

Abstract
Background
The proinflammatory prostaglandin E2 (PGE2) fluctuates over time in the cerebrospinal fluid of patients with Alzheimer's disease (AD), but the cerebral distribution and expression patterns of microsomal prostaglandin-E synthase (mPGES)–1 have not been compared with those of normal human brains.
Methods
Middle frontal gyrus tissue from AD and age-matched control brains was analyzed by Western blot, immunofluorescence, and immunohistochemistry with mPGES-1–specific antibodies.
Results
Western blotting revealed that mPGES-1 expression was significantly elevated in AD tissue. Furthermore, immunofluorescence of mPGES-1 was observed in neurons, microglia, and endothelial cells of control and AD tissue. Although mPGES-1 was consistently present in astrocytes of control tissue, it was present in only some astrocytes of AD tissue. Immunohistochemical staining suggested that mPGES-1 was elevated in pyramidal neurons of AD tissue when compared with controls.
Conclusions
The results suggest that mPGES-1 is normally expressed constitutively in human neurons, microglia, astrocytes, and endothelial cells but is up-regulated in AD.
Keywords: β-Amyloid, Cyclooxygenase, Middle frontal gyrus, Neuroinflammation, PGES
1. Background
Prostaglandin E2 (PGE2), the so-called proinflammatory end product of the arachidonic acid pathway, has been found to be elevated in cerebrospinal fluid early in Alzheimer's disease (AD) but to be decreased in end-stage AD [1]. Interestingly, nonsteroidal anti-inflammatory drugs (NSAIDs) are able to reduce PGE2 production and have been shown to reduce the risk of AD [2–4]. Nevertheless, in clinical studies, cyclooxygenase (COX)-2 inhibitors and NSAIDs have not been effective in slowing or reversing AD progression or cognitive decline [2,4–7]. NSAIDs might prevent or reduce the amyloid burden and oxidative damage caused by pro-amyloidogenic EP2 stimulation via PGE2 [8] and enhancement of microglial amyloid-β (Aβ) phagocytosis [9]. However, once Aβ has been deposited, microglia produce various proinflammatory cytokines such as interleukin-1β (IL-1β), which is believed to drive additional Aβ and neurofibrillary tangle production [10]. Such a feed-forward progression might be the reason that NSAIDs cannot slow or reverse AD. Also, elevated levels of Aβ can cause cholinergic hypofunction, a characteristic of AD [11]. With diminished vasodilatory effects of acetylcholine, cerebral blood flow can also be reduced; such hypoperfusion has been described in AD [12]. Conversely, other in vitro studies have shown PGE2 or PGE1 to be neuroprotective against Aβ [13,14] and glutamate-induced toxicity [15]. Thus, regulating PGE2 production might either increase or decrease neuron survival, depending on the location of the synthesis enzymes and levels attained.
Three types of terminal prostaglandin-E synthases (PGES), which catalyze the synthesis of PGE2, have been characterized: cytosolic PGES (cPGES), which is glutathione (GSH)-dependent, expressed constitutively in the cytoplasm, and is reported to be coupled preferentially with COX-1; microsomal PGES-2 (mPGES-2), which is cytoplasmic and perinuclear, GSH-independent, constitutively expressed, and functionally coupled with COX-1 and COX-2 [16]; and mPGES-1, which is a GSH-dependent, perinuclear, membrane-bound trimer [17] that is functionally coupled to COX-2 in preference to COX-1 [18].
mPGES-1 is thought to be linked to the COX-2 down-stream pathway, so investigating mPGES-1 expression patterns in humans might provide clues to the pathogenesis of AD and help identify specific pharmacotherapeutic targets. Because COX-2 inhibition could block the generation of other arachidonic acid metabolites, such as PGD2, PGF2α, PGI2, and thromboxanes, inhibition of a specific PGES could be an alternative therapy that lacks the detrimental effects associated with COX-2 inhibition.
mPGES-1 is induced in the delayed phase of inflammation, with activation time occurring in the course of approximately 1 hour. It is involved in wakefulness, inflammation, hyperalgesia, pain, fever, and cancer [16,19]. Its expression, together with that of COX-2, is induced by proinflammatory stimuli such as IL-1β and has been suggested to be down-regulated by anti-inflammatory glucocorticoids, docosahexaenoic acid, and eicosapentaenoic acid [16,20]. Although previous investigators have reported mPGES-1 protein expression and localization in animals, little is known about its expression patterns in the central nervous system, especially in the human brain. Studies regarding the brain have shown that mPGES-1 is involved in synaptic transmission [21], contributes to microglia-specific lipopolysaccharide-induced PGE2 production [22], is induced in Aβ1−40-treated rat astrocytes [23], and, unlike other PGES, is induced in endothelium via IL1-β [24], not by phosphatidylserine in microglia during apoptosis [25].
Because mPGES-1 is thought to preferentially couple to COX-2, published reports documenting COX isoform localization in human control and AD cortical tissue provided a basis for comparison to our study [26]. Here, we used immunofluorescence, immunohistochemical, and Western blot techniques to examine the localization and immunointensity of mPGES-1 in the middle frontal gyrus (MFG) of human brains from normal subjects and those with advanced AD.
2. Methods
2.1. Subjects
This study was conducted on postmortem human age-matched control and AD brains obtained from the Johns Hopkins Brain Resource Center within 4.0 to 19.0 hours of death. Informed consent was obtained from patients or relatives before death. We examined a total of 24 postmortem brains whose demographics and clinical and neuropathologic diagnoses are presented in Table 1.
Table 1.
