

Summary
Patterning of the terminal regions of the Drosophila embryo relies on the gradient of phosphorylated ERK/MAPK (dpERK), which is controlled by the localized activation of the Torso receptor tyrosine kinase [1–4]. This model is supported by a large amount of data, but the gradient itself has never been quantified. We present the first measurements of the dpERK gradient and establish a new intracellular layer of its regulation. Based on the quantitative analysis of the spatial pattern of dpERK in mutants with different levels of Torso as well as the dynamics of the wild-type dpERK pattern, we propose that the terminal-patterning gradient is controlled by a cascade of diffusion-trapping modules. A ligand-trapping mechanism establishes a sharply localized pattern of the Torso receptor occupancy on the surface of the embryo. Inside the syncytial embryo, nuclei play the role of traps that localize diffusible dpERK. We argue that the length scale of the terminal-patterning gradient is determined mainly by the intracellular module.
Results and Discussion
The terminal regions of the fruit fly embryo are patterned by the gradient ERK/MAPK signaling, which depends on the localized activation of the Torso receptor tyrosine kinase (RTK) and controls the expression of at least two genes with distinct expression thresholds [1]. Torso is activated by a locally processed ligand and is required both for signal transduction and for restricting the extracellular ligand diffusion [2–4]. Genetic experiments established that the pattern of Torso activation is spatially restricted, but the extent of this restriction remained unclear. The pattern of Torso occupancy and activation could be smoothly varying along the surface of the embryo and then simply mapped onto the intracellular gradient of MAPK activation [5]. Alternatively, the pattern of Torso occupancy could be strongly localized at the poles of the embryo, and the smooth dpERK gradient would have to be established by the intracellular diffusion and reaction processes [2–4]. To investigate the mechanisms that establish positional information in the terminal system, we developed assays for quantifying the dpERK gradients in space and time (Figure 1) and across multiple genetic backgrounds (Figure 2A).
Figure 1. Quantifying the dpERK Gradients in Syncytial Drosophila Embryos.
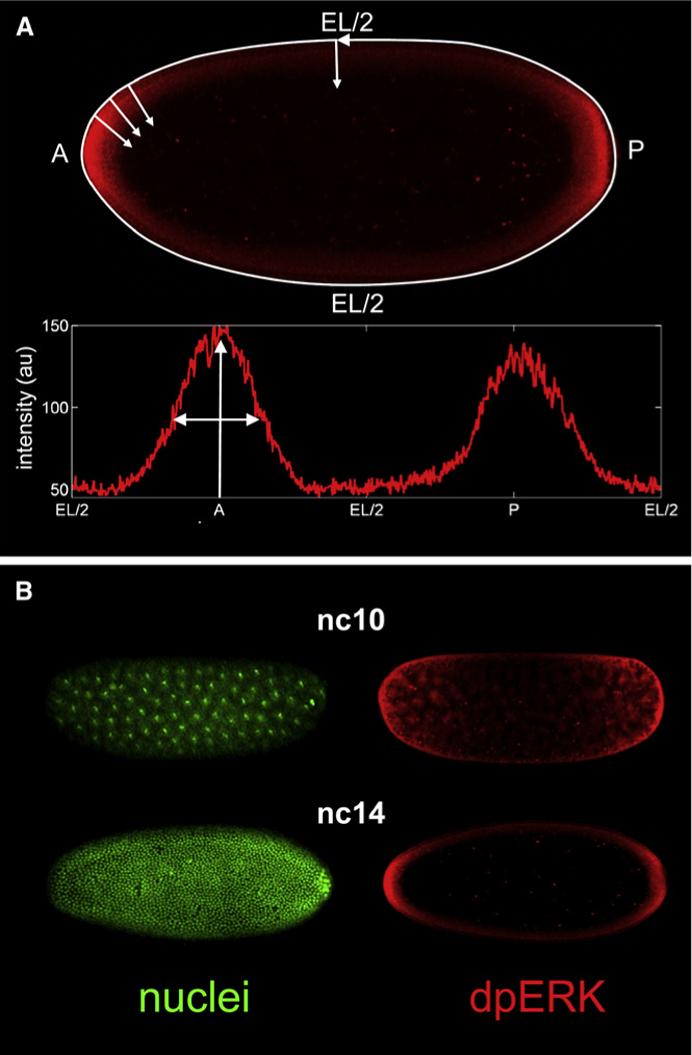
We use an antibody against the double phosphorylated MAPK (dpERK) to visualize the spatial pattern of MAPK phosphorylation along the embryo. The timings of nuclear divisions were characterized in detail, so we use the nuclear density as a marker of time. Thus, a double staining of the dpERK and nuclei provides simultaneous information about the age of the embryo and the spatial pattern of MAPK phosphorylation (B). Details of the assays and gradient quantification procedure are described in Supplemental Data.
