

Abstract
The extracellular environment controls many cellular activities thereby linking external material cues to internal cell function. By better understanding these processes, synthetic extracellular material niches can be tailored to present cells with highly regulated physical and/or chemical cues that promote or suppress selected cell functions. Here, poly(ethylene glycol) (PEG) hydrogels were functionalized with fluvastatin-releasing grafts and growth factor binding heparin domains to enable the dynamic exchange of information between the material and cells from the outside-in and inside-out (i.e., bidirectional signaling). By incorporating a fluvastatin-releasing graft and carefully controlling the dose and temporal release, materials were designed to promote bone morphogenic protein (BMP2) and alkaline phosphatase (ALP) production by human mesenchymal stem cells (hMSCs). When the release of fluvastatin was controlled to occur over 2 weeks, BMP2 and ALP production was increased 2.2-fold and 1.7-fold, respectively, at day 28 compared to hMSCs cultured in the absence of fluvastatin. By introducing a heparin functionality into the gel to sequester and localize the hMSC-produced BMP2, the osteogenic differentiation of hMSCs was further augmented over fluvastatin delivery alone. Osteopontin and core binding factor α1 gene expression was 6-fold and 4-fold greater for hMSCs exposed to fluvastatin in the presence of the heparin functionalities, respectively. These results demonstrate how multifunctional gels that interact with cells in a bidirectional manner can efficiently promote selected cell functions, such as the osteogenic differentiation of hMSCs.
1. Introduction
Hydrogels are an attractive class of materials for encapsulating cells and creating niches that promote tissue regeneration. The highly swollen environment allows facile transport of nutrients and wastes, supporting the long-term survival of numerous cell types,[1,2] while the structural properties provide a three-dimensional framework that directs cellular interactions and matrix deposition.[3,4] The biochemical hydrogel microenvironment can be designed to promote or suppress specific cellular activities, and recent efforts have focused on increasingly sophisticated methods to incorporate biological signals that are presented at the right time, in the right place, and at the right dose.[5]
Numerous examples exist where the hydrogel environment is tailored to provide cells with a physical or chemical cue, enabling the transmission of outside-in signals. The identification of small oligopeptide sequences within extracellular matrix (ECM) adhesion proteins, such as the ubiquitous Arg-Gly-Asp-Ser (RGDS), initiated the creation of ligand-functionalized, highly-defined synthetic ECM analogs capable of integrin interactions and subsequent intracellular signaling.[6] Alternative approaches exploit the ability of cells to modify their material environment and focus on strategies to sequester cell-secreted proteins. For example, a peptide sequence that specifically sequesters BMP2 was identified and induced dystrophic calcification when injected into mouse muscle.[7] Also, a fibronectin-binding peptide has been identified.[8] Beyond this type of unidirectional exchange of information, materials have been designed that present cells with growth factors that can be released upon demand. This approach is similar to the mechanism of the natural ECM where modulation of tissue dynamics is achieved through the ECM’s ability to bind, store, and release growth factors. Lutolf et al. described a synthetic matrix where a growth factor is bound to a synthetic material matrix and released through cell-mediated proteolytic cleavage.[9]
While complexity can provide advantages in designing niches to promote tissue regeneration, oftentimes assembling the simplest biomaterial system is a successful strategy, recognizing that not every aspect of the extracellular environment needs to be recapitulated. Significant advances are often achieved by reproducing one or two factors in the right context. For example, Richardson et al.[10] developed a polymeric scaffold that was designed to deliver vascular endothelial growth factor (VEGF), which stimulates outgrowth of endothelial channels, followed sequentially by second-phase delivery of platelet-derived growth factor (PDGF)-BB, which in turn stabilizes the nascent vessels by recruiting smooth muscle cells. Delivery of these molecules in tandem, rather than singularly, resulted in synergistically enhanced vessel formation while VEGF or PDGF-BB delivery alone resulted in fragile vascularity or no significant vessel formation.[10]
Clearly, application of factors known to play roles in tissue development and remodeling can play an important role in advancing the goals of functional tissue formation. The challenging aspect, however, is identifying those factors and presenting them in the appropriate context. Here, we demonstrate a simple, but versatile approach to the design of gel systems that modulate the activity of human mesenchymal stem cells (hMSCs), such as osteogenic differentiation, through a dynamic exchange of information between the cell and its material environment. In general, induction of the osteoblast phenotype requires interactions between osteoprogenitors and a recognizable ECM.[11] The ECM of bone is a reservoir of endogenous growth factors,[12] localizing growth factor activity, preventing growth factor degradation, and often enhancing growth factor binding to cell surface receptors,[13] all affecting autocrine signaling. One of these molecules, BMP2, binds to receptors on the surface of MSCs and is known to promote osteogenic differentiation.[14]
Natively, bone morphogenetic proteins (BMPs) are synthesized and secreted by osteprogenitor cells and stored in bone matrix and released during bone remodeling.[15,16] However, therapeutic doses of recombinant BMP are often several orders of magnitude higher than that found in the local ECM;[17] the effects of context and presentation can be quite dramatic. Thus, strategies that stimulate osteoprogenitor cells to produce and release BMPs are emerging, and statins are one class of small molecules known to stimulate osteoblast BMP2 production.[18]
To capture some of these critical events in the differentiation of osteoprogenitor cells and bone regeneration, hMSCs were encapsulated in a hydrogel environment containing two specific functionalities. The first functionality was a fluvastatin-containing tether that subsequently releases its functionality in a manner to promote the secretion of BMP2 by hMSCs. The second functionality was a large polysaccharide, heparin, that binds cell-secreted BMP2, increasing its local availability and sustaining autocrine signaling effects. These two functionalities, immobilized in a material matrix, participate in the regulation of a complex cascade of events where the process is initiated and sustained quite simply by the release of a small, potent therapeutic molecule, fluvastatin. Subsequent events exploit the ability of hMSCs to temporally and spatially regulate the modification and remodeling of their surrounding material microenvironment through the wide range of interactions of cell-secreted proteins with heparin.[19]
2. Results and Discussion
Statins (e.g., fluvastatin) have recently been reported to stimulate osteoprogenitor production of BMP2; statins are small, potent molecules that necessitate controlled and localized delivery strategies to sustain therapeutic efficacy. Here, three different macromolecular monomers were synthesized to control the release of fluvastatin through increasing incorporation of hydrolytically degradable ester bonds via lactic acid (LA) functionalities grafted from poly(ethylene glycol) monomethacrylate (PEGMMA). Specifically, PEG526MMA-2LA-fluvastatin, PEG526MMA-4LA-fluvastatin, PEG526MMA-6LA-fluvastatin were examined (526 is the average molecular weight (Da) of the PEG and 2, 4, and 6 correspond to theoretical numbers of LA repeat units based on reaction stoichiometry). The actual number of LA repeat units in the monomers was analyzed with proton nuclear magnetic resonance spectroscopy, and results are listed in Table 1. The release of fluvastatin from hydrogels formed from the different fluvastatin monomers copolymerized with PEG4600DM was measured, and results are shown in Figure 1. The rate of release of fluvastatin from the PEG526MMA-2LA-fluvastatin and the PEG526MMA-4LA-fluvastatin hydrogels was slower than the PEG526MMA-6LA-fluvastatin. Approximately 90% release was reached in 16 days for the 2LA linker, 9 days for the 4LA linker, and 4 days for the 6LA linker. This trend agrees with the hypothesis that increasing the number of lactic acid units in the LA block increases the probability that the fluvastatin is unbound and releasable at any given time. However, no statistical difference was observed in the cumulative release profile between the PEG526MMA-2LA-fluvastatin and PEG526MMA-4LA-fluvastatin. This may be related to the experimentally measured LA lengths being fairly close in number (1.7 vs. 3.2), and the fact that these values are average block lengths and a distribution inherently exists. However, cumulative release is found to be statistically different at times greater than 3 days between the n=4 and n=6 macromers. This is explained by the NMR-predicted block lengths for the n=4 versus n=6 macromers indicating that that the actual block length averages are 3.2 and 5.5, respectively, a difference of 2.3 LA. The n=2 block length averages 1.7 by NMR prediction, a difference only of 1.5 LA in comparison to the n=4 macromer. Therefore, assuming similar distributions exist, the n=4 and n=6 release studies can yield statistically different results while the n=2 and n=4 macromers may not.
