derived growth factor receptor (PDGFR), which is needed for kinase activation and mitogenic signaling. PAG, a ubiquitously expressed member of the transmembrane adaptor protein family, is known to negatively regulate SFK signaling though binding to Csk. We report that PAG modulates PDGFR levels in caveolae and SFK mitogenic signaling through a Csk-independent mechanism. Regulation of SFK mitogenic activity by PAG requires the first N-terminal 97 aa (PAG-N), which include the extracellular and transmembrane domains, palmitoylation sites, and a short cytoplasmic sequence. We also show that PAG-N increases ganglioside GM1 levels at the cell surface and, thus, displaces PDGFR from caveolae, a process that requires the ganglioside-specific sialidase Neu-3. In conclusion, PAG regulates PDGFR membrane partitioning and SFK mitogenic signaling by modulating GM1 levels within caveolae independently from Csk.
Introduction
Cytoplasmic tyrosine kinases of the Src family (Src family protein tyrosine kinases [SFK]) play important roles in signal transduction induced by a large number of extracellular stimuli including growth factors, integrins, and cytokines (Martin, 2001). For example, they control cellular responses induced by PDGF such as mitogenesis, survival, and cytoskeleton rearrangement. In contrast to several mitogens, PDGF-induced SFK activation does not have an impact on the Ras–MAPK pathway but, rather, controls c-myc expression that is needed for cell cycle progression (Bromann et al., 2004). Several Src substrates promoting mitogenic signaling have been characterized, including Shc, Stat3, Vav2, and Abl (Bromann et al., 2004). Rac has been further identified as an important element of Abl signaling, and we have shown that Src-induced c-myc expression and cell cycle progression need Rac/Jun N-terminal kinase and Rac–NADPH oxidase signaling pathways (Boureux et al., 2005). Intriguingly, requirement of SFK is also dependent on functional p53 (Broome and Courtneidge, 2000; Furstoss et al., 2002). Although not completely understood, data from Niu et al. (2005) suggest that Stat3 mediates p53 down-regulation, an event which is required for late G1 progression. Nevertheless, additional regulatory mechanisms might be envisaged.
How Src induces signaling specificity is not clearly established. Although phosphorylation of appropriate substrates may be part of the mechanism, spatial regulation could also be implicated. Indeed, SFK localization at focal adhesions is crucial for cell adhesion and migration (Frame, 2004), and accumulation in plasma membrane lipid rafts is apparently important for T and B cell receptor activation (Janes et al., 1999). Lipid rafts are somewhat controversial structures, defined as microdomains enriched in cholesterol and sphingolipids which concentrate and regulate signaling proteins at the plasma membrane (Horejsi, 2005). In nonlymphoid cells, this function may be partially ensured by caveolae. These membrane invaginations are composed of cholesterol, sphingolipids, structural proteins of the caveolin family, and signaling molecules. They play crucial roles in signal transduction induced by growth factors (Pike, 2005). In fibroblasts, they are highly abundant, regulate the localization of PDGF receptors (PDGFR), and contain probably >25% of the SFK present in these cells (Veracini et al., 2006). We have recently reported that these domains regulate SFK mitogenic signaling through SFK–PDGFR complex formation. In contrast, caveolae do not control PDGF-induced SFK signaling leading to F-actin assembly for dorsal ruffles formation (Veracini et al., 2006). Therefore, proteins that regulate tyrosine kinases in such membrane domains may play important roles in signaling specificity.
Src activity is subjected to a strict control based on intramolecular interactions of its SH2 domain with pTyr527 (phosphorylated Tyr527; referred to the avian Src) and of the SH3 domain with a linker domain between the SH2 and the catalytic core that keep the kinase in a closed and inactive conformation (Boggon and Eck, 2004). Phosphorylation of Tyr527 at the C terminus by the cytoplasmic tyrosine kinase Csk is crucial for SFK inactivation. Indeed, Csk inactivation in mice causes aberrant SFK activity and lethality during embryogenesis (Imamoto and Soriano, 1993; Nada et al., 1993). However, how Csk phosphorylates SFK in vivo is only partially understood. Although Src is a very good substrate in vitro, it does not interact efficiently with Csk in vivo because of the fact that Src is at the plasma membrane, whereas Csk is strictly cytosolic. Thus, in vivo activity requires membrane-associated Csk binders that allow its recruitment to subcellular compartments where SFK reside. For example, paxillin recruits Csk to the focal adhesions that may regulate SFK adhesive and migratory functions (Baumeister et al., 2005), whereas Dok-1 induces Csk translocation to the plasma membrane for mitogenic regulation (Zhao et al., 2006). This regulation is governed by a Csk-SH2 pTyr-dependent mechanism, and phosphorylation of Csk-binding proteins is triggered by SFK, which defines a negative-feedback regulatory loop.
Recently, a novel group of signaling proteins has emerged that may also be involved in SFK signaling and its regulation during immunoreceptor stimulation. This family, called TRAPs (transmembrane adaptor proteins), currently comprises seven members, the founder being LAT (the linker of activation of T cells; Horejsi et al., 2004). They all contain a short extracellular single transmembrane and a long intracellular domain with tyrosine motifs that potentially interact with SH2 domains of other signaling proteins. For example, LAT comprises four Tyr that, when phosphorylated by ZAP70 (a downstream effector of the SFK Lck and Fyn), create binding sites for PLCγ and Grb2, which are both required for efficient T cell activation. Another member of this family, PAG (phosphoprotein associated with glycosphingolipid-enriched microdomains; Brdicka et al., 2000), also called Cbp (Csk-binding protein; Kawabuchi et al., 2000), is ubiquitously expressed, unlike other members of the family whose expression is mostly restricted to hematopoietic tissues. PAG contains 10 Tyr residues in the cytoplasmic domain, 9 of which are SFK phosphorylation sites, 2 poly-Pro motifs for interaction with SH3-containing proteins, and 2 potential palmitoylation sites implicated in lipid rafts localization (Horejsi et al., 2004). It may allow Csk recruitment in these structures through interaction of Csk-SH2 with pTyr317. Thus, PAG may define an additional negative regulatory loop toward SFK in membrane domains such as caveolae (Shima et al., 2003). In this paper, we have addressed the role of PAG in the regulation of SFK mitogenic signaling. We show that this adaptor, independently of binding to Csk, regulates PDGFR partitioning at the plasma membrane through the ganglioside GM1.
Results
PAG negatively regulates Src mitogenic signaling
We first addressed the role of Csk in the PDGF-induced Src mitogenic signal transduction pathway. To this end, we transfected NIH 3T3 cells with a Csk-expressing construct. After serum starvation, we stimulated the transfected cells with PDGF for cell cycle reentry and monitored de novo DNA synthesis by BrdU incorporation into the nuclei. PDGF stimulation induced a 50% BrdU incorporation in control cells, which was inhibited by Csk overexpression (Fig. 1 A). DNA synthesis was restored by coexpression of the transcription factor Myc or the active RacV12 mutant of the Rho family of small GTPases, two elements of the Src pathway (Bromann et al., 2004; Boureux et al., 2005). In contrast, the active RasL61 mutant could not overcome the G1 block induced by Csk, confirming the Ras-independent nature of the Src pathway (Bromann et al., 2004). We concluded that Csk can regulate the PDGF-induced Src mitogenic function.
Figure 1.

PAG inhibits PDGF-induced Src mitogenic signaling. (A) Csk overexpression inhibits PDGF-induced Src mitogenic signaling. Quiescent NIH 3T3 seeded on coverslips and transfected, or not, with the indicated constructs were stimulated, or not, with 25 ng/ml PDGF in the presence of BrdU for 18 h. (B) PAG inhibits PDGF-induced Src mitogenic signaling. Quiescent NIH 3T3 seeded onto coverslips and transfected, or not, with the indicated constructs were treated as in A. (C) PAG does not inhibit mitogenesis in p53-deficient cells. p53+/+ and p53−/− MEF transfected, or not, with a PAG-encoding construct were stimulated, or not, with PDGF in the presence of BrdU. (D) PAG does not affect SrcY527F-driven DNA synthesis. Quiescent NIH 3T3 cells transfected with the indicated constructs were incubated with BrdU for 18 h. Cells were then fixed and processed for immunofluorescence. The percentage of transfected cells that incorporated BrdU was calculated as described in Material and methods. Results are expressed as the mean ± SD of three to five independent experiments.