(A) Top: spatial pattern of MAPK phosphorylation, detected with the dpERK antibody (red), in a representative cycle 14 embryo. Bottom: quantified dpERK gradient. A, anterior pole; P, posterior pole; EL/2, half of the egg length.
(B) Simultaneous visualization of nuclei (green, top view) and MAPK phosphorylation (red, mid-sagital view) in the representative cycle 10 and 14 embryos (top and bottom, respectively).
Figure 2. The dpERK Gradients in Embryos with Different Levels of Receptor Expression.
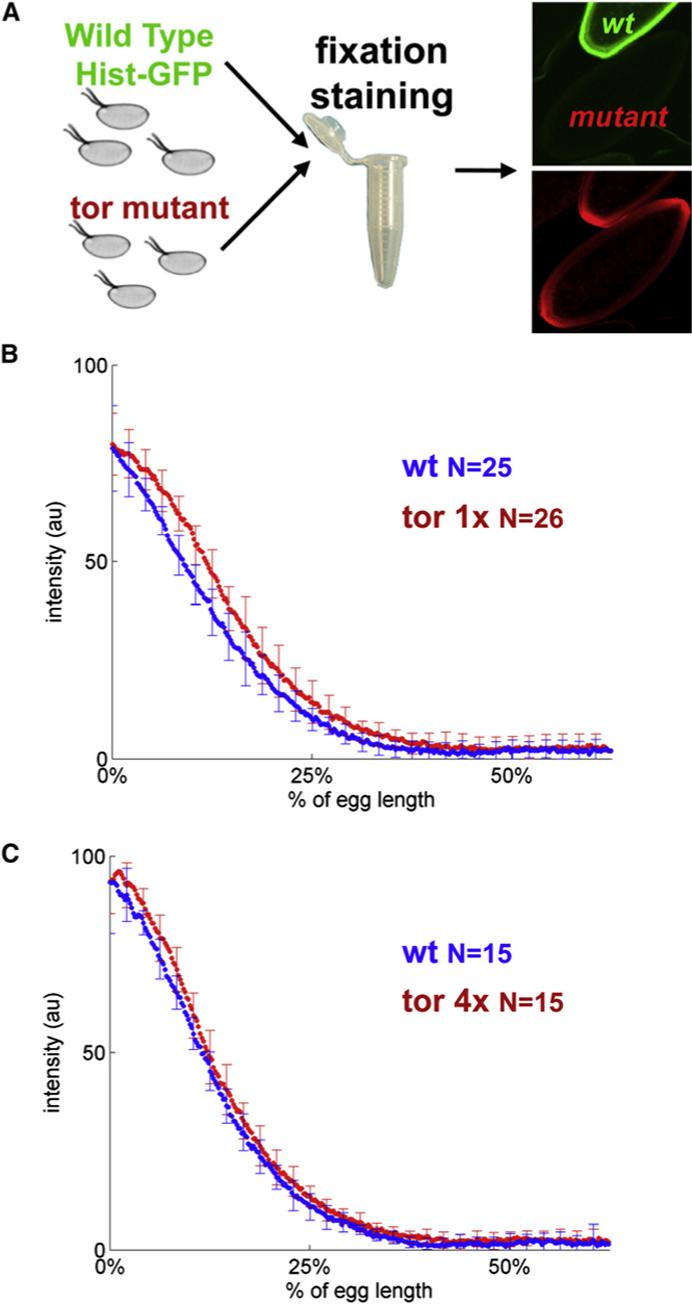
(A) Summary of the experimental procedure for comparing the gradients across genetic backgrounds: wild-type and mutant embryos are taken through the same fixation and staining procedure and imaged on the same microscope slide. In these experiments, embryos from a given mutant background are mixed and processed together with progeny of wild-type flies carrying the histone-GFP fusion transgene.