Demographics and stages of control and AD cases with results of immunohistochemistry immunointensities*
| Diagnosis | Case No. | Age (y) | Sex | PMT (h) | CERAD | Braak Stage | IHC mPGES-1 Immunointensity† |
|---|---|---|---|---|---|---|---|
| Control | C1 | 74 | M | 4 | II | + | |
| Control | C2 | 71 | F | 16 | 0 | − | |
| Control | C3 | 87 | M | 8 | II | nd | |
| Control | C4 | 80 | F | 8 | 0 | nd | |
| Control | C5 | 68 | M | 10 | II | ++ | |
| Control | C6 | 94 | M | 16 | III | nd | |
| Control | C7 | 80 | M | 22 | IV | nd | |
| Control | C8 | 92 | F | 6 | I | + | |
| Control | C9 | 91 | F | 8 | I | + | |
| Sporadic AD | AD1 | 79 | M | 10.5 | C | VI | nd |
| Sporadic AD | AD2 | 82 | F | 6 | C | VI | ++ |
| Sporadic AD | AD3 | 85 | M | 3.5 | C | VI | nd |
| Sporadic AD | AD4 | 89 | M | 9.5 | C | VI | +++ |
| Sporadic AD | AD5 | 80 | F | 12 | C | V | + |
| Sporadic AD | AD6 | 72 | F | 10 | C | VI | nd |
| Sporadic AD | AD7 | 54 | F | 14.5 | C | VI | nd |
| Sporadic AD | AD8 | 62 | F | 11 | C | VI | + |
| Sporadic AD | AD9 | 84 | F | 5 | C | V | nd |
| Sporadic AD | AD10 | 72 | F | 19 | C | VI | +++ |
| Familial AD | AD11 | 63 | M | 9 | C | VI | ++ |
| Familial AD | AD12 | 74 | F | 5.5 | C | VI | ++ |
| Familial AD | AD13 | 78 | M | 4 | C | VI | ++ |
| Familial AD | AD14 | 92 | F | 8 | C | VI | +++ |
| Familial AD | AD15 | 94 | F | 4 | C | VI | − |
Abbreviations: PMT, postmortem time; IHC, immunohistochemistry; nd, not determined.
Scoring for CERAD and Braak: Control, CERAD negative and Braak 0—IV; senile dementia, CERAD negative and Braak IV; probable AD, CERAD B and Braak IV—V; definite AD, CERAD C and Braak V—VI.
Immunointensity was determined visually and coded according to the following criteria: −, negligible staining; +, low immunointensity; ++, moderate immunointensity; +++, high immunointensity.
Among all subjects studied, 9 were clinically classified as controls, free of any neurologic or cognitive abnormalities, 10 were sporadic AD, and 5 were familial AD. All subjects were used for Western blot analysis, but immunohistochemical analysis involved only five control (three men, 2 women; mean age, 79.6 ± 1.5 years), five sporadic AD (one man, four women; mean age, 77.0 ± 4.6 years), and five familial AD (two men, three women; mean age, 80.2 ± 5.8 years) cases. There was no significant difference in the mean ages of the three groups, but the mean postmortem time of the familial AD group (6.1 ± 1.0 hours) was significantly lower than that of the control group (10.8 ± 1.0 hours) and the sporadic AD group (11.5 ± 2.1 hours).
2.2. Autopsies and neuropathologic evaluations
Coronal blocks of the brains' right hemispheres were fixed in buffered paraformaldehyde at 4°C and then snap-frozen in methylbutane. The left hemispheres were fixed in buffered formalin for 2 weeks, dissected into tissue blocks, and then embedded in paraffin. Tissue blocks of the MFG were cut into 10-μm sections and mounted onto Superfrost/Plus microscope slides (Fisher, Pittsburgh, PA) for immunostaining. The MFG was chosen for this study because in advanced stages of AD, the Aβ plaques and neurofibrillary tangles are particularly evident in the cortical tissue.
The Consortium to Establish a Registry of Alzheimer's Disease (CERAD) criteria were used to establish definite or probable cases of AD [27]. Braak Alzheimer classification [28], which stages AD by the changes in quantity and distribution of neurofibrillary tangles, was used to further classify AD severity. For this study, patients with Braak scores of 3 or higher with a CERAD score of C were considered to have AD. Aβ and tau immunostaining and the modified Bielschowsky silver-staining techniques were used to confirm the diagnosis.
2.3. Western blot analysis
Paraffinized tissue, as described above, was melted and then homogenized via sonication in tissue lysis buffer (Cell Signaling Technology, Beverly, MA) containing complete protease inhibitor (Roche, Indianapolis, IN), 10 mmol/L NaF (Sigma, St Louis, MO), and 1 nmol/L dimethyl sulfoxide (Sigma). After the BCA assay (Pierce, Rockford, IL) was used to quantify protein concentration, equal amounts of protein were loaded onto 12% gels (Invitrogen, Carlsbad, CA), separated by sodium dodecylsulfate–polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes (BA83 0.2 μm; Bio-Rad, Hercules, CA). Membranes were then visualized with Ponceau S solution (Sigma) to verify that equal amounts of protein were loaded in each lane. Membranes were then blocked with 5% skim milk in phosphate-buffered saline containing 0.1% Tween 20 for 45 minutes. Blots were washed and incubated with rabbit anti-mPGES-1 (1:500; Agrisera, Vännäs, Sweden; #AS03031) or rabbit anti-actin (1:500; Sigma; #A-2066) overnight at 4°C and then with goat anti-rabbit secondary antibody (1: 500; Vector Laboratories, Burlingame, CA) for 1 hour at room temperature. Enhanced chemiluminescence (ECL; Amersham Biosciences, Piscataway, NJ) was used to visualize the immunoreactive bands.