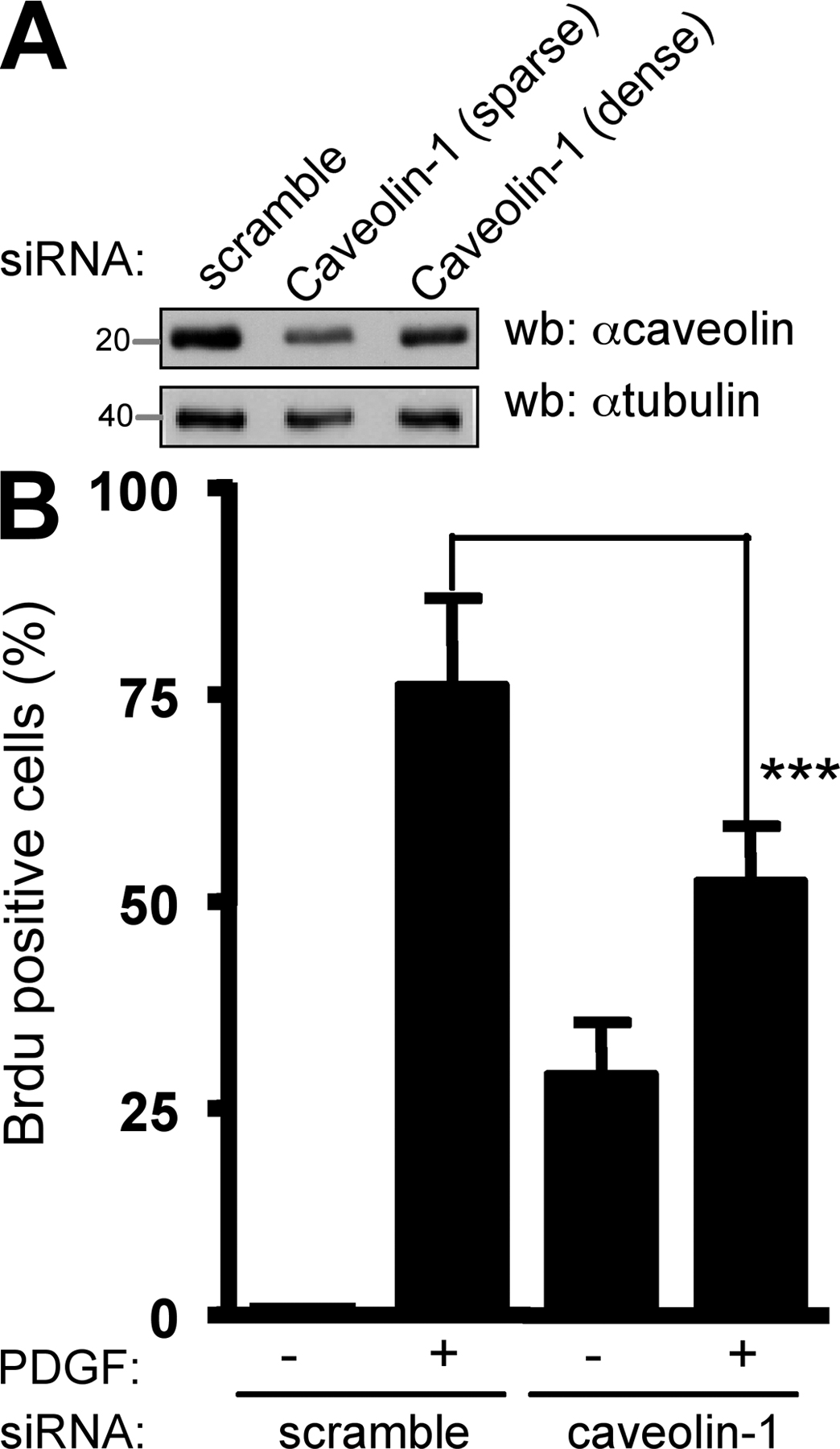
Our previous study suggested that Src mitogenic signaling was initiated in caveolae (Veracini et al., 2006). To confirm this data, we analyzed mitogenesis in cells with reduced level of caveolin. Although caveolin-1 knockdown promoted DNA synthesis by itself in 25% of NIH 3T3 cells, it significantly reduced PDGF mitogenic response in comparison to control cells, confirming the role of caveolin in this signaling pathway (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200705102/DC1). The residual mitogenic response could be attributed to the moderate efficacy of our siRNA to reduce plasma membrane caveolin-1 levels in cells cultured at high density (Galbiati et al., 1998) as required for efficient induction of quiescence. Nevertheless, we do not exclude the involvement of additional membrane domains in this cellular response. We were next interested in the adaptors that target Csk to these domains, particularly PAG. PAG overexpression strongly inhibited DNA synthesis induced by PDGF (Fig. 1 B). As for Csk, active RacV12 and Myc rescued the PDGFR signaling that was blocked by PAG. Myc effect was specific as it was not observed by expressing Fos. Moreover, although PAG inhibited the mitogenic response induced by PDGF in p53+/+ mouse embryonic fibroblasts (MEF), this inhibitory effect was not observed in p53−/− MEF (Fig. 1 C). A comparable result was described in NIH 3T3 cells that coexpressed the adenovirus E1B (early 1B) 55K protein, which inhibits p53 transactivation (Yew and Berk, 1992). These findings are consistent with the notion that PAG might inhibit the Src mitogenic pathway. Interestingly, PAG did not affect the capacity of oncogenic SrcY527F per se to induce DNA synthesis (Fig. 1 D), indicating that it may regulate signaling upstream of SFK, probably via a PDGF-induced SFK activation.
PAG mitogenic inhibition is independent of its interaction with Csk
We next asked whether PAG function was mediated by its association with Csk. Surprisingly, we found that the PAG Y317F, a mutant which did not interact with Csk (Brdicka et al., 2000; Kawabuchi et al., 2000), could still inhibit mitogenesis (Fig. 2 B, left). To confirm this observation, we coexpressed PAG with an inactive form of Csk, Csk-K−. By associating with PAG, Csk-K− was expected to prevent recruitment of endogenous Csk to membrane domains and, therefore, to overcome PAG inhibitory effect. As this was not the case, we concluded that the PAG antimitogenic effect is independent of its interaction with Csk. PAG contains nine additional Tyr residues that, when phosphorylated, create binding sites for other SH2-containing signaling proteins, including RasGAP and the adaptor Sam68 (Smida et al., 2007). Hence, we investigated whether one of these Tyr residues played a role in PAG antimitogenic effect. However, we found that all mutants in which a single Tyr residue was replaced by Phe were still inhibitory (unpublished data). We also tested the activity of the PAG-N mutant fused to the GFP, which comprises only the first 97 aa encompassing the extracellular/transmembrane domains, and a short cytoplasmic sequence without any obvious binding motifs for signaling proteins. Remarkably, PAG-N was sufficient to mimic the antimitogenic effect of full length PAG. Moreover, coexpression of PAG-N with Myc or RacV12 restored the mitogenic response of these cells upon PDGF stimulation (Fig. 2 B, right). Conversely, expression of PAG-C [98–433] fused to GFP, which could still associate with Csk, did not affect mitogenesis (Fig. 2, B [right] and C [middle and right]). We obtained similar data when PAG-N was transduced by retroviral infection, which induced moderate protein expression and, thus, excluded a nonspecific inhibitory effect caused by high protein expression (Fig. 3 C). In conclusion, PAG-N is sufficient to inhibit Src mitogenic signaling.
Figure 2.

PAG mitogenic inhibition is independent of Csk but requires PAG palmitoylation sites. (A) Schematic of the PAG constructs used. The extracellular (extra), transmembrane (TM), and cytoplasmic domains are indicated as well as the Csk (Y317) binding site and the GFP fused to PAG sequences. (B) PAG mitogenic inhibition does not require binding to Csk. Quiescent NIH 3T3 cells were transfected with the indicated constructs and stimulated, or not, with PDGF in the presence of BrdU. (C, left) Comparison of the levels of PAG and PAGY317F. (C, right) PAG-N does not bind to Csk unlike PAG-C. Protein levels were assessed by Western blotting with the indicated antibodies from whole cell lysates (WCL) of HEK 293 cells expressing the shown constructs. Csk–PAG complex formation was assessed by coimmunoprecipitation with an anti-Csk antibody. Molecular masses are indicated in kilodaltons. (D) Schematic of the PAG-N constructs used. Palmitoylation sites are indicated. (E) Levels of PAG-N and PAG-NCC/AA in whole cell lysates (top) and in CEF (bottom). HEK 293 cells transfected with the indicated constructs were lysed with Triton X-100 and homogenized with a Dounce, followed by sucrose gradient fractionation. Fractions were then directly analyzed by Western blotting with anti-GFP and anti-caveolin antibodies, as indicated. The respective proteins as well as the fractions' numbers are shown. Molecular masses are indicated in kilodaltons. (F) PAG-N mitogenic inhibition requires the palmitoylation sites. Quiescent NIH 3T3 cells transfected with the indicated constructs were stimulated, or not, with PDGF in the presence of BrdU. Cells were then fixed and processed for immunofluorescence. The percentage of transfected (or infected) cells that incorporated BrdU was calculated as described in Material and methods. Results are expressed as the mean ± SD of three to five independent experiments.