(B and C) Comparison of the dpERK gradients in embryos with different copy numbers of Torso. Numbers indicate the numbers of embryos quantified in each of the genetic backgrounds. Note that the y axes in (B) and (C) are not normalized to 100. After the normalization procedure, the amplitudes of the average profiles were rescaled to the mean peak value of the raw profiles independently for each genetic background. The differences in the amplitudes between the two wild-type profiles in (B) and (C) are due to variations in fixation and staining conditions; the need for a pairwise comparison of gradients in different backgrounds is described in (A).
To test whether the spatial pattern of Torso occupancy is smoothly varying or strongly localized, we analyzed the dpERK patterns in embryos with altered levels of Torso expression. If the pattern of Torso occupancy is long ranged [2, 6], it should become steeper upon increase in the level of the Torso expression and more shallow in embryos with lowered level of Torso. If, on the other hand, the pattern of Torso occupancy is already strongly localized and is a simple image of the spatial profile of ligand release, it should be insensitive to the level of cell-surface receptors. A similar approach was used to explore patterning gradients in other ligand-receptor systems [7–10].
We found that the dpERK gradients in embryos with one functional copy of Torso and those with two extra copies of Torso (four copies in total) are essentially indistinguishable from the wild-type dpERK pattern [11, 12] (Figures 2B and 2C). This result, based on the direct quantitative and statistical analysis of the dpERK gradients, is consistent with the results of the previous experiments that demonstrated that the transcriptional targets of Torso and terminal structures are unaffected by changes in the level of receptor expression [3, 4]. This strongly supports the hypothesis that the terminal system operates in a regime where the Torso receptors are in excess and the ligand molecules are trapped close to the point of their release, as suggested in [3, 4]. Given the smoothly varying pattern of dpERK, this implies that positional information in the terminal system is determined mainly inside the embryo. In this sense, the terminal gradient is different from the one that patterns the dorsoventral axis of the embryo, which is controlled by the extracellular reaction-diffusion processes [13].
Here we identify the nuclear trapping of dpERK as a mechanism responsible for the intracellular spatial processing of the terminal signal. This conclusion is based on the analysis of the dynamics of the wild-type dpERK gradient (Figure 3). We found that between nuclear cycles 10 and 14, the dpERK levels are amplified at the termini and attenuated in the subterminal regions of the embryo (Figure 3). As shown in the Supplemental Data (available online), the observed dynamics of the dpERK gradient is consistent with a model where dpERK is a diffusible molecule, which is trapped and dephosphorylated by the nuclei (Figure 4A). A uniform increase in the nuclear density would increase the trapping of the dpERK molecules at the poles and prevent their diffusion to the middle of the embryo (Figures 4B and 4C). This is consistent with biochemical and imaging data showing that dpERK rapidly translocates to the nucleus, which can also serve as a compartment of dpERK dephosphorylation [14–16]. In addition, our model makes a testable prediction about the dynamics of the nucleocytoplasmic (N/C) ratio of phosphorylated MAPK.
Figure 3. Dynamics of the Wild-Type dpERK Gradients.