Table 1.
Lactic acid repeat units per monomer (n in Figure 1) and the kinetic constant of hydrolysis of the LA block
| Monomer Acronym | Lactic acid units per monomer (n in Figure 1)a | mg statin/mg producta | k′ (hr−1)b |
|---|---|---|---|
| PEG526MMA-2LA-fluvastatin | 1.7 | 0.417 | 0.005 |
| PEG526MMA-4LA-fluvastatin | 3.2 | 0.357 | 0.009 |
| PEG526MMA-6LA-fluvastatin | 5.5 | 0.086 | 0.022 |
Determined by NMR (Bruker 500 MHz)
Pseudo first-order hydrolysis rate constant determined from fluvastatin release data (Figure 1)
Figure 1.
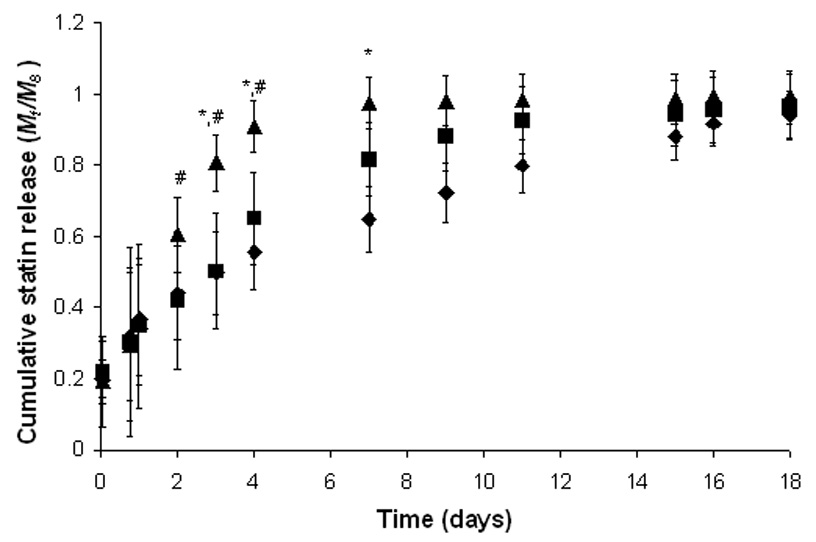
Cumulative fluvastatin release (Mt/M∞) as a function of time for hydrogels synthesized through the copolymerization of PEG526MMA-nLA-fluvastatin (n=2: ♦, n=4: ■, n=6: ▲) with a 10 wt% solution of PEG4600DM. Release profiles were measured in PBS at sink conditions. Gels (d=5 mm, t=2 mm) initially contained ~500 µg of statin. * and # - statistically different cumulative release values between n=4 and n=6 and n=2 and n=6, respectively, at particular times (p<0.05).
The degradation of tethered LA grafts in hydrogel environments similar to those present in PEG526MMA-nLA-fluvastatin has been thoroughly characterized,[20,21] and degradation of the LA block follows a pseudo first-order process, given the high water content in the gel:
| (Equation 1) |
Here, nPLA is the number of degradable PLA blocks in the gel; t is the degradation time; and k’ is the pseudo first-order kinetic constant for hydrolysis of the block. Given that the diffusion timescale (tD~200 min) is orders of magnitude faster than the hydrolysis reaction timescale, k’ can be estimated from the fluvastatin concentration present in the solution. The calculated hydrolysis rate constants are reported in Table 1 and are consistent with previous reports for PLA blocks in PEG gels.[20,21] In sum, the timescale for degradation-controlled release of the fluvastatin (trxn = 1/k’) ranges from 45 h to 110 h to 200 h as dictated by the length of the degradable block from 6 to 4 to 2, respectively.
As the LA blocks are hydrolyzed, fluvastatin is released and available for uptake by cells. During this release, several products can result, free fluvastatin and various fluvastatin-LA conjugates.[21–23] Fluvastatin-LA conjugates may not be internalized by cells as readily as free fluvastatin or may suffer from decreased biologic activity. If this occurs, the collective dose of the fluvastatin would be lowered, and the potential for fluvastatin to escape the gel environment and influence peripheral cells would exist. However, cells produce enzymes such as esterases and lipases capable of cleaving any residual LA units present on fluvastatin after it enters the cell.[22] Therefore, if cells can uptake the fluvastatin-LA conjugates, there is a high probability that it will be rapidly converted to fluvastatin and have a similar effect as free fluvastatin.
The uptake rate of hMSCs, estimated by using an analogous molecule to fluvastatin (cortisol), was found to be ~0.2 fg/cell/hr. Accounting for differences in the uptake rate and release rate (Figure 1), the concentration of fluvastatin within a three-dimensional hydrogel over time was estimated. To maintain the maximum fluvastatin concentration below a toxic level (~500 nM) while simultaneously providing a fluvastatin concentration near the therapeutic dose (~100 nM) for a sustained time period, the slowest releasing monomer, PEGMMA-2LA-fluvastatin, with a loading of ~0.2 µg/gel was chosen for subsequent experiments and corresponds to an average dose within the hydrogel environment of ~180 nM over two weeks of in vitro culture, where the highest dose at the onset of release is ~450 nM and the lowest dose at the end of release is ~10 nM.
The effect of fluvastatin delivery on three-dimensionally cultured hMSCs and their activity was evaluated as a function of time for the PEGMMA-2LA-fluvastatin gel system. RGDS was incorporated as pendant tethers at a concentration of 5 mM into all gel formulations to maintain hMSC survivability.[23] ALP and BMP2 production was monitored at 2, 10, and 28 days, and the results shown in Figure 2 were normalized to levels of metabolic activity as a relative measure of cell number. ALP production (Figure 2A) increased with culture time in the presence and absence of fluvastatin. However, by day 28 ALP production was 2.2-fold greater for hMSCs that were cultured in fluvastatin-releasing gels versus cells that were cultured in the absence of fluvastatin.
Figure 2.