Figure 3.

PAG-N does not regulate growth factor signaling outside caveolae. (A) PAG-N does not affect PDGF-induced dorsal ruffles' formation. (A, left) An example of circular dorsal ruffle formation assessed by actin staining in NIH 3T3 cells infected with the indicated retroviruses, stimulated or not with PDGF for 10 min. (A, right) Statistical analysis of dorsal ruffle formation in cells infected with the shown retroviruses, treated, or not, with 5 μM SU6656 for 30 min and stimulated with PDGF for 10 min. (B) EGF-induced mitogenic response is not regulated by caveolae in NIH 3T3 cells. Quiescent cells transfected with Cav3-DGV were stimulated with 25 ng/ml PDGF or 100 ng/ml EGF in the presence of BrdU. (C) PAG-N does not affect EGF-induced DNA synthesis. The percentage of transfected (or infected) cells that incorporated BrdU or that formed dorsal ruffles was calculated as described in Material and methods. Results are expressed as the mean ± SD of three to five independent experiments. **, P < 0.01 (using Student's t test).
PAG-N does not have an impact on growth factor signaling outside caveolae
We next investigated how membrane partitioning of PAG-N affects its activity. To this aim we used caveolae-enriched fractions (CEF) purified from HEK 293 cells expressing PAG-N. A Triton X-100 cell lysate was fractionated through a sucrose gradient and CEF were isolated in the light fractions (2–4) as shown by the bulk of caveolin (Veracini et al., 2006). As shown in Fig. 2 E, PAG-N was localized in CEF. Mutation of the lipid acylation sites Cys39 and Cys42 into Ala (PAG-N CC/AA) was sufficient to abrogate this localization. Interestingly, these mutations also impaired the capacity of PAG-N to inhibit mitogenesis (Fig. 2 F), suggesting that targeting to caveolae and/or other membrane domains is required for PAG-N function. We then asked whether PAG-N could also regulate growth factor signaling outside membrane microdomains. First, we examined the effect of PAG-N on PDGF-induced dorsal ruffle formation, which is known to be independent of caveolae (Veracini et al., 2006). We observed that 30% of control NIH 3T3 cells exhibited at least one dorsal ruffle 5 min after PDGF stimulation (Fig. 3 A) and that this response was not affected by overexpression of PAG-N. We then analyzed the mitogenic response induced by EGF in NIH 3T3 cells. Although the dominant-negative Cav-3DGV inhibited PDGF mitogenic response (Veracini et al., 2006), it did not affect the one induced by EGF (Fig. 3 B). This indicated that, in NIH 3T3 cells, EGF does not require caveolae and/or cholesterol-enriched membrane domains for mitogenesis. Interestingly, PAG-N also had no effect on EGF cellular response, whereas it still inhibited the one elicited by PDGF (Fig. 3 C). We concluded that PAG-N function is tightly linked to the signaling processes dependent on membrane domains such as caveolae.
PAG-N displaces PDGFR from caveolae, leading to the prevention of Src mitogenic signaling
We next addressed the molecular mechanism by which PAG-N inhibits SFK mitogenic function in greater detail. Because PAG-N may affect signaling upstream of SFK, we hypothesized that it could inhibit SFK-PDGFR coupling in caveolae. This idea was first evaluated in CEF of HEK 293 cells expressing human PDGFR β, with or without PAG-N. Surprisingly, PAG-N reduced PDGFR level in CEF by 50%, whereas SFK concentration remained unchanged (Fig. 4 A, left). Moreover, PAG-N did not affect the total level of the receptor (Fig. 4 A, right), thus excluding any protein degradation mechanism. The specificity of this inhibition was also illustrated by the inability of PAG-N CC/AA to displace the receptor from CEF. We confirmed these observations by electron microscopy using immunogold labeling of PDGFR in caveolae of NIH 3T3 cells infected with mock or PAG-N retroviruses. Indeed, although 50% of caveolae were positive for PDGFR immunolabeling in control cells, <10% were positive in PAG-N–expressing cells (Fig. 4 B). In contrast, PAG-N did not affect the caveolar immunogold labeling of caveolin, thus confirming the specificity of this effect (Fig. 4 C). We concluded that PAG-N specifically excludes PDGFR from caveolae.
Figure 4.

PAG-N excludes PDGFR from caveolae. (A) PAG-N reduces the level of PDGFR in CEF of HEK 293 cells expressing PDGFR. Cells were transfected with the indicated constructs and lysed with 1% Triton X-100 lysis buffer followed by sucrose gradient fractionation. Levels of PDGFR, caveolin, and SFK in CEF are shown. Levels of PDGFR and PAG mutants in a whole cell lysate (WCL) are also shown. Molecular masses are indicated in kilodaltons. (B) An example (arrows indicated PDGFR-labeled caveolae; left) and statistical analysis (mean ± SD; right) of PDGFR immunogold labeling in caveolae of NIH 3T3 cells infected with the indicated constructs are shown. (C) PAG-N does not affect caveolin localization in caveolae. An example (arrows indicate caveolin-labeled caveolae; left) and statistical analysis (mean ± SD; right) of caveolin immunogold labeling in caveolae of NIH 3T3 cells infected as indicated are shown. **, P < 0.01 (using Student's t test).
We then addressed the biological consequence of PDGFR caveolar depletion on Src mitogenic signaling. Although PDGFR association with SFK was observed in CEF from PDGF-stimulated control cells, complex formation was largely reduced in cells infected with PAG-N (Fig. 5 A). PAG-N also increased the association of SFK with caveolin, which is thought to participate in its catalytic inactivation (Li et al., 1996). Reduction of SFK–PDGFR complex formation was also suggested by a decrease of PDGF-induced SFK activation as well as phosphorylation of the Src mitogenic substrates Stat3, Tyr705, Shc, Tyr239, and Tyr240 (Bromann et al., 2004) in cells expressing PAG-N (Fig. 5, B and C). Conversely, activation of ERK1 and 2 upon PDGF stimulation was not modified (Fig. 5 D), indicating that PAG-N does not inhibit all signaling pathways emanated from the activated receptor but only the caveolae-regulated signaling.
Figure 5.

PAG-N inhibits SFK mitogenic signaling. (A) PDGFR–SFK complex formation is reduced in CEF of NIH 3T3 cells infected with PAG-N. Quiescent cells infected with the indicated retroviruses were stimulated with PDGF for 10 min. SFK association with caveolin and active PDGFR in CEF were evaluated by Western blotting of immunoprecipitated kinases with the indicated antibodies. Level of immunoprecipitated SFK is also shown. (B) Src mitogenic signaling is affected by PAG-N. SFK activity was measured by Western blotting with anti-pY416 antibody (left) or by in vitro kinase assay using denatured enolase (right) from immunoprecipitated kinases of the indicated cells stimulated or not with PDGF. Quantification (mean ± SD) of SFK activity was expressed as the ratio between active SFK and total SFK levels. (C) PAG-N inhibits phosphorylation of SFK mitogenic substrates. Levels of Stat3 and Shc phosphorylated on Tyr705 (left) or on Tyr239 and Tyr240 (right) are shown for cells infected with the indicated retroviruses. (D) PAG-N does not affect PDGF-induced Erk1-2 activation. Lysates of the indicated cells stimulated, or not, with PDGF for the indicated times were subjected to Western blotting with anti–phospho-Erk and anti-Erk–specific antibodies. Molecular masses are indicated in kilodaltons. *, P < 0.05 (using Student's t test).