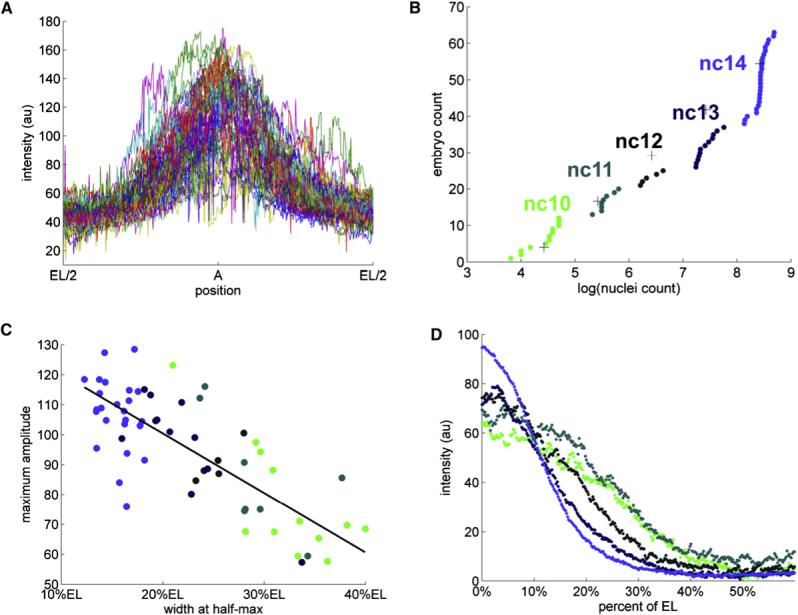
(A) Superposition of the unnormalized anterior dpERK gradients from a collection of 63 embryos. Even though the fixation, staining, and imaging conditions for all of these embryos were identical, the raw intensity profiles varied considerably. The spatially averaged variability was ∼80%, almost an order of magnitude greater than the variability associated with imaging of a single embryo (see Supplemental Data for details).
(B) Cumulative distribution function (c.d.f.) of nuclear densities from the same collection of embryos. Clear breaks in the c.d.f. correspond to the consecutive doublings in the number of syncytial nuclei. Different colors denote five temporal classes of embryos, corresponding to the nuclear cycles 10, 11, 12, 13, and 14. See Supplemental Data for details of clustering of embryos according to their nuclear densities.The absolute value of nuclei count correspond to the number of nuclei in a fixed window size (see Figure S3 for details).
(C) A significant fraction of high variability is explained by the dynamic changes of the dpERK profiles over consecutive nuclear divisions. The heights and widths of the unnormalized gradients are anticorrelated; in addition, both of these parameters are correlated with the nuclear density. In this image, each embryo is represented by a circle, colored according to its nuclear cycle.
(D) A gradient normalization procedure, applied to the embryos that have been grouped according to their age, reveals that the anterior gradients get progressively “taller” and “thinner” as the nuclear density increases. The dpERK profiles from the same temporal classes are strikingly similar (mean spatial variability of 6.9% for nuclear cycle 14). For example, the profiles from cycle 14 embryos are as similar as the ones obtained by repetitive imaging of the same embryo (mean spatial variability of 6%). See Supplemental Data for the details of normalization procedure.
Figure 4. Spatial Regulation of the dpERK Gradient by Syncytial Nuclei.

(A) dpERK is a diffusible molecule that shuttles in and out of the nuclei and can be dephosphorylated in either of these compartments.
(B) As a result of a spatially uniform increase in the nuclear density, dpERK is trapped close to the poles of the embryo, where it is generated by the locally activated Torso receptors. This limits the extent of its diffusion and lowers the dpERK level in the middle of the embryo.
(C) A mathematical model, described in the Supplemental Data, can predict gradient sharpening in response to a uniform change in nuclear density. Ctot(x)is the sum of the nuclear and cytoplasmic levels of dpERK: Ctot(x) = Cc(x) + Cn(x). The amplitude and decay length of the signal are increased and decreased, respectively, by a uniform increase in the nuclear density.