2.4. Immunofluorescence
Double immunofluorescence was conducted on frozen tissue sections from two control brains. Sections (20 μm) were fixed in 4% paraformaldehyde for 30 minutes and blocked in 10% normal goat serum (NGS; Cayman Chemical, Ann Arbor, MI) for 4 hours at room temperature. Sections were then incubated overnight at 4°C with primary antibodies against mPGES-1 (1:1000; Cayman Chemical; #160140) and other cell markers, including microglia (rat anti-CD11b, 1:1000; Serotec, Kidlington, Oxford, UK; #MCA711), astrocytes (rat anti-GFAP, 1:500; Zymed, San Francisco, CA; #13−0300), neurons (mouse anti-SMI-32, 1:1000; Sternberger Monoclonals, Lutherville, MD; SMI32), endothelium (mouse anti-von Willebrand factor, 1:200; Vector Laboratories; #VP-V687), smooth muscle cells (mouse anti-smooth muscle actin, 1:200; Zymed; #18−0106), and Aβ1−42 (6E10, 1:5000; Signet, Dedham, MA; #9392). Sections used as negative controls were incubated in 2% NGS in tris-buffered saline (TBS, pH 7.3) instead of primary antibody. Sections were subsequently incubated in corresponding fluorescent secondary antibodies: red Cy3-conjugated AffiniPure goat anti-mouse immunoglobulin G (1:200; Jackson IR Laboratories, West Grove, PA; #115−165−166) or goat anti-rat immunoglobulin G (1:200; Jackson IR Laboratories; #115−165−167), respective to the host of the primary antibody, and green Alexa Fluor 488–conjugated goat anti-rabbit immunoglobulin G (1:500; Molecular Probes, Carlsbad, CA; #A11034;) at room temperature for 4 hours in the dark. Vectashield dry mount hardset media (Vector Laboratories) were used to mount the coverslips. Sections were stored away from light at 4°C.
2.5. Immunohistochemistry
Tissue sections were deparaffinized with 30 minutes of 60°C heat and three 3-minute immersions in xylene (Fisher). Sections were then rehydrated by three 3-minute changes each of 100% and 95% ethanol, respectively, and washed with TBS. To enhance detection, slides were immersed in boiling 1 N citric acid (pH 6.0), microwaved for 6 minutes, and then cooled in distilled water. Endogenous peroxidases were quenched by exposing sections to 0.3% H2O2 (Sigma) and 1% methanol in TBS for 30 minutes. A circle of liquid blocker was drawn around each section with a Super Pap pen (Daido Sangyo Company, Tokyo, Japan) to create a waterproof barrier to prevent leakage of applied reagents. Sections were blocked in 10% NGS and 3% Triton X-100 in TBS for 4 hours at room temperature and incubated overnight at 4°C in rabbit polyclonal antibody against mPGES-1 (1:1000; Cayman Chemical; #160140). Corresponding negative control sections were incubated with 2% NGS in TBS without primary antibody. After being washed in TBS, sections were subsequently incubated in biotinylated goat anti-rabbit secondary antibody (1:500 TBS; Vector Laboratories) for 1 hour and in Avidin D-horseradish peroxidase (HRP; ABC Elite; Vector Laboratories) for 1 hour followed by incubation in diaminobenzidine tetrahydrochloride (DAB; Vector Laboratories) for 5 minutes. To stain nonspecific nuclei, sections were counterstained by immersion in distilled water for 5 minutes, hematoxylin for 20 minutes, and then in running water for 15 minutes. Sections were subsequently dehydrated in three 3-minute rinses of 95% ethanol, 100% ethanol, and xylene. Per-mount mounting medium (Fisher) was used for mounting coverslips.
2.6. Visualization of immunohistochemistry and immunofluorescence
A Nikon Eclipse TE-2000-E inverted microscope (Nikon, Nanjing, China) was used to visualize the sections. For immunohistochemistry, the SPOT RT monochrome camera (Diagnostic Instruments, Inc, Sterling Heights, MI) and Metavue imaging software (Molecular Devices, Sunnyvale, CA) were used to photograph microglia, neurons, astrocytes, and smooth muscle cells, and a Nikon DXM 1200F digital camera (Nikon, Tokyo, Japan) with Nikon ACT-1 Version 2.62 software was used to photograph endothelial cells. The Nikon microscope, digital camera, and software were used to photograph all immunohistochemical sections.
3. Results
3.1. mPGES-1 protein expression in AD versus control brains
Western blot analysis showed that mPGES-1 protein expression level in the MFG appeared to be present at low to negligible levels in most normal age-matched controls but was up-regulated in most AD samples (Figure 1A). Quantification by calculating the ratio of mPGES-1 to actin band intensity confirmed that the mean level of mPGES-1 was significantly greater (P < .001) in the AD samples than in the control samples (Figure 1B).
Fig 1.
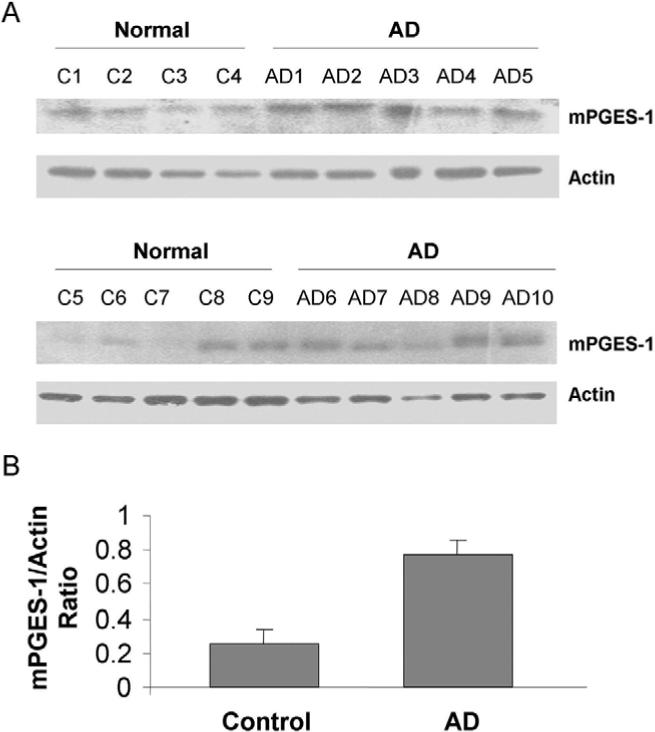
Differential expression of mPGES-1 in human brain MFG. (A) Western blot analysis showed that mPGES-1 protein was expressed in the MFG of AD (n = 10) and age-matched control (n = 9) brains. (B) After normalization to actin, the mean expression of mPGES-1 in the AD brains was shown to be significantly greater than that of the control brains; **P < .001.