PAG-N displaces PDGFR from caveolae via GM1
We then investigated how PAG-N depletes PDGFR from caveolae. Because PAG-N does not interact physically with PDGFR (unpublished data), we reasoned that it may affect caveolae properties necessary for PDGFR localization. Indeed, we observed a drastic redistribution of caveolin at the periphery of PAG-N–expressing cells. This was confirmed by electron microscopy, which showed that 70% of mature caveolae were connected to the surface of cells expressing PAG-N as compared with 25% in mock-infected cells (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200705102/DC1). This finding suggested that PAG-N modulates caveolae properties and/or endocytosis. In addition to caveolin, lipid constituents, such as cholesterol and ganglioside GM1, also play important roles in caveolae functions (Sharma et al., 2004). Remarkably, we observed a large accumulation of GM1 at the surface of cells expressing PAG-N (Fig. 6 A). This accumulation was dependent on the membrane domain localization of the adaptor, as the mutant PAG-N CC/AA did not modify GM1 membrane level. A moderate but significant accumulation of cholesterol was also observed at the surface of cells stably expressing PAG-N in comparison to mock-infected cells (unpublished data), confirming that PAG-N alters caveolae constituents. We then analyzed the effect of such modifications in membrane lipid constituents on PDGFR membrane partitioning. Previous reports suggested that PDGF localization could be regulated by GM1 (Mitsuda et al., 2002). Therefore, we hypothesized that exclusion of PDGFR from caveolae by PAG-N was mediated by GM1. Indeed, exogenous GM1 reduced PDGFR accumulation in CEF from HEK 293 cells expressing the human receptor (Fig. 6 B). This finding was further confirmed by electron microscopy on NIH 3T3 cells, where immunogold labeling of PDGFR in caveolae was reduced by 80% upon addition of exogenous GM1 (Fig. 6 C). We then asked whether GM1 could also influence PDGF-induced mitogenesis. Exogenous GM1 reduced PDGF mitogenic response by 70% (Fig. 6 D). This inhibition was reversed by expression of Myc or RacV12, two elements of the Src pathway (Bromann et al., 2004; Boureux et al., 2005). In contrast, it was not modified by expression of the constitutively active RasL61 mutant or the transcription factor Fos, demonstrating that GM1 inhibitory effect was independent of Ras. We concluded that an excess of GM1 at the plasma membrane delocalizes PDGFR from caveolae and inhibits PDGF mitogenic response via inhibition of an Src-dependent signaling pathway.
Figure 6.

PAG-N displaces PDGFR from caveolae and inhibits Src mitogenic signaling via GM1. (A) PAG-N increases GM1 content at the cell surface. Representative example of quiescent NIH 3T3 cells transfected with the indicated constructs and stained for GM1 at the cell surface with CTxB–Alexa 594. Arrows indicate the GM1 staining of transfected cells. (B) Exogenous GM1 reduces PDGFR level in CEF. HEK 293 cells expressing PDGFR were treated at 4°C with 10 μM GM1 for 30 min and then fractionated through a sucrose gradient. Levels of PDGFR and caveolin in CEF are shown as well as the level of PDGFR in whole cell lysates. Molecular masses are indicated in kilodaltons. (C) PAG-N depletes caveolar PDGFR via GM1. Representative example (left) and statistical analysis (right) of NIH 3T3 cells treated with GM1 or ethanol (vehicle) and then subjected to PDGFR immunogold labeling. Arrows highlight PDGFR-positive caveolae. (D) GM1 regulates PDGF-induced Src mitogenic signaling. Quiescent NIH 3T3 cells transfected with the indicated constructs were incubated, or not, with 10 μM GM1 at 4°C for 30 min and stimulated with PDGF at 37°C for 18 h in the presence of BrdU. Cells were then fixed and processed for immunofluorescence. The percentage of transfected cells that incorporated BrdU was calculated as described in Material and methods. Results are expressed as the mean ± SD of three to five independent experiments. **, P < 0.01 (using Student's t test).
The ganglioside-specific sialidase Neu-3 mediates PAG-N–induced GM1 antimitogenic effects
We next sought to try to understand how PAG-N induces cell surface GM1. The effect of PAG-N CC/AA suggested a role for membrane domain localization in this molecular process. Furthermore, we found that caveolin-1 knockdown also reduced the capacity of PAG-N to impact on GM1 (Fig. 7, A and B), suggesting that caveolin or caveolae integrity might be also required. The ganglioside-specific sialidase Neu-3 allows the conversion of di- and trisialidated gangliosides to monosialidated glycolipids such as GM1. Interestingly, this enzyme is localized at the external leaflet of the plasma membrane and interacts with caveolin (Wang et al., 2002; Papini et al., 2004). We thus investigated whether Neu-3 was involved in PAG-N–induced GM1 effects. Neu-3 knockdown strongly reduced GM1 cell surface accumulation elicited by PAG-N (Fig. 7, A–C). We confirmed this result using 2-deoxy-2,3-didehydro-N-acetylneuraminic acid (DANA), which specifically inhibits cell surface–localized Neu-3 (Da Silva et al., 2005). DANA treatment largely reversed both PAG-N–induced GM1 cell surface accumulation and PDGFR displacement from caveolae, confirming Neu-3 knockdown experiments (Fig. 7, A, B, and D). We then addressed the biological impact of Neu-3 activity on PAG-N antimitogenic effect. Neu-3 inhibition, either with DANA (Fig. 7 E, left) or through Neu-3 siRNA transduction (Fig. 7 E, right), largely reversed PAG-N mitogenic inhibition. Collectively, these data point to GM1 as a mediator of PAG-N antimitogenic function. Moreover, these observations stress that Neu-3 controls PAG-N–induced GM1 antimitogenic effects.
Figure 7.

The ganglioside sialidase Neu-3 mediates PAG-N–induced GM1 antimitogenic effects. (A and B) PAG-N–induced GM1 cell surface accumulation requires caveolin and Neu-3 activity. An example (A) of cell surface GM1 level in PAG-N–expressing cells with knocked-down caveolin-1 or Neu-3 is shown. PAG-N–expressing cells were transfected with the indicated siRNA or treated with DANA, as indicated, and were stained with CTxB–Alexa 594. (B) Statistical analysis of the percentage of cells exhibiting specific staining (GM1-positive cells) is shown. (C) Knockdown of Neu-3 mRNA levels. Results are shown as the mean ± SD of three independent experiments. (D) PDGFR immunogold labeling in PAG-N–expressing cells that were treated with 10 μM DANA. A representative example (left) and the statistical analysis (right) are shown. Arrows highlight PDGFR-positive caveolae. (E) PAG-N mitogenic inhibition is reversed by Neu-3 inactivation. NIH 3T3 cells, infected, or not, with the indicated retroviruses, were transfected with the indicated siRNAs (right), serum starved for 30 h with or without 10 μM DANA (left), and then stimulated or not with PDGF in the presence of BrdU. Cells were then fixed and processed for immunofluorescence. The percentage of transfected cells that incorporated BrdU was calculated as described in Material and methods. Results are expressed as the mean ± SD of three to five independent experiments. *, P < 0.05 (using Student's t test).
PAG antimitogenic function is also mediated by a Neu3–GM1 pathway
We next asked whether full-length PAG could also inhibit PDGF mitogenic response through a similar mechanism. First, we found a similar GM1 accumulation at the cell surface of cells expressing PAG, which was abolished by the Neu-3 cell surface inhibitor DANA (Fig. 8 A). PAG also decreased PDGFR localization in caveolae, which was fully restored by lowering GM1 cell surface level via Neu-3 inhibition with DANA (Fig. 8 B). Most importantly, DANA restored PDGF mitogenic response in cells expressing PAG (Fig. 8 C). Thus, we concluded that PAG mitogenic inhibition is ensured by its N terminus and implicates a Neu-3–GM1 mechanism.
Figure 8.

PAG antimitogenic function is also mediated by a Neu3–GM1 pathway. (A) PAG induces cell surface GM1 accumulation mediated by Neu-3. A representative example of cell surface GM1 expression in NIH 3T3 cells transfected with a full-length PAG construct (GFP) and treated or not with DANA as indicated. (B) PAG displaces PDGFR from caveolae. Shown are a representative example (left) and the statistical analysis (right) of PDGFR staining by immunogold with the anti-PRC antibody in caveolae of cells infected with a PAG-expressing retrovirus and treated, or not, with DANA. Arrows highlight PDGFR-positive caveolae. (C) PAG mitogenic inhibition requires Neu-3 activity. (C, left) NIH 3T3 cells infected with the indicated retroviruses were treated, or not, with 10 μM DANA and then stimulated, or not, with PDGF in the presence of BrdU. Cells were then fixed and processed for immunofluorescence. The percentage of transfected cells that incorporated BrdU was calculated as described in Material and methods. Results are expressed as the mean ± SD of three independent experiments. (C, right) The level of expressed PAG. Molecular mass is indicated in kilodaltons.