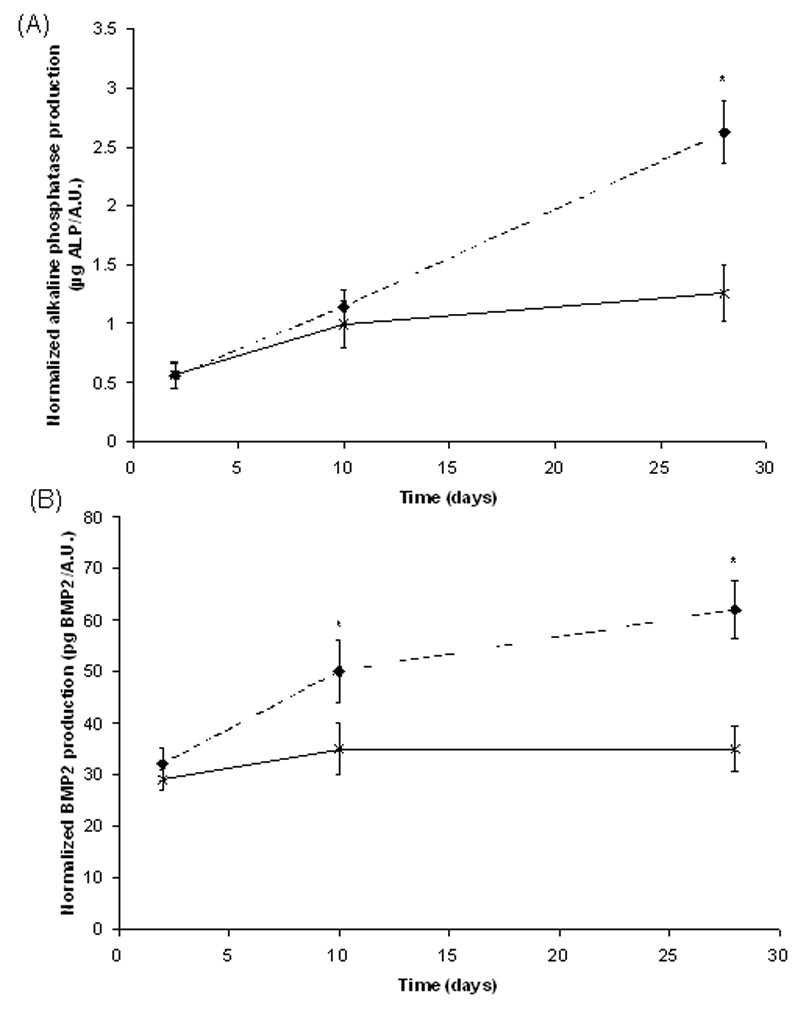
ALP and BMP2 production normalized to metabolic activity (AlamarBlue) of hMSCs encapsulated in RGDS-functionalized hydrogels in the absence of fluvastatin (–x–) or with releasing fluvastatin (--♦--), n=4 samples/condition. * p<0.05 of sample versus control (no delivered fluvastatin) at that time.
BMP2 production (Figure 2B) by hMSCs also increased in both culture conditions over the 28 day study. The greatest BMP2 production was found for cells cultured in fluvastatin-releasing hydrogels, which is 1.7-fold greater than the levels produced by hMSCs cultured in the absence of released fluvastatin at day 28. These results suggest that fluvastatin is being released at an appropriate dose and in an active form, leading to an increase in ALP and BMP2 production after 10 days of culture, which are both indicative of hMSC osteogenic differentiation. Induction of osteogenic differntiation of hMSCs begins with increases in the transcriptional factor core binding factor alpha 1 (CBFA1), which regulates the osteogenic lineage. After 7–10 days after upregulation of CBFA1, osteogenic proteins such as ALP, BMP2, bone sialoprotein, and osteopontin are augmented.[24]
While external delivery of fluvastatin at a controlled dose and rate can affect hMSCs ALP and BMP2 production, a means to sequester the cell-produced BMP2 would enable localized availablility and autocrine signaling similar to the native ECM. In addition, the biological activity of BMP2 may be enhanced in the presence of sulfated polysaccharides, suggesting that heparin can enhance the biological activity of BMPs by continuously presenting the ligands to their signaling receptors expressed on cell membranes.[25] With this in mind, introduction of heparin functionalities to the hydrogels was explored as a means to sequester hMSC-produced BMP2.
To formulate heparin-functionalized PEG hydrogels, heparin was modified with methacrylate groups to allow for copolymerization with PEG4600DM. Heparin possesses specific binding interactions with many proteins, of which BMP2 is one. The ability of free and copolymerized methacrylate-modified heparin to retain its binding properties with BMP2 was examined. When BMP2 was combined with heparin or methacrylated heparin, decreased staining intensity for BMP2 on native PAGE gels was observed with increasing degree of heparin functionalization, indicating BMP2 binding to methacrylated heparin was less efficient than BMP2 binding to native heparin. Results are shown in Figure 3A and quantified in Figure 3B as a percent of the control (native heparin) binding. The degree of BMP2 association with methacrylated heparin was dependent on the extent of methacrylation of heparin, with a higher degree of modification corresponding to a decreased BMP2 binding.
Figure 3.
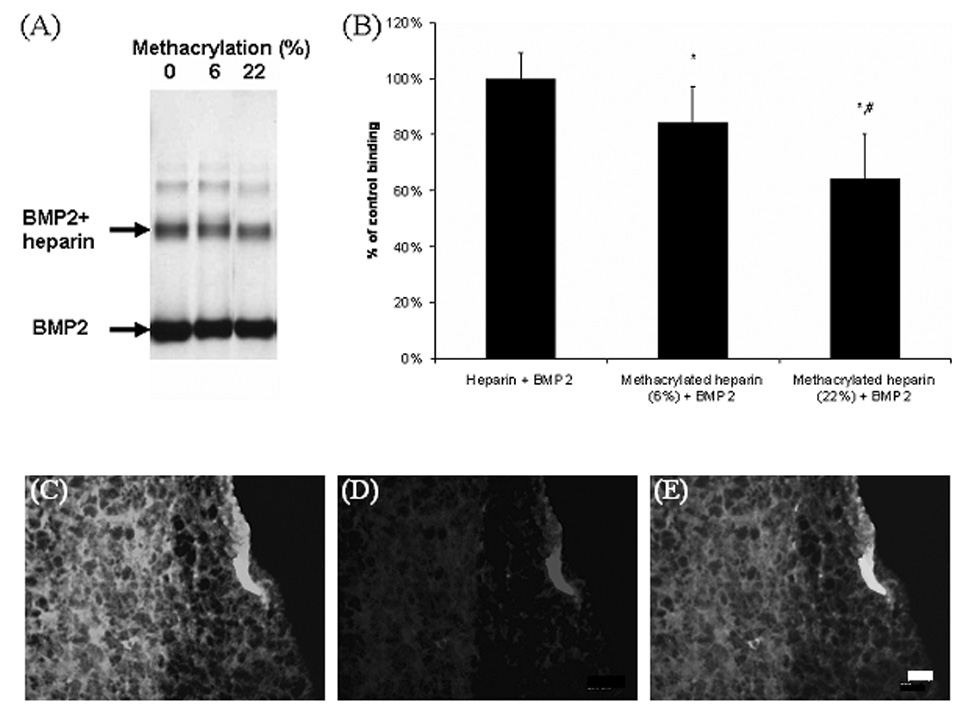
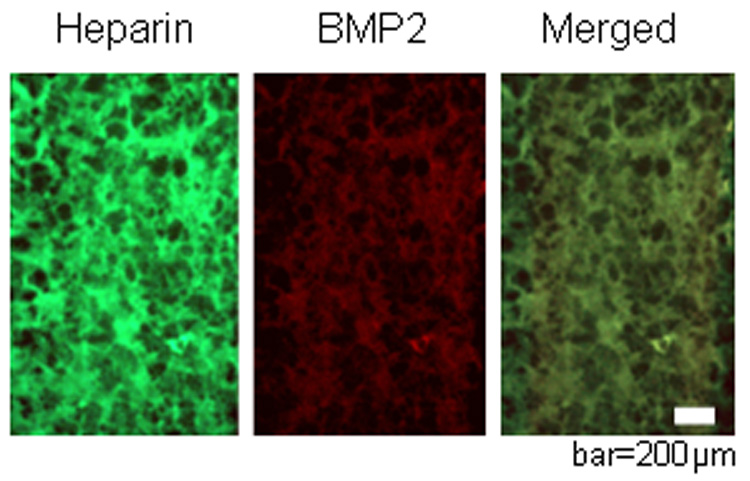
Native PAGE gel (A) and semi-quantitative analysis (B) demonstrated that BMP2 binding to heparin decreased with increasing extent of methacrylation (% methacrylation in parentheses). * p< 0.05 versus no methacrylation and # p<0.05 versus 6% methacrylation, n=3 samples/condition. Representative images showing that methacrylated heparin, labeled with Oregon green and copolymerized with PEG4600DM is uniformly distributed (C), and retained its ability to specifically bind to BMP2 (red) (D) as qualitatively shown by the merged image, where the colocalized fluorescence appears orange (E). Control PEG hydrogels showed no staining (not pictured), indicating no BMP2 sequestering, bar=200 µm.