3.2. Immunohistochemical analysis of mPGES-1
mPGES-1 immunostaining was negligible in the control cases (Figure 2B), except in case C5, in which large pyramidal neurons were positively stained. In contrast, tissue from all sporadic AD and familial AD cases showed positive staining for mPGES-1, mainly in large pyramidal neurons, as represented in Figure 2D and F, respectively. Among the different subjects, mPGES-1 staining appeared to vary in intensity in sporadic AD cases, whereas familial AD cases appeared to have a more consistent level of intensity. No distinctive immunostaining patterns were observed regarding age, gender, or postmortem time.
Fig 2.
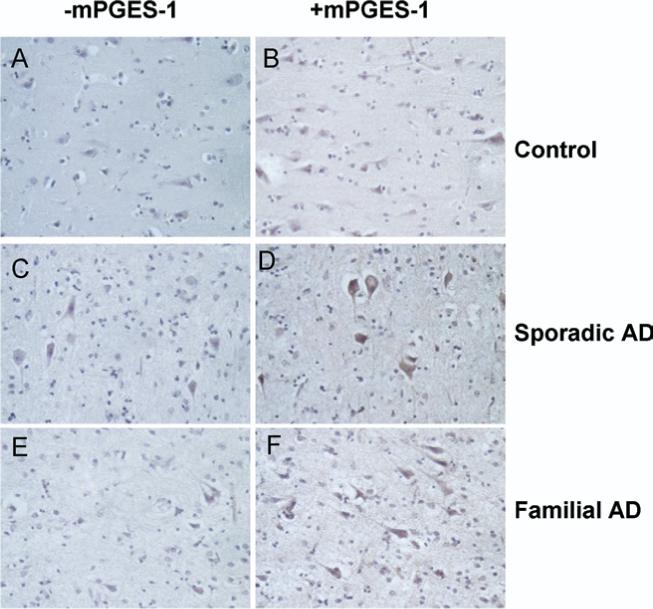
Immunohistochemical study of mPGES-1 in AD. (A) Negative control normal brain; (B) a normal brain positively immunostained with anti–mPGES-2. (C) Negative control sporadic AD brain tissue; (D) mPGES-1–stained sporadic AD brain tissue. (E) Negative control familial AD brain tissue; (F) positively immunostained familial AD brain tissue. Neurons displayed visible detectable immunostaining. Original magnification, 40×.
3.3. mPGES-1 immunofluorescent reactivity
In control brain sections, mPGES-1 colocalized with neurons (Figure 3A–C), microglia (Figure 3D–F), astrocytes (Figure 3G–I), and endothelial cells (Figure 3J–L), but not with smooth muscle cells (Figure 3M–O). In addition, mPGES-1 colocalized with Aβ1−42 (Figure 3P–R). Similar to the control sections, AD brain sections revealed mPGES-1 colocalization with neurons (Figure 4A–C), microglia (Figure 4D–F), endothelial cells (Figure 4G–I), and with astrocytes in some areas (Figure 4J, K, L), but not with smooth muscle cells (Figure 4M–O). Also, in AD tissue, Aβ1−42 colocalized with mPGES-1 (Figure 4P–R). Negative control brain sections showed some red and green nonspecific and/or autofluorescent staining (data not shown), but it was distinguishable from immunostained samples.
Fig 3.
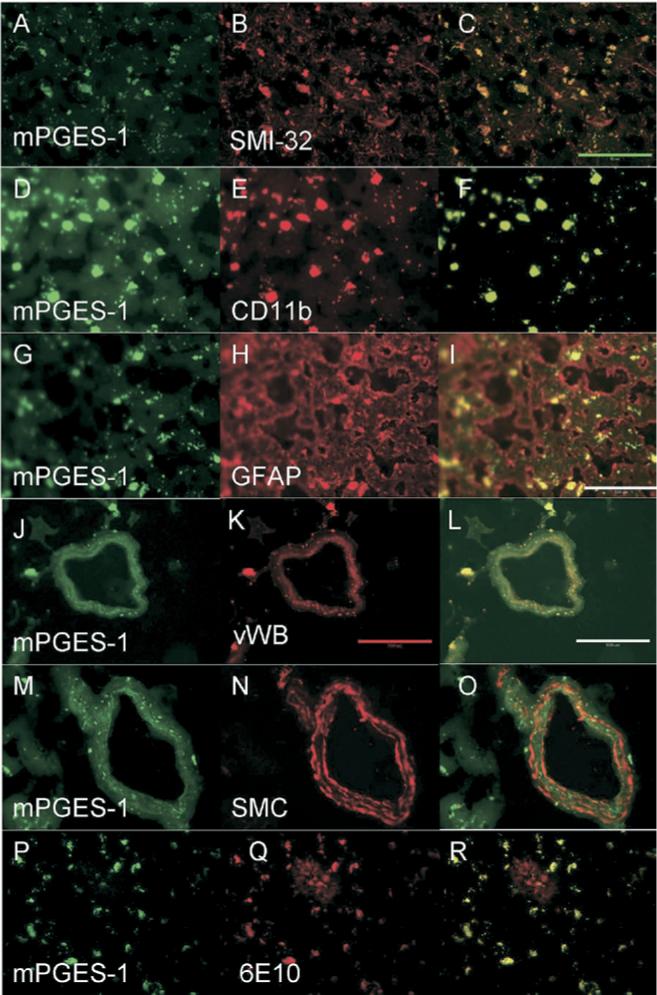
Microsomal PGES-1 (green) colocalization with various cell types (red) in normal brain. Yellow areas show overlap of mPGES-1 with the cell type, indicating colocalization. mPGES-1 colocalized with neurons labeled by SMI-32 antibody (A–C), with microglia labeled by CD11b antibody (D–F), with astrocytes labeled by GFAP antibody (G–I), and with endothelial cells labeled by vWB antibody (J–L). mPGES-1 did not colocalize with smooth muscle cells labeled by SMC antibody (M–O). Aβ1−42, which was stained with 6E10 antibody, colocalized with mPGES-1 in microglial-shaped cells (P–R). The plaque itself (red area seen in R) did not remarkably colocalize with mPGES-1. Green scale bar = 50 μm; red/white scale bar = 100 μm.