Endogenous PAG regulates PDGF mitogenic activity via a GM1-dependent mechanism
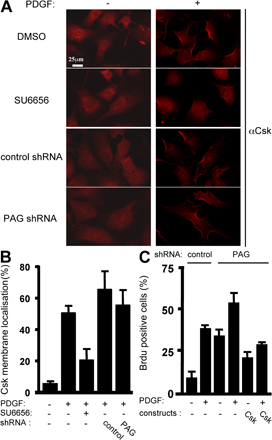
Finally, we asked whether endogenous PAG had a similar role. To this end, mouse NIH 3T3 cells were stably transfected with a vector expressing a small hairpin RNA that reduced PAG level in CEF by 80%, as compared with mock-transfected cells (Fig. 9 A). We observed that GM1 accumulation at the cell surface was significantly reduced in PAG knocked-down cells (Fig. 9 B). Remarkably, PDGFR caveolar distribution was significantly increased in cells with reduced PAG, as revealed by the appearance of multi-labeled PDGFR beads in caveolae, whereas caveolin was not (Fig. 9 C). In accordance with our hypothesis, exogenous GM1 largely restored PDGFR monolabeling in caveolae. We then addressed the effect of endogenous PAG on mitogenesis. PAG down-regulation induced an increase in BrdU incorporation both in the presence and absence of growth factors (Fig. 9 D), indicating that endogenous PAG negatively regulates mitogenesis. Interestingly, increase in BrdU incorporation was abrogated by exogenous GM1, which is in agreement with the hypothesis of a PAG–GM1 signaling pathway regulating mitogenesis. We then analyzed the molecular determinants involved in endogenous PAG biological activity by introducing human PAG mutants that were not targeted by the mouse shRNA sequence (Fig. 9 E). Human PAG restored DNA synthesis to the level observed in control cells, indicating that the biological effect was not caused by a side target of the shRNA. Similar results were obtained with PAG Y317F and PAG-N, which do not bind to Csk, unlike PAG-C. These data are consistent with the idea that the N-terminal sequence is responsible for the mitogenic inhibitory activity of endogenous PAG. We also found that increased DNA synthesis after PAG down-regulation was inhibited by AG-1296, a PDGFR inhibitor (Kovalenko et al., 1997), but not by Iressa, a specific inhibitor of EGF receptors (Fig. 9 F; Herbst et al., 2004). SFK and caveolae were also implicated in this cellular response, as revealed by the inhibitory effect of the Src inhibitor SU6656 and the Cav3-DGV mutant, respectively. Accordingly, basal PDGFR and ERK activities were enhanced in these cells (Fig. 9 G). Hence, an increased PDGFR signaling mediated by its membrane redistribution may be responsible for the promotion of DNA synthesis in cells with reduced PAG. Finally, we also wished to confirm the Csk-independent nature of endogenous PAG activity. A 10-min PDGF stimulation induced Csk membrane recruitment in 50% of cells, as revealed by Csk immunostaining at the plasma membrane (Fig. S3, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200705102/DC1). PAG silencing had no effect on Csk relocalization and mitogenic activity (Fig. S3 C), as expected. Nevertheless, the specific SFK inhibitor SU6656 strongly reduced Csk relocalization, confirming the existence of a distinct SFK substrate for Csk membrane translocation and activity.
Figure 9.

Endogenous PAG regulates PDGF mitogenic activity by a GM1-dependent mechanism. (A) PAG silencing in NIH 3T3 cells. Levels of PAG and caveolin in CEF from cells expressing the indicated shRNAs. (B) PAG knockdown reduces GM1 cell surface level. A representative example of cell surface GM1 level in NIH 3T3 cells expressing the indicated shRNAs is shown. (C, left) PAG knockdown increases PDGFR caveolar distribution, but this effect is reversed by GM1 addition. PDGFR and caveolin immunogold labeling in cells expressing PAG shRNA and treated, or not, with GM1. Arrows highlight caveolin or PDGFR-labeled caveolae. (C, right) statistical analysis of the percentage of caveolae that were multilabeled with the anti-PDGFR antibody (αPRC) in the indicated cells treated, or not, with GM1. (D) PAG knockdown increases DNA synthesis that is dependent on GM1 and PAG-N. NIH 3T3 cells infected with the indicated retroviruses were treated with 10 μM GM1 or vehicle during starvation and then stimulated, or not, with PDGF in the presence of BrdU. (E) PAG negative regulation of DNA synthesis induction requires PAG-N. NIH 3T3 cells, infected with the indicated retrovirus, were transfected with different human PAG coding vectors. (F) PAG negative regulation of DNA synthesis requires PDGFR. NIH 3T3 cells, infected with the indicated viruses, were transfected with Cav3-DGV or treated with the indicated reagents and then incubated with BrdU in the absence of PDGF for 18 h. Cells were then fixed and processed for immunofluorescence. The percentage of transfected cells that incorporated BrdU was calculated as described in Material and methods. Results are expressed as the mean ± SD of three to five independent experiments. (G) PDGFR and Erk activation in cells expressing the indicated shRNAs. Levels of phosphorylated and total PDGFR and Erk were assessed by Western blotting with the indicated antibodies. Molecular masses are indicated in kilodaltons.*, P < 0.05; **, P < 0.01 (using Student's t test).
Discussion
PAG was originally identified as a Csk-binding protein and a phosphoprotein associated with glycosphingolipid-enriched microdomains (i.e., membrane rafts). So far, PAG functions were mostly related to Csk activation (Horejsi et al., 2004). Recently, additional PAG-interacting proteins with signaling functions have been reported, including the E3 ligase SOCS1, which is important for SFK degradation (Ingley et al., 2006), the negative Ras regulator RasGAP (Smida et al., 2007), and SFK as well (Oneyama et al., 2008; Solheim et al., 2008; Tauzin et al., 2008). In this paper, we have uncovered a novel property of this transmembrane protein, unrelated to its association with Csk or any other signaling protein yet implicating SFK regulation. By controlling GM1 levels at the plasma membrane, PAG regulates PDGFR partitioning in caveolae and its association with SFK for mitogenic signaling. Recent data have shown that PAG can sequester and affect signaling of oncogenic SFK in membrane domains and this implicates a phosphotyrosine-dependent mechanism (Oneyama et al., 2008). This may not occur in PDGF-stimulated fibroblasts, probably because of the inefficiency of endogenous SFK to phosphorylate PAG in these conditions.
How GM1 regulates PDGFR levels in caveolae is not clearly established, but the simplest model would implicate a competition mechanism. In addition, one might speculate that there is repulsion between the GM1 carbohydrate moiety and an epitope in the extracellular domain of PDGFR, as was recently found for the ganglioside GM3 and the insulin receptor (Kabayama et al., 2007). Thus, the balance between the two constituents would regulate PDGFR concentration in these membrane domains. Additionally, GM1 may also regulate PDGFR catalytic activity, as suggested by Mistuda et al. (2002), and by the increased PDGFR activity observed in PAG-depleted cells with reduced GM1 level. Therefore, by regulating GM1 level at the plasma membrane, PAG may have an impact on PDGFR mitogenic signaling via both receptor membrane partitioning and catalytic activity (Fig. 10).
Figure 10.

A model for how PAG regulates PDGFR-induced DNA synthesis. Under normal conditions, PDGF-induced receptor activation triggers its association with SFK in caveolae, leading to Src mitogenic signaling, which is required for induction of DNA synthesis. An increase in PAG level induces Neu-3 sialidase activity that leads to GM1 accumulation and PDGFR exclusion from caveolae. This, in turn, prevents Src mitogenic signaling and induction of DNA synthesis. Conversely, a reduction of PAG level lowers Neu-3 sialidase activity and GM1 cell surface accumulation, allowing an increase in PDGFR caveolar localization. This induces PDGFR activation and Src mitogenic signaling even in the absence of PDGF.