To assess the ability of heparin-functionalized gels to bind BMP2, Oregon green labeled methacrylated heparin was copolymerized with PEG4600DM. The resulting gels fluoresce green (pictured in Figure 3C) and were also shown to specifically retain noncovalently bound BMP2 as evidenced by colocalization of green (heparin, 3C) and red (BMP2, 3D) and the overlayed fluorescent staining of the heparin-functionalized hydrogel, appearing orange (Figure 3E). To demonstrate that the proteins were binding specifically to heparin contained in the gels, negative control gels (PEG only) were also stained and showed no significant staining of BMP2 (not pictured). These results indicate that BMP2 can be successfully sequestered in PEG hydrogels through heparin functionalization. These results are similar to those explored previously by Masters et al.[26] where methacrylated hyaluronic acid retained the ability to bind fibronectin in solution and when polymerized to form a hydrogel. In addition, two other heparin-binding proteins, basic fibroblast growth factor and fibronectin, were found to associate and bind with methacrylated heparin copolymerized with PEG4600DM.[27]
To test the combined effects of fluvastatin delivery and heparin functionalization on BMP2 production and local concentration, encapsulated hMSCs were cultured in vitro in heparin-functionalized hydrogels with and without grafted fluvastatin. At various time points (2, 10, and 28 days), media was collected and analyzed for released BMP2. Figure 4 shows the BMP2 production normalized to metabolic activity as a relative measure of viable cells. For comparison, results for BMP2 production in the absence of heparin is included. BMP2 production by hMSCs increased in all culture conditions over the 28 day study. BMP2 production was affected similarly for both fluvastatin-releasing culture systems, causing about a 2-fold increase over the same gel chemistry in the absence of fluvastatin at day 28. Interestingly, the RGDS-functionalized, fluvastatin-releasing hydrogels led to a larger increase in measured BMP2 in solution compared to heparin-functionalized counterparts at day 28. This higher level likely indicates that BMP2 is being sequestered within the heparin-functionalized hydrogel, thereby decreasing the BMP2 levels in media slightly. There is not likely to be a significant reduction in solution BMP2 concentration due to the limited number of BMP2-sequesterin heparin sites within the hydrogel. Those sites, even when occupied with BMP2, will not drastically change the solution concentration of BMP2 but will dramatically alter the local concentration within the gels and we hypothesized on the encapsulated hMSC osteogenic differentiation. To test this hypothesis, the influence of the gel functionality on encapsulated hMSC osteogenic differentiation was examined.
Figure 4.
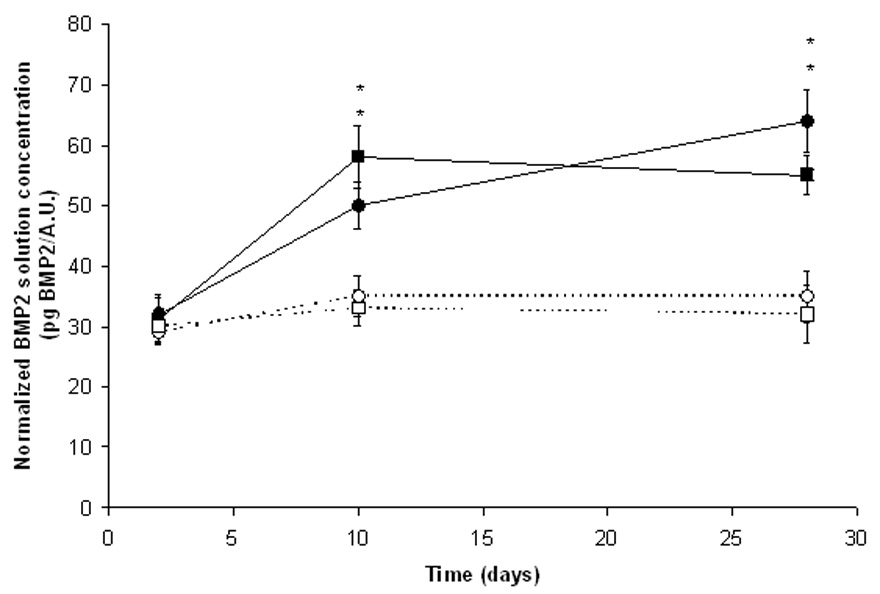
BMP2 production normalized to levels of metabolic activity (AlamarBlue) of hMSCs encapsulated in RGDS-functionalized hydrogels in the absence of fluvastatin (--○--) or with releasing fluvastatin (–●–) or encapsulated in heparin-functionalized hydrogels in the absence of fluvastatin (--□--) or with releasing fluvastatin (–■–), n=4 samples/condition. * p<0.05 of sample versus control (same chemistry but no delivered fluvastatin) at that time.
To further test the complex regulation and signaling provided by the heparin-functionalized, fluvastatin-releasing hydrogels, osteopontin (OPN) and core binding factor α1 (CBFA1) gene expression profiles were analyzed for hMSCs encapsulated in four hydrogel formulations: RGDS-functionalized hydrogels; RGDS-functionalized, fluvastatin-releasing hydrogels; heparin-functionalized hydrogels; and heparin-functionalized, fluvastatin-releasing hydrogels. Gene expression results, normalized to GAPDH, are shown in Figure 5. OPN is expressed in developing bone cells during early stages of osteogenesis prior to mineralization,[28] whereas CBFA1 is a transcription factor implicated in osteogenic differentiation. Expression of CBFA1 is specific to the osteogenic lineage, and although it is present in chondrogenic mesenchymal stem cells, it will remain activated later in development only in osteogenic cells.[29] Therefore, upregulation of OPN and CBFA1 in hMSCs is indicative of osteogenic differentiation.
Figure 5.