(D) Splitting the dpERK gradients into the nuclear and cytoplasmic parts. Nuclear gradients are extracted from the histone-GFP embryos; see the Supplemental Data for the definition and processing of nuclear and cytoplasmic gradients.
(E) The N/C ratio is constant throughout the embryo and increases between cycles 13 and 14. For details of the statistical analysis, see the Supplemental Data.
(F) Defects in the nuclear density are correlated with the defects in the dpERK pattern in shkl embryos. The two images correspond to the two different focal planes of the same embryo.
(G) The average cycle 14 wild-type gradient was subtracted from the quantified posterior dpERK gradient in the shkl embryo, revealing the correlation between the nuclear density and dpERK.
Let the concentrations of cytoplasmic and nuclear dpERK at a point x in the embryo be denoted by Cc = Cc(x) and Cn = Cn(x), respectively. With the system at steady state, as suggested by the low variability of the dpERK gradients at any given nuclear density (Figure 3D and Figure S10), the rate of nuclear import of dpERK is balanced by the rates of its nuclear export and dephosphorylation. Modeling these processes as first-order reactions with the rate constants k+ (nuclear import), k− (nuclear export), and kn (dephosphorylation in the nucleus), we arrive at the following expression for the nucleocytoplasmic (N/C) ratio of the dpERK:
The rate constants k− and kn reflect the properties of a single nucleus and a single dpERK molecule; therefore, we assume that they do not depend on the number of nuclei in the system. On the other hand, the trapping rate constant k+, which depends both on the nuclear import rate constant and nuclear density, will increase with every nuclear division. As a consequence, the N/C ratio should be an increasing function of the nuclear density. Because the right side of the equation above does not depend on x, the N/C ratio should be constant throughout the embryo.
We tested this prediction by quantifying the nuclear and cytoplasmic levels of dpERK in cycle 13 and 14 embryos (Figures 4D and 4E). Plotting the nuclear and cytoplasmic profiles against each other gives a clear linear relationship, as predicted by the simple formula above (Figure 4E). Furthermore, the nucleocytoplasmic ratio clearly increases between these two nuclear cycles (see Supplemental Data and Figure S6): the N/C ratio is ∼1.4 and ∼2 at nuclear cycles 13 and 14, respectively. These measurements show that the nuclear trapping rate is indeed an increasing function of the nuclear density, as predicted by the model.
The observed N/C ratios show that a significant fraction of total dpERK nuclear. As a consequence, defects in the nuclear density should generate clear defects in the gradient. This can be tested in mutants with “holes” in the nuclear density in blastoderm embryos. For example, in shakleton (shkl) embryos, the migration of nuclei to the poles is delayed and a number of embryos exhibit major disruptions in nuclear density [17]. As predicted by the model, shkl embryos show striking disruptions in the dpERK gradient (Figure 4F). The quantified posterior gradient of this particular mutant embryo shows a clear local correlation with the nuclear distribution, emphasizing the role of the nuclei at this stage (Figure 4G). In early embryos, the gradient is more extended, presumably reflecting the lack of the nuclei at the poles (Figure S12). We found similar defects in the giant nuclei (gnu) mutant embryos, which show a different type of defect in nuclear organization (see Figure S13) [18]. These results support the model in which the syncytial nuclei play an important role in shaping the dpERK gradient.
To summarize, we propose that the dpERK gradient is controlled by a cascade of at least two diffusion-trapping modules. In the extracellular compartment, a ligand-trapping mechanism, identified in previous studies, establishes a sharp gradient of Torso receptor occupancy [2]. A similar mechanism regulates the dpERK gradient inside the embryo, where syncytial nuclei act as traps that localize diffusible dpERK. At this time, we cannot rule out that the observed sharpening of the dpERK gradient can be modulated also by changes in the spatial distribution of the Torso ligand, but currently there are no data in support of this mode of regulation.