One important issue raised by these observations is to understand how PAG regulates GM1 accumulation. A first hypothesis implies that, by associating with GM1, it allows lipid delivery to the cell surface, as was recently described for syntaxin-6, a t-SNARE (target membrane–soluble N-ethylmaleimide attachment protein receptor) involved in membrane fusion events along the secretory pathway (Choudhury et al., 2006). Indeed, syntaxin-6 controls the level of GM1 in caveolae and we observed that its overexpression also inhibits PDGF-induced Src mitogenic signaling (unpublished data). However, syntaxin-6 functions were not affected by PAG down-regulation (unpublished data), suggesting that these two proteins regulate mitogenesis and GM1 accumulation through separate mechanisms. A second hypothesis implies that PAG has an impact on GM1 biosynthesis. This notion is supported by the important role of the membrane domain–localized sialidase Neu-3 (Wang et al., 2002; Papini et al., 2004) on PAG activity. Thus, PAG may have an impact on Neu-3 by modulating its expression, localization, enzymatic activity, and/or substrate availability. We also demonstrated that PAG functions require palmitoylation of the protein for its localization in membrane domains (Uittenbogaard and Smart, 2000). Surprisingly, these properties were not observed in any other member of the TRAP family we tested (i.e., LAT, SIT, TRIM, and NTAL; unpublished data), including the ones that also harbor palmitoylation sites (LAT and NTAL). Therefore, additional mechanisms must be required for full PAG activity. Accordingly, a detailed mutagenesis analysis identified Phe 25, 32, and 35 as important determinants of PAG-N functions, which define a consensus sequence for binding to caveolin-1 (XΦXXXXΦXXΦ, where Φ corresponds to Phe, Tyr, or Trp, and X to any other amino acid; Couet et al., 1997; Jiang et al., 2006). However, the endogenous proteins were found to interact weakly in cells (unpublished data), suggesting that this sequence might have additional roles.
Moreover, the present findings raise the issue of the nature of the membrane-anchored Csk binders involved in the regulation of Src mitogenic signaling. Zhao et al. (2006) clearly implicated the adaptor protein Dok-1. Using Src phosphorylation on Tyr450, Dok-1 recruits Csk to the cell periphery for regulation of Src-induced c-myc expression and cell cycle progression. However, they did not investigate whether Dok-1 regulates signaling in caveolae. Additional Csk-binding proteins could be involved in the regulation of SFK mitogenic signaling. One obvious candidate may be caveolin-1 itself, when it is phosphorylated on Tyr14 by SFK (Lee et al., 2000).
Finally, our study raises important questions about PAG signaling specificity and PAG regulation. Our data show that PAG action is restricted to specific membrane domains. Therefore, we could also expect that PAG impact on receptor signaling regulated by these membrane domains. For instance, NGF receptors have been reported to require GM1 for neurite outgrowth (Mutoh et al., 1995), and PAG, which is highly expressed in the brain (Kawabuchi et al., 2000), may also contribute to the ability of NGF to induce neurites. Additional work is certainly needed to define the regulation of this novel PAG function. Regulation of protein expression could be part of the mechanism, as suggested by our gene silencing experiments. Another level of regulation may implicate deacylation on Cys39 and Cys42 (Greaves and Chamberlain, 2007). By removing palmitoyl residues, PAG will be excluded from membrane domains and will not have an impact on GM1 at the cell surface. Although not predicted, phosphorylation at the N terminus could also influence PAG antimitogenic function. Clearly, further investigations are required to unravel the molecular mechanism by which this GM1-mediated PAG function is regulated.
Materials and methods
DNA constructs, antibodies, and reagents
Constructs expressing PAG and PAGY317F have been described in Brdicka et al., (2000). PAG-N and PAG-C correspond to human PAG [1–97] and PAG [98–433] sequences that have been subcloned in frame into pEGFP-N2 and pEGFP-C2 vectors, respectively. PAG-N CC/AA (PAG-N C39C42/A39A42) and PAG-N 3F/3A (PAG-N F25F32F35/A25A32A35) mutants were generated using PAG-N as template and the Quick Change Site Directed mutagenesis kit (Stratagene) and subcloned into the pBabe retroviral vector. Constructs expressing Myc, Fos, E1B, and SrcY527F have been described in Furstoss et al., (2002) and Franco et al. (2006), Rac V12 in Boureux et al. (2005), Csk in Bénistant et al. (2001), and Cav3 DGV and Cav1-GFP in Veracini et al. (2006). Construct encoding RasL61 was obtained from P. Fort (Centre de Recherche en Biochimie Macromoléculare, Montpellier, France), PDGFR was obtained from A. Kazlauskas (Harvard Medical School, Boston, MA), and syntaxin6 was obtained from S.A. Tooze (London Research Institute, London, United Kingdom). The shRNA specific for mouse PAG (GTCCAAGTCCACTTCTGCC) was cloned into pSUPER vector. A pSuper construct expressing a siRNA against the unrelated human protein interferon receptor 1 was used as a control (gift from A. Blangy and G. Uzé, Centre de Recherche en Biochimie Macromoléculare, Montpellier, France). siRNAs specific for mouse Caveolin-1 (AAGCAAGTGTATGACGCGCAC), mouse Neu-3 (AAGGATTAACCTAGGCATCTA), and corresponding scramble siRNAs were obtained from QIAGEN. Neu-3 mRNA levels were measured by real-time quantitative PCR (Boureux et al., 2005) using the following primers: forward, 5′-GCCCGTCTCTGCTTCCTTT-3′; and reverse, 5′-CTTCCCGAGTGTAGCTGGATG-3′. Anti-SFK (cst1), -PDGFRβ (PRC), and -myc epitope (9E10) antibodies were described in Boureux et al. (2005) and Franco et al. (2006). The anti-pY416Src antibody was obtained from Invitrogen, anti–pan-caveolin from BD Biosciences, anti-ERKs (SC-94) and -Csk from Santa Cruz Biotechnology, Inc., anti-pY705Stat3 from Cell Signaling Technology, anti-Stat3 from Millipore, anti–phospho-ERKs from New England Biolabs, Inc., and anti-BrdU from Becton Dickinson. Anti-Flag antibody and filipin were obtained from Sigma-Aldrich, anti-4G10 and anti-tubulin were obtained from P. Mangeat and N. Morin, respectively (Centre de Recherche en Biochimie Macromoléculare, Montpellier, France). Antibodies coupled to Fluorescein isothiocyanate and Texas Red isothiocyanate were obtained from Sigma-Aldrich, Rhodamine phalloidin and cholera toxin subunit B (CTxB)–Alexa Fluor 594 were obtained from Invitrogen, BrdU from Boehringer Ingelheim, PDGF-BB from Abcys, and EGF and DANA from Sigma-Aldrich. SU6656 and AG-1296 were obtained from EMD, GM1 from Avanti Polar Lipids, and Iressa was a gift of F. Cruzalgui (Servier Laboratories, Paris, France). Protein A gold conjugates were obtained from the Cell Microscopy Center at Utrecht Medical School (Utrecht, The Netherlands).
Cell culture and transfections
NIH 3T3, p53+/+ MEF, p53−/− MEF, and HEK 293 cells were cultured, transfected, and infected as described in Franco et al. (2006) and Veracini et al. (2006). HEK 293 cells stably expressing PDGFRβ were obtained by infection of retroviruses expressing the human receptor. Transient transfections were performed with Lipofectamine Plus Reagent (Invitrogen) or jetPEI (Polyplus-transfection) according to the manufacturer's instructions. For biochemical analysis, cells were transfected 40 h before lysis. For DNA synthesis experiments, cells seeded onto glass coverslips were serum starved in medium containing 0.25% serum supplemented with insulin-transferrin-selenium buffer (Invitrogen) for 30 h and then stimulated, or not, with the indicated mitogens.
BrdU incorporation and immunofluorescence
DNA synthesis in quiescent fibroblasts seeded onto glass coverslips was monitored by adding 0.1 mM BrdU to the medium for 18 h. For drug treatments, cells were incubated with the indicated drug (or vehicle) 1 h before stimulation, except for DANA which was added during serum starvation. For GM1 experiments, cells were treated with 10 μM GM1 for 30 min at 4°C before stimulation. Cells were then fixed and analyzed for BrdU incorporation and dorsal ruffle formation as in Veracini et al. (2006). The percentage of transfected cells that incorporated BrdU was calculated by the following formula: % of BrdU-positive cells = [number of BrdU-positive transfected cells]/[number of transfected cells] × 100. The percentage of transfected cells that formed dorsal ruffles was calculated by the following formula: % of cells with dorsal ruffle = [number of ruffles-positive transfected cells]/[number of transfected cells] × 100. For each coverslip ∼150–200 cells were analyzed. Cell surface GM1 and cholesterol levels were visualized on PFA-fixed cells by incubation with 0.1 μg/ml CTxB–Alexa 594 and 5 μg/ml filipin, respectively, in PBS–10% FCS. Fluorescent labeling was observed with a motorized microscope (DMRA; Leica) using a 40 or 63× plasmapochromatic oil immersion objective (1.32 NA) in Mowiol. Acquisition was performed with a camera (coolSNAP HQ; Photometrics) driven by the MetaMorph 6.2r4 acquisition software (MDS Analytical Technologies).