Fig 4.
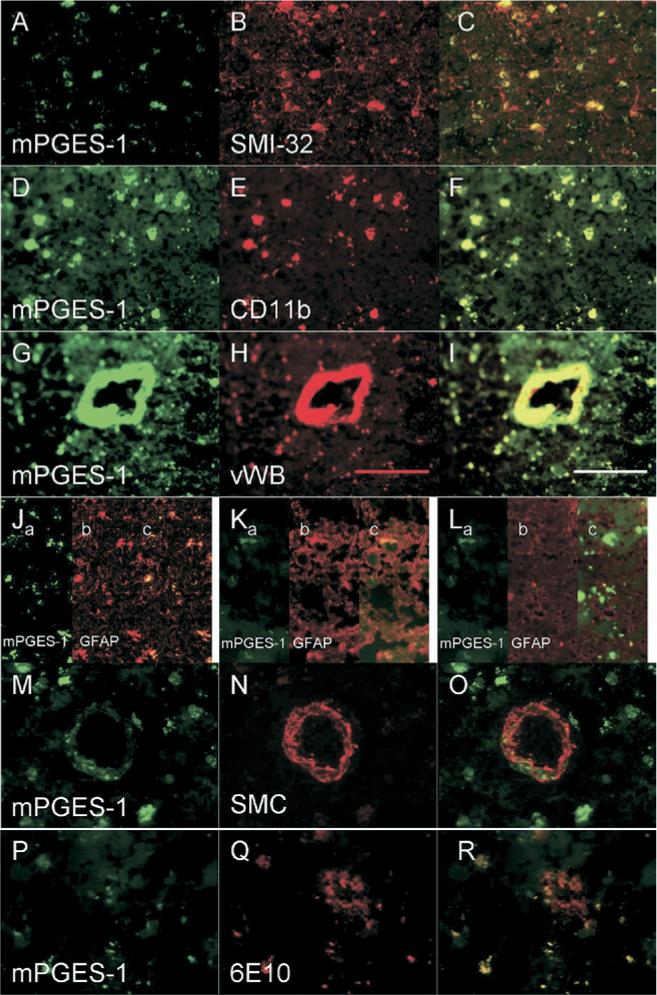
Microsomal PGES-1 (green) colocalization with various cell types (red) in AD brain. Staining was similar to that of control brain. mPGES-1 colocalized with neurons labeled by SMI-32 antibody (A–C), microglia labeled by CD11b antibody (D–F), and endothelial cells labeled by vWB antibody (G–I). mPGES-1 colocalized with astrocytes (labeled by anti-GFAP) in some areas (Jc, Kc) but not in others (Lc). mPGES-1 did not colocalize with smooth muscle cells labeled by SMC antibody (M–O). Aβ1−42, which was stained with 6E10 antibody, colocalized with mPGES-1 in microglial-shaped cells (P–R). The plaque itself (red area seen in R) did not colocalize with mPGES-1. Scale bar = 100 μm.
4. Discussion
In this study, immunohistologic results indicated that mPGES-1 was present in the microglia, neurons, astrocytes, and endothelial cells of all the brains analyzed. Importantly, we showed for the first time via Western blot analysis that mPGES-1 was detectable at low levels in most age-matched control MFG brain regions, but interestingly it was up-regulated in the AD brain homogenates. Furthermore, immunohistochemical staining of mPGES-1 was more intense in the pyramidal neurons of the AD tissue than of the control tissue. This increase in mPGES-1 levels in AD could potentially be a target for a more focused approach to limiting the prostaglandin-mediated neuroinflammatory cascade in AD.
In addition to the increase in the mPGES-1 in the MFG of AD brains, we observed that mPGES-1 colocalized with Aβ in microglia-shaped cells in control and AD brains. This finding implies a potential role for mPGES-1 in microglial phagocytosis of Aβ plaques, a result consistent with that of a previous study [29]. In addition, our results indicate that mPGES-1 is constitutively expressed in human microglia. Furthermore, in rat microglial cultures it has been reported previously that mPGES-1 can be induced in response to proinflammatory stimulants, such as lipopolysaccharide, but not by purported apoptosis products like phosphatidylserine.
In support of our findings, it should be noted that Sang et al [21] also reported that mPGES-1 was constitutively present in the postsynaptic dendrites of rats, and Vasquez-Tello et al [30] reported that normal adult rat brain cortex constitutively expressed neuronal perinuclear mPGES-1 and COX-2 (see Table 2 for comparison studies). The latter group also showed mPGES-1 and COX-2 colocalization in neurons by using immunofluorescence microscopy in the cortex and hippocampus of adult rat brain sections. Because the antibodies we used are known to be immunoreactive in humans, some of the variations in the reported findings could be attributed to slight differences in the antibodies used or differences between animals and humans in mPGES-1 expression.