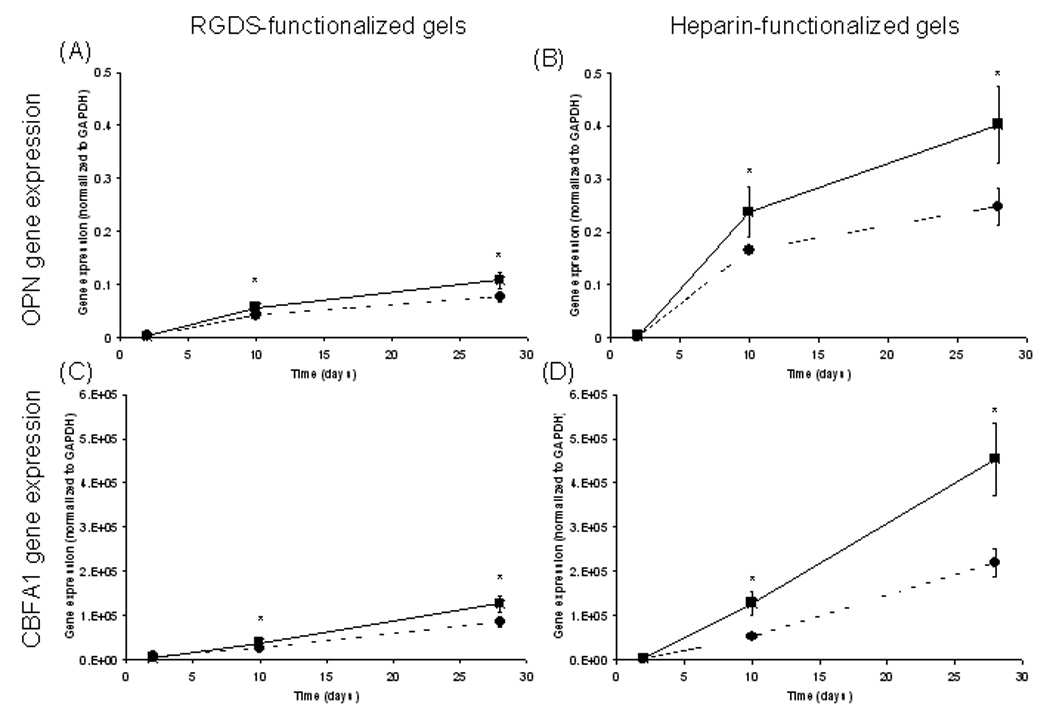
hMSC OPN (A+B) and CBFA1 (C+D) gene expression normalized to levels of GAPDH. hMSCs were cultured in the presence (–■–) or absence (--●--) of fluvastatin in RGDS-functionalized hydrogels, in RGDS-functionalized, fluvastatin-releasing hydrogels, in heparin-functionalized hydrogels, or in heparin-functionalized, fluvastatin-releasing hydrogels. n = 4 samples/condition. * p<0.05 of sample versus control (same chemistry but no delivered fluvastatin) at that time.
OPN (Figure 5A+B) gene expression was at the same level at day 2 and increased over the time course of the study for all treatments. About a 4-fold increase in gene expression was found at day 10 for cells encapsulated in fluvastatin-delivering gels (both RGDS- and heparin functionalities) over cells cultured in the absence of the fluvastatin-delivering monomer. OPN gene expression increased yet again at day 28, when cells encapsulated in the heparin-functionalized, fluvastatin-delivering system exhibited nearly a 6-fold greater gene expression than the RGDS-functionalized, fluvastatin delivering hydrogel. hMSCs cultured in the heparin-functionalized hydrogel in the absence of fluvastatin showed about 4-fold OPN gene expression than the RGDS-functionalized, fluvastatin-delivering hydrogels at day 28.
CBFA1 (Figure 5C+D) gene expression of hMSCs was at the same level, regardless of treatment, at culture day 2 and increased for all treatments over the 28 day culture period. The greatest increases in hMSC CBFA1 gene expression were found for heparin-functionalized, fluvastatin-releasing hydrogels where expression levels were 3-fold and 2-fold that of hMSCs cultured in heparin-functionalized hydrogels in the absence of delivered fluvastatin for culture days 10 and 28, respectively. In addition, hMSCs in heparin-functionalized hydrogels in the absence of delivered fluvastatin exhibited ~2-fold greater CBFA1 gene expression when compared with cells in RGDS-functionalized, fluvastatin-delivering hydrogels. Previously, heparin functionalized hydrogels, alone, were found to increase osteogenic differentiation hMSCs.[30] The importance of sulfated glycosaminoglycans for osteogenic differentiation is inherent to their ability to bind most of the growth factors (e.g., FGFs, TGF-β1, BMP2 and 4, IGF-II) involved in the regulation of cells of the osteoblast lineage.[19]
BMPs act on MSCs through heterodimeric type I/type II receptor complexes, which cause the activation of intracellular Smad proteins as well as mitogen-activated protein (MAP) kinase pathway.[31,32] Upon BMP binding to the type I/type II receptor, an intracellular signal is generated and translated to the nucleus through phosphorylation cascades to regulate the gene expression of Smad proteins.[33] These heterodimeric Smad complexes then affect regulation of specific gene transcription in MSCs[34] by binding to the CBFA1 transcription factor.[35] In the system presented here, upregulation of osteogenic differentiation is likely to proceed through this pathway, as the heparin functionality sequesters hMSC-produced BMP2, facilitating both BMP2 availability and subsequent BMP2 receptor activation.[36]
In addition, BMP production by osteoprogenitor cells or mature osteoblasts may be important for propagation of the BMP signal to neighboring cells during bone formation and regeneration, and may explain how hMSC-produced BMP2, as studied here, can initiate a sustained osteogenic response, although recombinant BMPs, once implanted, have only a short in vivo half-life.[37] These findings, taken together, outline the importance of autocrine signaling, as well as matrix interactions, in the osteogenic differentiation of progenitor cells.
3. Conclusions
The development of a multifunctional gel as a mimic of the native ECM with which the host interacts physiologically and that can respond appropriately to elicit a desired morphogenetic response is shown here. By inducing and sequestering BMP2 similar to the native bone matrix and allowing for autocrine signaling, an increased rate of differentiation is achieved. Heparin-functionalized, fluvastatin-releasing hydrogels may be useful in bone tissue engineering applications by providing an environment capable of bidirectional signaling, mimicking native bone healing and remodeling processes.
4. Experimental
Materials
Materials were purchased from Sigma-Aldrich unless otherwise specified.
Synthesis of macromers
The synthesis of PEG4600DM was performed as described elsewhere.[38] NMR analysis on the PEGDM revealed an average of 88% methacrylation based on the ratio of the integrals for the PEG protons and the terminal vinyl protons. 1H NMR (CDCl3): δ=3.6 (ether protons of PEG), 5.5 (terminal vinyl proton), 6.2 (terminal vinyl proton).
The synthesis of PEGMMA-nLA-fluvastatin was performed as described elsewhere.[39] Depending on the ratio of lactides to PEG526MMA (1:1, 1:2, or 1:3), different products containing 2 (PEG526MMA-2LA), 4 (PEG526MMA-4LA), or 6 (PEG526MMA-6LA) lactic acid repeats were synthesized. 1H NMR (CDCl3) analysis indicated the average number of LA units based on the ratio of the integrals for the PEG backbone protons and the LA backbone protons. The addition of fluvastatin (i.e., milligrams fluvastatin per milligrams total product) was based on the ratio of phenyl protons and terminal vinyl protons. 1H NMR (CDCl3): δ=3.6 (ether protons of PEG), 5.0 (proton on LA backbone), 5.5 (terminal vinyl proton), 6.2 (terminal vinyl proton), 7.3 (phenyl protons).