Transmission electron microscopy (TEM)
TEM was performed on thin sections of epon-embedded cells. Monolayers of cells grown in 100-mm dishes were fixed with 2.5% glutaraldehyde in 0.1 M Na cacodylate buffer, pH 7.4, postfixed in 1% OsO4 in the same buffer, dehydrated through graded series of ethanol, removed from the dish by propylene oxide, and pelleted, followed by epon embedding. Thin sections were stained with uranyl acetate and lead citrate and observed with a transmission electron microscope (1200 EX; Jeol). TEM was performed on ultrathin cryosections and after immunogold labeling. Cells were fixed with 4% PFA in 0.1 M phosphate buffer, pH 7.2, for 48 h at 4°C. After fixation, cells were scraped off, let to sediment for 30 min at RT, microfuged for 1 min, and then embedded in 5% gelatin in PBS for 30 min at 37°C. After cooling on ice and trimming, cell pellets were infused overnight in 2.3 M sucrose, mounted on aluminum stubs, and frozen in liquid nitrogen. Ultrathin sections were cut with microtomes (UCT and EMFCS; Leica) and a Diamond knife, collected with 2.3 M sucrose, and mounted on Parlodion-coated copper or nickel grids. Sections were incubated with rabbit polyclonal anti-PDGFR and -caveolin antibodies diluted 1:200 in PBS–10% FCS for 30 min, and then with protein A gold conjugates diluted 1:70 in PBS–10% FCS for 30 min to 1 h. Sections were then embedded in methyl cellulose uranyl acetate before drying and observation with a transmission electron microscope (EM10 CR; Carl Zeiss, Inc.).
Biochemistry
Cell lysis, immunoprecipitation, Western blotting, and in vitro kinase assays were performed as in Veracini et al. (2006) and results were quantified using the ImageQuant TL software (Molecular Dynamics). For fractionation experiments, 108 cells were incubated with vehicle or 25 ng/ml PDGF for 10 min at 37°C, rinsed with PBS, scraped in 1 mM of ice-cold PBS-containing vanadate, and pelleted. Pellets were then suspended in ice-cold 2× lysis buffer containing 1% Triton X-100, 10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 75 U/ml aprotinin, and 1 mM vanadate for 20 min. Cell suspensions were homogenized in a tight-fitting Dounce homogenizer with 10 strokes and centrifuged for 5 min at 1,300 g to remove nuclei and large cellular debris. Supernatants were fractionated through a 5–42.5% wt/vol sucrose gradient in 4-ml tubes. Nine fractions were collected from the top to the bottom of the gradient. In some experiments, fractions two through four were pooled and diluted fivefold in 25 mM MOPS, pH 6.5, and 150 mM NaCl containing 1% Triton X-100 and centrifuged at 50,000 g at 4°C for 30 min. Pellets were then suspended in dissociation buffer containing 10 mM Tris, pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.5 mM EGTA, 1 mM vanadate, 50 mM NaF, 75 U/ml aprotinin, 1% NP 40, and 60 mM octylglucoside and incubated for 1 h on ice before SFK immunoprecipitation with the anti-cst1 antibody.
Online supplemental material
Fig. S1 shows the effect of a siRNA specific for caveolin-1 on the caveolin-1 level and on the mitogenic response induced by PDGF in NIH 3T3 cells. Fig. S2 illustrates the effect of PAG-N on caveolae localization as shown by caveolin-1 immunostaining and the percentage of caveolae connected at the cell surface. Fig. S3 shows the effect of PAG on PDGF-induced Csk membrane recruitment as well as the effect of Csk overexpression on mitogenesis of cells with reduced PAG. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200705102/DC1.
Supplementary Material
Acknowledgments
We thank Dr. A. Blangy (Centre de Recherche en Biochimie Macromoléculare, Montpellier), F. Cruzalegui (Servier Laboratories, Paris, France), Dr. A. Kazlauskas (Harvard Medical School, Boston USA), Dr. S. Tooze (Cancer Research UK, London Research Institute, London, UK), and Dr. G. Uzé (Centre National de la Recherche Scientifique UMR5124, Montpellier, France) for various reagents and Franck Godiard and Samira Haddouche (SCME-UM2) and members of the Montpellier RIO imaging platform for their technical assistance.
This work was supported by the Centre National de la Recherche Scientifique, Association de Recherche contre le Cancer, Association for International Cancer Research, Institut National du Cancer, and Ministère Français des Affaires Etrangères. L. Veracini was supported by the Ligue Nationale Contre le Cancer and Association de Recherche contre le Cance charities and C. Benistant and S. Roche by Institut National de la Santé et de la Recherche Médicale.
Abbreviations used in this paper: CEF, caveolae-enriched fractions; CTxB, cholera toxin subunit B; DANA, 2-deoxy-2,3-didehydro-N-acetylneuraminic acid; MEF, mouse embryonic fibroblast; PDGFR, PDGF receptor; SFK, Src family protein tyrosine kinases; TEM, transmission electron microscopy.
References
- Baumeister, U., R. Funke, K. Ebnet, H. Vorschmitt, S. Koch, and D. Vestweber. 2005. Association of Csk to VE-cadherin and inhibition of cell proliferation. EMBO J. 24:1686–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénistant, C., J. Bourgaux, H. Chapuis, N. Mottet, S. Roche, and J. Bali. 2001. The C-terminal Src kinase is a tumour antigen in human carcinoma. Cancer Res. 61:1415–1420. [PubMed] [Google Scholar]
- Boggon, T.J., and M.J. Eck. 2004. Structure and regulation of Src family kinases. Oncogene. 23:7918–7927. [DOI] [PubMed] [Google Scholar]
- Boureux, A., O. Furstoss, V. Simon, and S. Roche. 2005. c-Abl tyrosine kinase regulates a Rac/JNK and a Rac/Nox pathway for DNA synthesis and c-myc expression induced by growth factors. J. Cell Sci. 118:3717–3726. [DOI] [PubMed] [Google Scholar]
- Brdicka, T., D. Pavlistova, A. Leo, E. Bruyns, V. Korinek, P. Angelisova, J. Scherer, A. Shevchenko, I. Hilgert, J. Cerny, et al. 2000. Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J. Exp. Med. 191:1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromann, P.A., H. Korkaya, and S.A. Courtneidge. 2004. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 23:7957–7968. [DOI] [PubMed] [Google Scholar]
- Broome, M.A., and S.A. Courtneidge. 2000. No requirement for src family kinases for PDGF signaling in fibroblasts expressing SV40 large T antigen. Oncogene. 19:2867–2869. [DOI] [PubMed] [Google Scholar]
- Choudhury, A., D.L. Marks, K.M. Proctor, G.W. Gould, and R.E. Pagano. 2006. Regulation of caveolar endocytosis by syntaxin 6-dependent delivery of membrane components to the cell surface. Nat. Cell Biol. 8:317–328. [DOI] [PubMed] [Google Scholar]
- Couet, J., S. Li, T. Okamoto, T. Ikezu, and M.P. Lisanti. 1997. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J. Biol. Chem. 272:6525–6533. [DOI] [PubMed] [Google Scholar]
- Da Silva, J.S., T. Hasegawa, T. Miyagi, C.G. Dotti, and J. Abad-Rodriguez. 2005. Asymmetric membrane ganglioside sialidase activity specifies axonal fate. Nat. Neurosci. 8:606–615. [DOI] [PubMed] [Google Scholar]
- Frame, M.C. 2004. Newest findings on the oldest oncogene; how activated src does it. J. Cell Sci. 117:989–998. [DOI] [PubMed] [Google Scholar]
- Franco, M., O. Furstoss, V. Simon, C. Benistant, W.J. Hong, and S. Roche. 2006. The adaptor protein Tom1L1 is a negative regulator of Src mitogenic signaling induced by growth factors. Mol. Biol. Cell. 26:1932–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furstoss, O., K. Dorey, V. Simon, D. Barilla, G. Superti-Furga, and S. Roche. 2002. c-Abl is an effector of Src for growth factor-induced c-myc expression and DNA synthesis. EMBO J. 21:514–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbiati, F., D. Volonte, J.A. Engelman, G. Watanabe, R. Burk, R.G. Pestell, and M.P. Lisanti. 1998. Targeted downregulation of caveolin-1 is sufficient to drive cell transformation and hyperactivate the p42/44 MAP kinase cascade. EMBO J. 17:6633–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves, J., and L.H. Chamberlain. 2007. Palmitoylation-dependent protein sorting. J. Cell Biol. 176:249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst, R.S., M. Fukuoka, and J. Baselga. 2004. Gefitinib–a novel targeted approach to treating cancer. Nat. Rev. Cancer. 4:956–965. [DOI] [PubMed] [Google Scholar]