Table 2.
Microsomal PGES-1 reference chart
| Species/Cell Type | mPGES-1 | Method |
|---|---|---|
| Human (periphery) | ||
| Endothelial cells | Absent | Northern blot [24], Western blot [47] |
| Endothelial cells | Present | Immunohistochemistry [47] |
| Smooth muscle cells | Present | Northern blot [24], Western blot, immunohistochemistry [47] |
| Rat (brain) | ||
| Endothelial cells | Present | Immunohistochemistry [30] |
| Astrocytes | Absent | Northern blot [23], Western blot [22] |
| Microglia | Present | Northern blot [22] |
| Microglia | Absent | Western blot [22,30,48] |
| Neurons | Present | Immunohistochemistry [21,30] |
| Neurons | Absent | Western blot [22] |
| Pig (brain) | ||
| Endothelial cells | Present | Western blot [30] |
Our results revealed that in large pyramidal neurons, immunostaining of mPGES-1 was more intense in AD tissue than in control tissue. Although some investigators have observed a similar pattern for COX-2 immunostaining [31], others have reported opposing findings especially in end-stage AD [26]. The results are highly relevant because the generation of PGE2 results from the combined actions of COX activity and PGE2 synthesis enzymes. Nonetheless, as our Western blot analysis confirmed up-regulation of mPGES-1 in AD cortical tissue, other studies have also shown a corresponding up-regulation of COX-2 [32,33]. In light of the functional coupling of COX-2 and mPGES-1 [34], COX-2 might colocalize with mPGES-1 in pyramidal neurons. Future detailed studies of colocalization with standard immunohistologic approaches and electron microscopy of COX isoforms and mPGES-1 in optimal human brain specimens (eg, shortest postmortem delay, optimal fixation) could support this hypothesis.
Human neuropathologic data are conflicting in regard to whether COX-2 is present in the astrocytes of AD patients [26,35]. Aβ fragments were found to induce COX-2 and PGE2 release in human primary midbrain astrocytes [36]. In addition, rat astrocytic mPGES-1 mRNA was reported to be elevated in response to Aβ stimulation [23]. Here we did not observe consistent mPGES-1 immunostaining in astrocytes of postmortem AD patients or in age-matched controls. In AD brains, astrocyte accumulation of Aβ1−42 could result from phagocytosis of degenerated dendrites. Some of these Aβ1−42-laden astrocytes could undergo lysis, leading to the formation of astrocyte-derived plaque formation [37]. This phenomenon could help explain the various immunostaining results of astrocytes that we observed. Our results also could have been affected by tau pathology, which can affect the processing of numerous proteins, particularly because most of our specimens were from patients with late-stage AD. For example, a previous study has suggested that tau pathology would develop after the increase in neuronal COX-2 expression during early stages [26], but this possibility needs to be confirmed by additional work.
In the early stages of AD, excess Aβ causes increased PGE2 production [13]. This PGE2 can act through various PGE2 binding sites, but mainly it acts via the PGE2 EP receptor family members (EP1−4). The complexity of the G-protein–coupled receptors has been highlighted in recent reviews [38–40]. For example, EP2 receptor sites, despite neuroprotective capabilities at low doses [13], have been found to create a positive feedback loop by leading to further Aβ production [8]. In astrocytes, the elevated PGE2 might also cause glutamate excitotoxicity via stimulation of the EP1 receptor [41–44]. Interestingly, in our studies mPGES-1 appeared to be up-regulated in the pyramidal neurons in advanced AD brains. Although pyramidal neurons generally are known to be vulnerable in AD etiopathology, it is possible that the remaining neurons might have developed resistance to cell death, and that an increase in mPGES-1 might be an attempt to generate prostanoids to stimulate a survival pathway. This possibility is supported by the multiple actions of PGE2, which, depending on the receptor being stimulated, might lead to toxicity, cell survival, or stimulation of a pathway to a decrease in the inflammatory cascade [38–40,45]. In addition, it has been suggested that metabolites of PGE2 might lead to cyclopentanone prostaglandins, which can also have intracellular protective effects [46]. This work is actively being pursued in various laboratories including our own.
In conclusion, we found that in normal brains, mPGES-1 is constitutively expressed in microglia, astrocytes, neurons, and endothelium but not in smooth muscle cells. In end-stage AD, mPGES-1 appears to be markedly elevated in pyramidal neurons while somewhat diminished or absent in astrocytes. Elucidating the patterns of expression and activity of mPGES-1 in various stages of AD can lead to more appropriate and selective pharmacologic alternatives to NSAIDs and COX-2 inhibitors and to a better understanding of the pathogenesis of AD.
Acknowledgments
This work was supported by NIH grants AG022971-02, -03, -04 (S.D., B.C.) and NS046400-02, -03, -04 (S.D.). Postmortem brain tissues were provided by the Johns Hopkins University Alzheimer's Disease Research Center (AG05146). We thank Eduardo Zamora, MD for his technical assistance and all members of the Doré lab for helpful discussions.