The dynamics of the dpERK gradient is qualitatively different from that of the Bicoid gradient, which remains stable during the last five nuclear divisions [19]. We have recently proposed that a stable gradient of Bicoid can be established in the absence of Bicoid degradation, because of the reversible trapping of Bicoid by an exponentially increasing number of nuclei [20]. We attribute the differences between the dynamics of the Bicoid and dpERK gradients to two effects. The first effect is due to the differences in the “chemistries” of the two systems: the morphogen in the terminal system is degraded (MAPK is dephosphorylated), whereas the anterior morphogen is stable (we propose that Bicoid is not degraded on time scale of the gradient formation). The second effect is due to the differences in the initial conditions: by the 10th nuclear division, which is the starting point of the activation of the terminal system, Bicoid gradient is essentially fully established. Thus, a common biophysical framework can describe the Bicoid and dpERK gradients. It remains to be determined whether the nuclear export affects the length scale of the Dorsal gradient, which patterns the dorsoventral axis of the embryo [21, 22].
Supplementary Material
Acknowledgments
We thank Eric Wiechaus, Thomas Gregor, Oliver Grimm, and Bill Bialek for helpful discussions; Ben-Zion Shilo for the dpERK staining protocol; Eric Wieschaus, Willis Li, and Oliver Grimm for fly stocks; and Joe Goodhouse for help imaging. We are grateful to Trudi Schupbach, Lea Goentoro, Kevin Moses, Ze'ev Paroush, Gerardo Jimenez, Jeremy Zartman, Jennifer Lippincott-Schwartz and Gary Struhl for comments on the manuscript. This work was supported by the P50 GM071508 and R01 GM078079 grants from the NIH. This was supported by the Intramural Research Program of the NIH, Center for Information Technology.
Footnotes
Supplemental Data
13 figures and Experimental Procedures are available at http://www.current-biology.com/cgi/content/full/18/12/915/DC1/.
References
- 1.Li WX. Functions and mechanisms of receptor tyrosine kinase Torso signaling: lessons from Drosophila embryonic terminal development. Dev. Dyn. 2005;232:656–672. doi: 10.1002/dvdy.20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casanova J, Struhl G. The torso receptor localizes as well as transduces the spatial signal specifying terminal body pattern in Drosophila. Nature. 1993;362:152–155. doi: 10.1038/362152a0. [DOI] [PubMed] [Google Scholar]
- 3.Casanova J, Struhl G. Localized surface-activity of Torso, a receptor tyrosine kinase, specifies terminal body pattern in Drosophila. Genes Dev. 1989;3:2025–2038. doi: 10.1101/gad.3.12b.2025. [DOI] [PubMed] [Google Scholar]
- 4.Sprenger F, Nusslein-Volhard C. Torso receptor activity is regulated by a diffusible ligand produced at the extracellular terminal regions of the Drosophila egg. Cell. 1992;71:987–1001. doi: 10.1016/0092-8674(92)90394-r. [DOI] [PubMed] [Google Scholar]
- 5.Furriols M, Sprenger F, Casanova J. Variation in the number of activated torso receptors correlates with differential gene expression. Development. 1996;122:2313–2317. doi: 10.1242/dev.122.7.2313. [DOI] [PubMed] [Google Scholar]
- 6.Berezhkovskii AM, Batsilas L, Shvartsman SY. Ligand trapping in epithelial layers and cell cultures. Biophys. Chem. 2004;107:221–227. doi: 10.1016/j.bpc.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Nellen D, Burke R, Struhl G, Basler K. Direct and long-range action of DPP morphogen gradient. Cell. 1996;85:357–368. doi: 10.1016/s0092-8674(00)81114-9. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Struhl G. Dual roles for patched in sequestering and transducing hedgehog. Cell. 1996;87:553–563. doi: 10.1016/s0092-8674(00)81374-4. [DOI] [PubMed] [Google Scholar]