The amino acid sequence RGDS was synthesized using solid phase methods on an ABI 433A Peptide Synthesizer (Applied Biosystems) and following procedures for HBTU activation coupling. Lyophilized RGDS was coupled to acrylated-PEG3400-NHS following a previously reported method,[40] purified using dialysis (MWCO~1000), and coupling analyzed by the reduction of the primary amine group on the peptide using the fluorescamine assay.
Heparin (MW ~ 16 kDa) was methacrylated following a previous method.[26] 1H NMR analysis (in D2O) revealed an average of 6 and 22% methacrylation, respectively, for two separate reactions, based on the ratio of the integrals for the methacrylate protons (δ=1.9) and the hydroxyl protons (δ=2.0). The percentage of methacrylation refers to the number of methacrylate groups per heparin disaccharide unit. With exception to the native PAGE (Fig. 3), the 22% methacrylated heparin was utilized for all experiments herein. Chemical structures of PEG4600DM, PEG526MMA-nLA-fluvastatin, acrylated-PEG3400-RGDS, and methacrylated heparin are shown in Figure 6.
Figure 6.
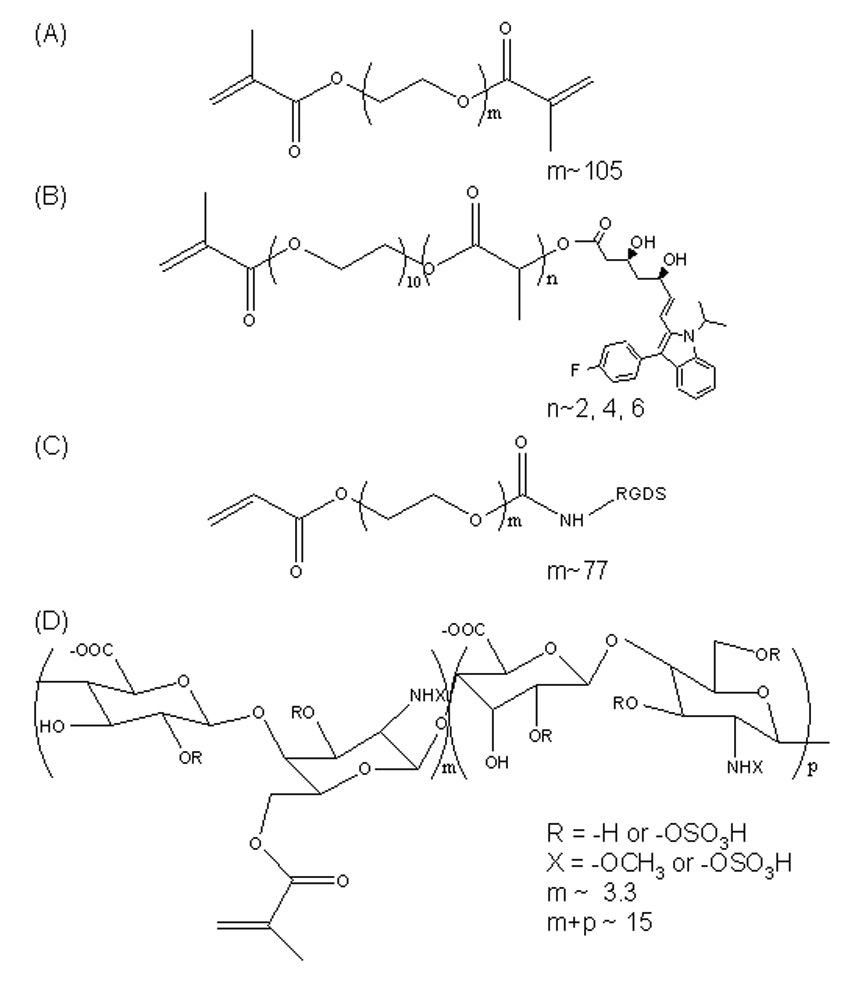
Structures of PEG4600DM (A), PEG526MMA-nLA-fluvastatin (B), acrylated-PEG3400-RGDS (C), and methacrylated heparin (D) used for hydrogel fabrication.
Copolymerization of PEG526MMA-LA-fluvastatin with PEG4600DM and degradation-controlled release of fluvastatin
PEG hydrogels with tethered fluvastatin were prepared by mixing 10 wt% PEGDM, 0.05 wt% I2959 (Ciba-Geigy) initiator, and PEG526MMA-nLA-fluvastatin in DMSO. A constant loading of ~12.5 mg/ml of PEG526MMA-nLA-fluvastatin was added to synthesize a series of gels used to examine the effect of lactic acid length on the fluvastatin release profile. The solution was polymerized under ultraviolet light (365 nm, ~5 mW/cm2) for 10 min in molds yielding disk-shaped hydrogels (d=5 mm, t=2 mm).
Each disk was added to 1 ml PBS, and fluvastatin release was followed over time at 37 °C. At each time point, the supernatant was collected and replaced with fresh PBS to ensure sink conditions. Fluvastatin release was quantified from the collected samples using high performance liquid chromatography (HPLC, Waters Delta Prep 4000). Briefly, 300 µl of each sample was injected into the HPLC inlet, and peaks were analyzed with an ultraviolet detector set at λ=310 nm (Waters 2487). The mobile phase consisted of 67% acetonitrile and 33% of 0.1 mM ammonium acetate, and the column was a Waters Nova-Pak C18 (3.9 mm × 150 mm). Fluvastatin concentration was calculated by integrating areas, and comparing this to a standard curve measuring samples of known concentration.
The uptake rate of fluvastatin by hMSCs was estimated for the closely related molecule, cortisol, due to the availability of an ELISA kit for cortisol and not for fluvastatin. hMSCs were cultured in the presence of 100 nM of cortisol. The media was sampled every 2–6 hours over 48 hours and analyzed via an ELISA for the residual concentration of cortisol in the media. Through a mass balance, the uptake rate of cortisol was estimated for hMSCs. Accounting for differences in the uptake rate and release rate (Figure 1), the concentration of fluvastatin within the hydrogel was estimated with respect to time.
Interactions of methacrylated heparin with BMP
Methacrylated heparin with different degrees of methacrylate modification and native heparin (as a control) were dissolved in PBS and combined with 25 µg/ml BMP2. These solutions were diluted 1:2 with native PAGE sample buffer (0.0625 M Tris-HCl pH 6.8, 30% (v/v) glycerol, and 0.1% (w/v) bromophenol blue in diH2O) and electrophoresed on a 4–15% Ready-Gel precast polyacrylamide Tris-HCl gels (Bio-Rad) using a vertical electrophoresis system (Mini-Protean II, Bio-Rad) at 150 V in native PAGE running buffer (3 g Tris base and 14.4 g glycine per 1 L diH2O). BMP2 bands were detected with a BioSafe Coomassie Stain (Bio-Rad), and band intensity was quantified using Kodak 1D software. BMP2 bands indicate free or unbound BMP2, while the BMP2 complexed with heparin appears as a higher molecular weight band. Results are reported as the difference in BMP2 band intensity of the BMP2 + methacrylated heparin sample from pure BMP2 divided by the difference in band intensity of the BMP2 + heparin sample from pure BMP2.