References
- 1.Combrinck M, Williams J, De Berardinis MA, Warden D, Puopolo M, Smith AD, et al. Levels of CSF prostaglandin E2, cognitive decline, and survival in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2006;77:85–8. doi: 10.1136/jnnp.2005.063131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Breitner JC, Welsh KA, Helms MJ, Gaskell PC, Gau BA, Roses AD, et al. Delayed onset of Alzheimer's disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol Aging. 1995;16:523–30. doi: 10.1016/0197-4580(95)00049-k. [DOI] [PubMed] [Google Scholar]
- 3.Zandi PP, Breitner JC. Do NSAIDs prevent Alzheimer's disease? and, if so, why? the epidemiological evidence. Neurobiol Aging. 2001;22:811–7. doi: 10.1016/s0197-4580(01)00297-4. [DOI] [PubMed] [Google Scholar]
- 4.Szekely CA, Thorne JE, Zandi PP, Ek M, Messias E, Breitner JC, et al. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology. 2004;23:159–69. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- 5.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996;47:425–32. doi: 10.1212/wnl.47.2.425. [DOI] [PubMed] [Google Scholar]
- 6.Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289:2819–26. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 7.Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, et al. Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62:66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- 8.Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, et al. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer's disease. J Neurosci. 2005;25:10180–7. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shie FS, Breyer RM, Montine TJ. Microglia lacking E prostanoid receptor subtype 2 have enhanced Abeta phagocytosis yet lack Abeta-activated neurotoxicity. Am J Pathol. 2005;166:1163–72. doi: 10.1016/s0002-9440(10)62336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griffin WS, Liu L, Li Y, Mrak RE, Barger SW. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation. 2006;3:5. doi: 10.1186/1742-2094-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giovannini MG, Scali C, Prosperi C, Bellucci A, Vannucchi MG, Rosi S, et al. Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol Dis. 2002;11:257–74. doi: 10.1006/nbdi.2002.0538. [DOI] [PubMed] [Google Scholar]
- 12.Paris D, Town T, Parker T, Humphrey J, Mullan M. A beta vasoactivity: an inflammatory reaction. Ann N Y Acad Sci. 2000;903:97–109. doi: 10.1111/j.1749-6632.2000.tb06355.x. [DOI] [PubMed] [Google Scholar]
- 13.Echeverria V, Clerman A, Doré S. Stimulation of PGE2 receptors EP2 and EP4 protects cultured neurons against oxidative stress and cell death following β-amyloid exposure. Eur J Neurosci. 2005;22:2199–206. doi: 10.1111/j.1460-9568.2005.04427.x. [DOI] [PubMed] [Google Scholar]
- 14.Yagami T, Nakazato H, Ueda K, Asakura K, Kuroda T, Hata S, et al. Prostaglandin E2 rescues cortical neurons from amyloid beta protein-induced apoptosis. Brain Res. 2003;959:328–35. doi: 10.1016/s0006-8993(02)03773-3. [DOI] [PubMed] [Google Scholar]
- 15.Cazevieille C, Muller A, Meynier F, Dutrait N, Bonne C. Protection by prostaglandins from glutamate toxicity in cortical neurons. Neurochem Int. 1994;24:395–8. doi: 10.1016/0197-0186(94)90118-x. [DOI] [PubMed] [Google Scholar]
- 16.Murakami M, Nakashima K, Kamei D, Masuda S, Ishikawa Y, Ishii T, et al. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem. 2003;278:37937–47. doi: 10.1074/jbc.M305108200. [DOI] [PubMed] [Google Scholar]
- 17.Murakami M, Kudo I. Recent advances in molecular biology and physiology of the prostaglandin E2-biosynthetic pathway. Prog Lipid Res. 2004;43:3–35. doi: 10.1016/s0163-7827(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 18.Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006;116:1391–9. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang ZL, Sato Y, Mochizuki T, Okada T, Qu WM, Yamatodani A, et al. Prostaglandin E2 activates the histaminergic system via the EP4 receptor to induce wakefulness in rats. J Neurosci. 2003;23:5975–83. doi: 10.1523/JNEUROSCI.23-14-05975.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forsberg L, Leeb L, Thoren S, Morgenstern R, Jakobsson P. Human glutathione dependent prostaglandin E synthase: gene structure and regulation. FEBS Lett. 2000;471:78–82. doi: 10.1016/s0014-5793(00)01367-3. [DOI] [PubMed] [Google Scholar]
- 21.Sang N, Zhang J, Marcheselli V, Bazan NG, Chen C. Postsynaptically synthesized prostaglandin E2 (PGE2) modulates hippocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J Neurosci. 2005;25:9858–70. doi: 10.1523/JNEUROSCI.2392-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikeda-Matsuo Y, Ikegaya Y, Matsuki N, Uematsu S, Akira S, Sasaki Y. Microglia-specific expression of microsomal prostaglandin E2 synthase-1 contributes to lipopolysaccharide-induced prostaglandin E2 production. J Neurochem. 2005;94:1546–58. doi: 10.1111/j.1471-4159.2005.03302.x. [DOI] [PubMed] [Google Scholar]
- 23.Satoh K, Nagano Y, Shimomura C, Suzuki N, Saeki Y, Yokota H. Expression of prostaglandin E synthase mRNA is induced in beta-amyloid treated rat astrocytes. Neurosci Lett. 2000;283:221–3. doi: 10.1016/s0304-3940(00)00926-5. [DOI] [PubMed] [Google Scholar]
- 24.Uracz W, Uracz D, Olszanecki R, Gryglewski RJ. Interleukin 1beta induces functional prostaglandin E synthase in cultured human umbilical vein endothelial cells. J Physiol Pharmacol. 2002;53:643–54. [PubMed] [Google Scholar]