- 9.Lecuit T, Cohen SM. Dpp receptor levels contribute to shaping the Dpp morphogen gradient in the Drosophila wing imaginal disc. Development. 1998;125:4901–4907. doi: 10.1242/dev.125.24.4901. [DOI] [PubMed] [Google Scholar]
- 10.Goentoro LA, Reeves GT, Kowal CP, Martinelli L, Schupbach T, Shvartsman SY. Quantifying the gurken morphogen gradient in Drosophila oogenesis. Dev. Cell. 2006;11:263–272. doi: 10.1016/j.devcel.2006.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cleghon V, Gayko U, Copeland TD, Perkins LA, Perrimon N, Morrison DK. Drosophila terminal structure development is regulated by the compensatory activities of positive and negative phosphotyrosine signaling sites on the Torso RTK. Genes Dev. 1996;10:566–577. doi: 10.1101/gad.10.5.566. [DOI] [PubMed] [Google Scholar]
- 12.Schupbach T, Wieschaus E. Maternal-effect mutations altering the anterior-posterior pattern of the Drosophila embryo. Rouxs Arch. Dev. Biol. 1986;195:302–317. doi: 10.1007/BF00376063. [DOI] [PubMed] [Google Scholar]
- 13.Moussian B, Roth S. Dorsoventral axis formation in the Drosophila embryo—shaping and transducing a morphogen gradient. Curr. Biol. 2005;15:R887–R899. doi: 10.1016/j.cub.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 14.Fujioka A, Terai K, Itoh RE, Aoki K, Nakamura T, Kuroda S, Nishida E, Matsuda M. Dynamics of the Ras/ERK MAPK cascade as monitored by fluorescent probes. J. Biol. Chem. 2006;281:8917–8926. doi: 10.1074/jbc.M509344200. [DOI] [PubMed] [Google Scholar]
- 15.Costa M, Marchi M, Cardarelli F, Roy A, Beltram F, Maffei L, Ratto GM. Dynamic regulation of ERK2 nuclear translocation and mobility in living cells. J. Cell Sci. 2006;119:4952–4963. doi: 10.1242/jcs.03272. [DOI] [PubMed] [Google Scholar]
- 16.Karlsson M, Mandl M, Keyse SM. Spatio-temporal regulation of mitogen-activated protein kinase (MAPK) signalling by protein phosphatases. Biochem. Soc. Trans. 2006;34:842–845. doi: 10.1042/BST0340842. [DOI] [PubMed] [Google Scholar]
- 17.Yohn CB, Pusateri L, Barbosa V, Lehmann R. l(3)malignant brain tumor and three novel genes are required for Drosophila germ-cell formation. Genetics. 2003;165:1889–1900. doi: 10.1093/genetics/165.4.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freeman M, Nüsslein-Volhard C, Glover DM. The dissociation of nuclear and centrosomal division in gnu, a mutation causing giant nuclei in Drosophila. Cell. 1986;46:457–468. doi: 10.1016/0092-8674(86)90666-5. [DOI] [PubMed] [Google Scholar]
- 19.Gregor T, Wieschaus E, McGregor AP, Bialek W, Tank DW. Stability and nuclear dynamics of the bicoid morphogen gradient. Cell. 2007;130:141–153. doi: 10.1016/j.cell.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coppey M, Berezhkovskii AM, Kim Y, Boettiger AN, Shvartsman SY. Modeling the Bicoid gradient: diffusion and reversible nuclear trapping of a stable protein. Dev. Biol. 2007;312:623–630. doi: 10.1016/j.ydbio.2007.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeLotto R, DeLotto Y, Steward R, Lippincott-Schwartz J. Nucleocytoplasmic shuttling mediates the dynamic maintenance of nuclear dorsal levels during Drosophila embryogenesis. Development. 2007;134:4233–4241. doi: 10.1242/dev.010934. [DOI] [PubMed] [Google Scholar]
- 22.Morisato D, Anderson KV. Signaling pathways that establish the dorsal-ventral pattern of the Drosophila embryo. Annu. Rev. Genet. 1995;29:371–399. doi: 10.1146/annurev.ge.29.120195.002103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.