The ability of the PEG:heparin copolymer hydrogels to non-covalently interact with BMP2 was determined. First, EDAC/sulfo-NHS coupling was used to fluorescently label a fraction of the remaining carboxylic acid groups of the methacrylated heparin. Oregon green cadaverine (Pierce), EDAC (Sigma), and sulfo-NHS (Pierce) were all added at 0.1 molar excess of originally available carboxylic acid groups of the heparin in 0.1 M sodium phosphate buffer and allowed to react on a shaker at room temperature and in the dark for 2 h. Unreacted Oregon green was removed through dialysis (MWCO = 1000). Hydrogels were fabricated from a solution containing 10 wt% of the PEG4600DM monomer with or without 0.1 mM of the labeled, methacrylated heparin in PBS and 0.05 wt% I2959. Solutions were polymerized in a sterile mold (d=5 mm, t=2 mm) with ~4 mW/cm2 of 365 nm. After fabrication, hydrogels were swelled over two weeks in a BMP2 solution (1 µg/ml in 0.1% sodium azide-containing PBS, recombinant human BMP2 from R&D Systems) while shaking. After the incubation period, which is sufficiently long to attain complete diffusion of the BMP2, gels were washed with copious amounts of PBS and fixed in 10% formalin overnight, transferred to 22 wt% sucrose for 72 hours, frozen in Cryo-gel (Instrumedics, Inc.), and cryosectioned (10 µm sections). The slides were blocked in 10% normal goat serum and 0.5% BSA for 30 min, incubated in primary antibodies (mouse anti-human BMP2, R&D Systems) at 1:1000 for 4 h, then incubated in secondary antibody (goat anti-mouse Alexa Fluor 633, Molecular Probes) at 1:750 for 2 h. Finally, the sections were mounted with Vectashield (Vector Labs) and imaged using conventional fluorescence microscopy (Nikon Eclipse TE300 and associated SPOT software).
hMSC cell culture
hMSCs were purchased from Cambrex and cultured in growth medium: low-glucose Dulbecco’s modified eagle medium (Gibco) supplemented with 10% FBS (Invitrogen), 1% penicillin/streptomycin (Gibco), 0.25% gentamicin (Gibco), and 0.25% fungizone (Gibco). hMSCs at passage 3 were used in these studies.
hMSC encapsulation
All hydrogels were formulated by dissolving PEG4600DM in PBS to achieve a final monomer concentration of 10 wt% and 0.05 wt% I2959 was added. hMSCs were combined with sterile macromer/initiator solutions (with 5 mM of acrylated-PEG3400-RGDS, with 0.1 mM methacrylated heparin, and/or 0.2 µg/ml of fluvastatin monomer) at a concentration of 25 × 106 cells/ml and photoencapsulated using a longwave ultraviolet lamp (UVP, model XX-20) at an intensity of ~4 mW/cm2 for 10 min.[41] Without any adhesive ligands, hMSCs are known to undergo apoptosis after < 2 weeks of culture.[23] Therefore, RGDS was incorporated into some of the gels to maintain hMSC viability.[23] Heparin has also been found to maintain the viability of hMSCs.[30] The resulting cell-hydrogel constructs were cultured in growth medium. Growth medium was changed every 2–3 days including 24 hours prior to each timepoint in the studies outlined herein.
Encapsulated hMSC BMP2 production
At various time points, (2, 10, and 28 days), media surrounding the encapsulated cells was exchanged. After 24 hours, the media was collected and stored at −80 °C until assayed. The stored media was thawed and assayed for BMP2 production with an ELISA (Quantikine, R & D Systems) using standard manufacturer’s instructions and normalized to a measure of cell viability, metabolic activity.
Metabolic activity of the encapsulated hMSCs, as analyzed by the AlamarBlue assay (Serotec), was used as an indirect measure of the number of living cells. At 2, 10, and 28 days, a 10 vol% solution of AlamarBlue in media was added to the constructs. Active mitochondria convert resazurin, the active ingredient in AlamarBlue, to resorufin, a fluorescent molecule. After 4 hours of incubation, the media/AlamarBlue solution was analyzed for fluorescence (Ex/Em=560/590 nm) with a plate reader (Victor2, Perkin Elmer). AlamarBlue readings were utilized to normalize BMP2 production in order to eliminate error due to cell number fluctuations.
Encapsulated hMSCs ALP production
ALP production was measured using an assay based on the change in absorbance of o-nitrophenol as it is enzymatically cleaved by ALP. Cell/hydrogel constructs were removed from in vitro culture at 2, 10, and 28 days, lysed with Cell-glo lysis buffer (Promega), and manually homogenized. The assay was performed by combining 100 µl of sample with 100 µl of the ALP substrate. By performing the assay using known concentrations of ALP in parallel with the samples, the concentrations of ALP for the samples were calculated and normalized to metabolic activity.
Encapsulated hMSC gene expression
Gene expression of encapsulated hMSCs was analyzed using reverse transcription polymerase chain reaction (RT-PCR). After 2, 10, and 28 days, constructs were removed from culture and rinsed three times with PBS. Total RNA was isolated using the TRI reagent and standard manufacturer’s protocols. After allowing the RNA pellet to dry, it was resuspended in nuclease-free water, and any residual genomic DNA in the samples was digested (DNase I, Invitrogen). RNA was then quantified using the RiboGreen assay (Invitrogen) based on the manufacturer’s instructions.
Reverse transcription was performed using the iScript cDNA Synthesis Kit (Bio-Rad). A 15 ng total RNA sample was used for the single strand cDNA synthesis. The reverse transcription reaction was incubated at 25 °C for 5 min, 42 °C for 30 min, and terminated at 85 °C for 5 min. PCR was conducted using the iCycler Real-Time PCR machine (Bio-Rad), and primers and probes were designed using the Beacon Designer primer design program. Primers (Invitrogen) and probes (Integrated DNA Technologies) for OPN (sense primer- 5’-ATTCTGGGAGGGCTTGGTTG-3’, anti-sense primer- 5’-TCTGGTCCCGACGATGCT-3’, probe- 5’-CTCTGCCTCCTCCTGCTGCTGCTG-3’), CBFA1 (sense primer- 5’-GGTATGTCCGCCACCACTC-3’, anti-sense primer- 5’-TGACGAACTCCCATAGTAGAGATA-3’, probe- 5’- CTACCACACCTACCTGCCACCACC-3’), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH: sense primer- 5’-GCAAGAGCACAAGAGGAAGAG-3’, anti-sense primer- 5’AAGGGGTCTACATGGCAACT-3’, probe- 5’-ACCCTCACTGCTGGGGAGTCC-3’) were used. The following PCR parameters were utilized: 95 °C for 90 sec followed by 45 cycles of 95 °C for 30 sec and 55 °C for 60 sec. Threshold cycle (CT) analysis was used to quantify PCR products, normalized to GAPDH.
Statistical analysis
Statistical analysis was performed using a one-way ANOVA with α=0.05. Data are presented as mean ± one standard deviation.
Figure 7.
Controlled delivery of fluvastatin, a small molecule that increases BMP2 production, combined with heparin functionalities (green) incorporated into a poly(ethylene glycol)-based hydrogel enables local sequestering of cell-produced BMP2 (red) to temporally and spatially regulate human mesenchymal stem cell osteogenesis.