- Horejsi, V. 2005. Lipid rafts and their roles in T-cell activation. Microbes Infect. 7:310–316. [DOI] [PubMed] [Google Scholar]
- Horejsi, V., W. Zhang, and B. Schraven. 2004. Transmembrane adaptor proteins: organizers of immunoreceptor signalling. Nat. Rev. Immunol. 4:603–616. [DOI] [PubMed] [Google Scholar]
- Imamoto, A., and P. Soriano. 1993. Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell. 73:1117–1124. [DOI] [PubMed] [Google Scholar]
- Ingley, E., J.R. Schneider, C.J. Payne, D.J. McCarthy, K.W. Harder, M.L. Hibbs, and S.P. Klinken. 2006. Csk-binding protein mediates sequential enzymatic down-regulation and degradation of Lyn in erythropoietin-stimulated cells. J. Biol. Chem. 281:31920–31929. [DOI] [PubMed] [Google Scholar]
- Janes, P.W., S.C. Ley, and A.I. Magee. 1999. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J. Cell Biol. 147:447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, L.Q., X. Feng, W. Zhou, P.G. Knyazev, A. Ullrich, and Z. Chen. 2006. Csk-binding protein (Cbp) negatively regulates epidermal growth factor-induced cell transformation by controlling Src activation. Oncogene. 25:5495–5506. [DOI] [PubMed] [Google Scholar]
- Kabayama, K., T. Sato, K. Saito, N. Loberto, A. Prinetti, S. Sonnino, M. Kinjo, Y. Igarashi, and J. Inokuchi. 2007. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proc. Natl. Acad. Sci. USA. 104:13678–13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabuchi, M., Y. Satomi, T. Takao, Y. Shimonishi, S. Nada, K. Nagai, A. Tarakhovsky, and M. Okada. 2000. Transmembrane phosphoprotein Cbp regulates the activities of Src-family tyrosine kinases. Nature. 404:999–1003. [DOI] [PubMed] [Google Scholar]
- Kovalenko, M., L. Ronnstrand, C.H. Heldin, M. Loubtchenkov, A. Gazit, A. Levitzki, and F.D. Bohmer. 1997. Phosphorylation site-specific inhibition of platelet-derived growth factor beta-receptor autophosphorylation by the receptor blocking tyrphostin AG1296. Biochemistry. 36:6260–6269. [DOI] [PubMed] [Google Scholar]
- Lee, H., D. Volonte, F. Galbiati, P. Iyengar, D.M. Lublin, D.B. Bregman, M.T. Wilson, R. Campos-Gonzalez, B. Bouzahzah, R.G. Pestell, et al. 2000. Constitutive and growth factor-regulated phosphorylation of caveolin-1 occurs at the same site (Tyr-14) in vivo: identification of a c-Src/Cav-1/Grb7 signaling cassette. Mol. Endocrinol. 14:1750–1775. [DOI] [PubMed] [Google Scholar]
- Li, S., J. Couet, and M.P. Lisanti. 1996. Src tyrosine kinases, Galpha subunits, and H-Ras share a common membrane-anchored scaffolding protein, caveolin. Caveolin binding negatively regulates the auto-activation of Src tyrosine kinases. J. Biol. Chem. 271:29182–29190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, G.S. 2001. The hunting of the Src. Nat. Rev. Mol. Cell Biol. 2:467–475. [DOI] [PubMed] [Google Scholar]
- Mitsuda, T., K. Furukawa, S. Fukumoto, H. Miyazaki, T. Urano, and K. Furukawa. 2002. Overexpression of ganglioside GM1 results in the dispersion of platelet-derived growth factor receptor from glycolipid-enriched microdomains and in the suppression of cell growth signals. J. Biol. Chem. 277:11239–11246. [DOI] [PubMed] [Google Scholar]
- Mutoh, T., A. Tokuda, T. Miyadai, M. Hamaguchi, and N. Fujiki. 1995. Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc. Natl. Acad. Sci. USA. 92:5087–5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nada, S., T. Yagi, H. Takeda, T. Tokunaga, H. Nakagawa, Y. Ikawa, M. Okada, and S. Aizawa. 1993. Constitutive activation of Src family kinases in mouse embryos that lack Csk. Cell. 73:1125–1135. [DOI] [PubMed] [Google Scholar]
- Niu, G., K.L. Wright, Y. Ma, G.M. Wright, M. Huang, R. Irby, J. Briggs, J. Karras, W.D. Cress, D. Pardoll, et al. 2005. Role of Stat3 in regulating p53 expression and function. Mol. Cell. Biol. 25:7432–7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oneyama, C., T. Hikita, K. Enya, M.W. Dobenecker, K. Saito, S. Nada, A. Tarakhovsky, and M. Okada. 2008. The lipid raft-anchored adaptor protein cbp controls the oncogenic potential of c-Src. Mol. Cell. 30:426–436. [DOI] [PubMed] [Google Scholar]
- Papini, N., L. Anastasia, C. Tringali, G. Croci, R. Bresciani, K. Yamaguchi, T. Miyagi, A. Preti, A. Prinetti, S. Prioni, et al. 2004. The plasma membrane-associated sialidase MmNEU3 modifies the ganglioside pattern of adjacent cells supporting its involvement in cell-to-cell interactions. J. Biol. Chem. 279:16989–16995. [DOI] [PubMed] [Google Scholar]
- Pike, L.J. 2005. Growth factor receptors, lipid rafts and caveolae: An evolving story. Biochim. Biophys. Acta. 1746:260–273. [DOI] [PubMed] [Google Scholar]
- Sharma, D.K., J.C. Brown, A. Choudhury, T.E. Peterson, E. Holicky, D.L. Marks, R. Simari, R.G. Parton, and R.E. Pagano. 2004. Selective stimulation of caveolar endocytosis by glycosphingolipids and cholesterol. Mol. Biol. Cell. 15:3114–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima, T., S. Nada, and M. Okada. 2003. Transmembrane phosphoprotein Cbp senses cell adhesion signaling mediated by Src family kinase in lipid rafts. Proc. Natl. Acad. Sci. USA. 100:14897–14902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smida, M., A. Posevitz-Fejfar, V. Horejsi, B. Schraven, and J.A. Lindquist. 2007. A novel negative regulatory function of PAG: blocking Ras activation. Blood. 110:596–615. [DOI] [PubMed] [Google Scholar]
- Solheim, S.A., K.M. Torgersen, K. Tasken, and T. Berge. 2008. Regulation of FynT function by dual domain docking on PAG/Cbp. J. Biol. Chem. 283:2773–2783. [DOI] [PubMed] [Google Scholar]
- Tauzin, S., H. Ding, K. Khatib, I. Ahmad, D. Burdevet, G. van Echten-Deckert, J.A. Lindquist, B. Schraven, N.U. Din, B. Borisch, and D.C. Hoessli. 2008. Oncogenic association of the Cbp/PAG adaptor protein with the Lyn tyrosine kinase in human B-NHL rafts. Blood. 111:2310–2320. [DOI] [PubMed] [Google Scholar]
- Uittenbogaard, A., and E.J. Smart. 2000. Palmitoylation of caveolin-1 is required for cholesterol binding, chaperone complex formation, and rapid transport of cholesterol to caveolae. J. Biol. Chem. 275:25595–25599. [DOI] [PubMed] [Google Scholar]
- Veracini, L., M. Franco, A. Boureux, V. Simon, S. Roche, and C. Benistant. 2006. Two distinct pools of Src family tyrosine kinases regulate PDGF-induced DNA synthesis and actin dorsal ruffles. J. Cell Sci. 119:2921–2934. [DOI] [PubMed] [Google Scholar]
- Wang, Y., K. Yamaguchi, T. Wada, K. Hata, X. Zhao, T. Fujimoto, and T. Miyagi. 2002. A close association of the ganglioside-specific sialidase Neu3 with caveolin in membrane microdomains. J. Biol. Chem. 277:26252–26259. [DOI] [PubMed] [Google Scholar]
- Yew, P.R., and A.J. Berk. 1992. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature. 357:82–85. [DOI] [PubMed] [Google Scholar]
- Zhao, M., J.A. Janas, M. Niki, P.P. Pandolfi, and L. Van Aelst. 2006. Dok-1 independently attenuates Ras/mitogen-activated protein kinase and Src/c-myc pathways to inhibit platelet-derived growth factor-induced mitogenesis. Mol. Cell. Biol. 26:2479–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}