- 25.Zhang J, Fujii S, Wu Z, Hashioka S, Tanaka Y, Shiratsuchi A, et al. Involvement of COX-1 and up-regulated prostaglandin E synthases in phosphatidylserine liposome-induced prostaglandin E(2) production by microglia. J Neuroimmunol. 2006;172:112–20. doi: 10.1016/j.jneuroim.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 26.Hoozemans JJ, Rozemuller AJ, Janssen I, De Groot CJ, Veerhuis R, Eikelenboom P. Cyclooxygenase expression in microglia and neurons in Alzheimer's disease and control brain. Acta Neuropathol (Berl) 2001;101:2–8. doi: 10.1007/s004010000251. [DOI] [PubMed] [Google Scholar]
- 27.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): part II—standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 28.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 29.Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer's disease patients. J Alzheimers Dis. 2005;7:221–32. doi: 10.3233/jad-2005-7304. discussion 55−62. [DOI] [PubMed] [Google Scholar]
- 30.Vazquez-Tello A, Fan L, Hou X, Joyal JS, Mancini JA, Quiniou C, et al. Intracellular-specific colocalization of prostaglandin E2 synthases and cyclooxygenases in the brain. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1155–63. doi: 10.1152/ajpregu.00077.2004. [DOI] [PubMed] [Google Scholar]
- 31.Hoozemans JJ, Veerhuis R, Rozemuller AJ, Arendt T, Eikelenboom P. Neuronal COX-2 expression and phosphorylation of pRb precede p38 MAPK activation and neurofibrillary changes in AD temporal cortex. Neurobiol Dis. 2004;15:492–9. doi: 10.1016/j.nbd.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 32.Kitamura Y, Shimohama S, Koike H, Kakimura J, Matsuoka Y, Nomura Y, et al. Increased expression of cyclooxygenases and peroxisome proliferator-activated receptor-gamma in Alzheimer's disease brains. Biochem Biophys Res Commun. 1999;254:582–6. doi: 10.1006/bbrc.1998.9981. [DOI] [PubMed] [Google Scholar]
- 33.Pasinetti GM, Aisen PS. Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer's disease brain. Neuroscience. 1998;87:319–24. doi: 10.1016/s0306-4522(98)00218-8. [DOI] [PubMed] [Google Scholar]
- 34.Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–92. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 35.Yermakova AV, O'Banion MK. Downregulation of neuronal cyclooxygenase-2 expression in end stage Alzheimer's disease. Neurobiol Aging. 2001;22:823–36. doi: 10.1016/s0197-4580(01)00303-7. [DOI] [PubMed] [Google Scholar]
- 36.Hull M, Muksch B, Akundi RS, Waschbisch A, Hoozemans JJM, Veerhuis R, et al. Amyloid beta peptide (25−35) activates protein kinase C leading to cyclooxygenase-2 induction and prostaglandin E2 release in primary midbrain astrocytes. Neurochem Int. 2006;48:663–72. doi: 10.1016/j.neuint.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 37.Nagele RG, D'Andrea MR, Lee H, Venkataraman V, Wang HY. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003;971:197–209. doi: 10.1016/s0006-8993(03)02361-8. [DOI] [PubMed] [Google Scholar]
- 38.Shie FS, Montine KS, Breyer RM, Montine TJ. Microglial EP2 as a new target to increase amyloid beta phagocytosis and decrease amyloid beta-induced damage to neurons. Brain Pathol. 2005;15:134–8. doi: 10.1111/j.1750-3639.2005.tb00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doré S. GPCR antagonists as an alternative to COX-2 inhibitors: a case for the PGE2 EP1 receptor. Trends Pharmacol Sci. 2006;27:458–60. doi: 10.1016/j.tips.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Zhu T, Gobeil F, Vazquez-Tello A, Leduc M, Rihakova L, Bossolasco M, et al. Intracrine signaling through lipid mediators and their cognate nuclear G-protein-coupled receptors: a paradigm based on PGE2, PAF, and LPA1 receptors. Can J Physiol Pharmacol. 2006;84:377–91. doi: 10.1139/y05-147. [DOI] [PubMed] [Google Scholar]
- 41.Gendron TF, Brunette E, Tauskela JS, Morley P. The dual role of prostaglandin E(2) in excitotoxicity and preconditioning-induced neuroprotection. Eur J Pharmacol. 2005;517:17–27. doi: 10.1016/j.ejphar.2005.05.031. [DOI] [PubMed] [Google Scholar]
- 42.Saleem S, Li R, Wei G, Doré S. Effects of EP1 receptor on cerebral blood flow in the middle cerebral artery occlusion model of stroke in mice. J Neurosci Res. 2007;85:2433–40. doi: 10.1002/jnr.21399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmad AS, Saleem S, Ahmad M, Doré S. Prostaglandin EP1 receptor contributes to excitotoxicity and focal ischemic brain damage. Toxicol Sci. 2006;89:265–70. doi: 10.1093/toxsci/kfj022. [DOI] [PubMed] [Google Scholar]
- 44.Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, et al. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–9. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- 45.Matsuoka T, Narumiya S. Prostaglandin receptor signaling in disease. ScientificWorldJournal. 2007;7:1329–47. doi: 10.1100/tsw.2007.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhuang H, Kim YS, Namiranian K, Doré S. Prostaglandins of J series control heme oxygenase expression: potential significance in modulating neuroinflammation. Ann N Y Acad Sci. 2003;993:208–16. doi: 10.1111/j.1749-6632.2003.tb07531.x. [DOI] [PubMed] [Google Scholar]
- 47.Giannoulias D, Alfaidy N, Holloway AC, Gibb W, Sun M, Lye SJ, et al. Expression of prostaglandin I(2) synthase, but not prostaglandin E synthase, changes in myometrium of women at term pregnancy. J Clin Endocrinol Metab. 2002;87:5274–82. doi: 10.1210/jc.2002-020521. [DOI] [PubMed] [Google Scholar]
- 48.Greco A, Ajmone-Cat MA, Nicolini A, Sciulli MG, Minghetti L. Paracetamol effectively reduces prostaglandin E2 synthesis in brain macrophages by inhibiting enzymatic activity of cyclooxygenase but not phospholipase and prostaglandin E synthase. J Neurosci Res. 2003;71:844–52. doi: 10.1002/jnr.10543. [DOI] [PubMed] [Google Scholar]