Acknowledgements
This work was supported by the National Institute of Health (DE016523) and the Department of Education’s GAANN program and a NSF-GRF (fellowships to DSWB).
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Ramamurthy A, Vesely I. Biomaterials. 2005;26:999. doi: 10.1016/j.biomaterials.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 2.Burdick JA, Anseth KS. Biomaterials. 2002;23:4315. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman AS. Adv. Drug Del. Rev. 2002;43:3. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 4.Lee KY, Mooney DJ. Chem. Rev. 2001;101:1869. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 5.Babensee JE, McIntire LV, Mikos AG. Pharm. Res. 2000;17:497. doi: 10.1023/a:1007502828372. [DOI] [PubMed] [Google Scholar]
- 6.Lutolf MP, Hubbell JA. Nat. Biotechnol. 2004;23:47. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 7.Behnam K, Phillips ML, Silva JDP, Brochmann EJ, Duarte MEL, Murray SS. J. Orthop. Res. 2005;23:175. doi: 10.1016/j.orthres.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Signas C, Raucci G, Jonsson K, Lindred P, Anantharamaiah GM, Hook M, Lindberg M. Proc. Natl. Acad. Sci. U.S.A. 1989;86:699. doi: 10.1073/pnas.86.2.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lutolf MP, Weber FE, Schmoekel HG, Schense JC, Kohler T, Muller R, Hubbell JA. Nat. Biotechnol. 2003;21:513. doi: 10.1038/nbt818. [DOI] [PubMed] [Google Scholar]
- 10.Richardson DM, Peters MC, Ennet AB, Mooney DJ. Nat. Biotechnol. 2001;19:1029. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- 11.Francheschi RT. Crit. Rev. Oral. Biol. Med. 1999;10:40. doi: 10.1177/10454411990100010201. [DOI] [PubMed] [Google Scholar]
- 12.Schonherr E, Hausser HJ. Dev. Immunol. 2000;7:89. doi: 10.1155/2000/31748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roghani M, Mansukhani A, Dellera P, Bellosta P, Basilico C, Rifkin DB, Moscatelli D. J. Biol. Chem. 1994;269:3976. [PubMed] [Google Scholar]
- 14.Lane JM. J. Bone Joint Surg. 2000;83A:S161. [PubMed] [Google Scholar]
- 15.Reddi AH, Huggins CB. Proc. Natl. Acad. Sci. U.S.A. 1972;69:1601. doi: 10.1073/pnas.69.6.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Groeneveld EHJ, Burger EH. Eur. J. Endocrinol. 2000;142:9. doi: 10.1530/eje.0.1420009. [DOI] [PubMed] [Google Scholar]
- 17.Service RF. Science. 2000;289:1498. doi: 10.1126/science.289.5484.1498. [DOI] [PubMed] [Google Scholar]
- 18.Mundy G, Garrett R, Harris S, Chan J, Chen D, Rossini G, Boyce B, Zhao M, Gutierrez G. Science. 1999;286:1946. doi: 10.1126/science.286.5446.1946. [DOI] [PubMed] [Google Scholar]
- 19.Canalis E. Skeletal Growth Factors. Philadelphia: Lippincott Williams & Williams; 2000. [Google Scholar]
- 20.Metters AT, Anseth KS, Bowman CN. Polymer. 2000;41:3993. [Google Scholar]
- 21.Bryant SJ, Anseth KS. J. Biomed. Mater. Res. A. 2003;64A:70. doi: 10.1002/jbm.a.10319. [DOI] [PubMed] [Google Scholar]
- 22.Carvalheira AF, Pearse AGE. Histochem. Cell Biol. 1967;8:175. doi: 10.1007/BF00306470. [DOI] [PubMed] [Google Scholar]
- 23.Nuttelman CR, Tripodi MC, Anseth KS. Matrix Biol. 2005;24:208. doi: 10.1016/j.matbio.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Stein GS, Lian JB. Endocrine Rev. 1993;14:424. doi: 10.1210/edrv-14-4-424. [DOI] [PubMed] [Google Scholar]
- 25.Takada T, Katagiri T, Ifuku M, Morimura N, Kobayashi M, Hasegawa K, Ogamo A, Kamijo R. J. Biol. Chem. 2003;278:43229. doi: 10.1074/jbc.M300937200. [DOI] [PubMed] [Google Scholar]
- 26.Masters KS, Shah DN, Walker G, Leinwand LA, Anseth KS. J. Biomed. Mater. Res. A. 2004;71A:172. doi: 10.1002/jbm.a.30149. [DOI] [PubMed] [Google Scholar]
- 27.Benoit DSW, Anseth KS. Acta Biomater. 2005;1:461. doi: 10.1016/j.actbio.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Mark MP, Prince CW, Oosawa T, Gay S, Bronckers AL, Butler WT. J. Histochem. Cytochem. 1987;35:707. doi: 10.1177/35.7.3295029. [DOI] [PubMed] [Google Scholar]
- 29.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Cell. 1997;89:747. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 30.Benoit DSW, Durney AR, Anseth KS. Biomaterials. 2007;1:66. doi: 10.1016/j.biomaterials.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 31.Lee KS, Hong SH, Bae SC. Oncogene. 2002;21:7156. doi: 10.1038/sj.onc.1205937. [DOI] [PubMed] [Google Scholar]
- 32.Kloen P, Di Paola M, Borens O, Richmond J, Perino G, Helfet DL, Goumans MJ. Bone. 2003;33:362. doi: 10.1016/s8756-3282(03)00191-1. [DOI] [PubMed] [Google Scholar]
- 33.Marrony S, Bassilana F, Seuwen K, Keller H. Bone. 2003;33:426. doi: 10.1016/s8756-3282(03)00195-9. [DOI] [PubMed] [Google Scholar]
- 34.Hu PP, Datto MB, Wang XF. Endocr Rev. 1998;19:349. doi: 10.1210/edrv.19.3.0333. [DOI] [PubMed] [Google Scholar]
- 35.Hanai J, Chen LF, Kanno T, Ohtani-Fujita N, Kim WY, Guo WH, Imamura T, Ishidou Y, Fukuchi M, Shi MJ, Stavnezer J, Kawabata M, Miyazono K, Ito Y. J. Biol. Chem. 1999;274:31577. doi: 10.1074/jbc.274.44.31577. [DOI] [PubMed] [Google Scholar]
- 36.Xiao G, Gopalakrishnan R, Jian D, Reith E, Benson MD, Franceschi KT. J. Bone Miner. Res. 2002;17:101. doi: 10.1359/jbmr.2002.17.1.101. [DOI] [PubMed] [Google Scholar]
- 37.Giannobile WV. Bone. 1996;19:23S. doi: 10.1016/s8756-3282(96)00127-5. [DOI] [PubMed] [Google Scholar]
- 38.Sawhney AS, Pathak CP, Hubbell JA. Macromolecules. 1993;26:581. [Google Scholar]
- 39.Benoit DSW, Nuttelman CR, Collins SD, Anseth KS. Biomaterials. 2006;36:6102. doi: 10.1016/j.biomaterials.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 40.Hern DL, Hubbell JA. J. Biomed. Mater. Res. 1998;39:266. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 41.Bryant SJ, Nuttelman CR, Anseth KS. J. Biomat. Sci. Polym. Ed. 2000;11:439. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]