

Enzymes in the non-heme diiron super-family perform key biological functions ranging from radical generation for DNA biosynthesis to regio- and stereo-selective oxidation of hydrocarbons at structurally similar active sites.1 The features that determine the outcome of O2 activation at these sites are unclear and are currently of great interest.
In the catalytic cycles of these enzymes, a diiron(II) center reacts with O2 to generate reactive intermediates that have thus far been observed in only a few cases.2,3 Among members of the sub-family of bacterial multi-component monooxygenase (BMM) enzymes,4 soluble methane monooxygenase is the only one for which intermediates have been observed. In this system, the reduced hydroxylase component, MMOH, reacts with O2 to generate spectroscopically characterized diiron(III) and diiron(IV) intermediates.3a,b Both of these species can oxidize hydrocarbons,3 which can enter the active site of MMOH via a string of hydrophobic pockets.5 In contrast, a large substrate-access channel is present in toluene/o-xylene monooxygenase hydroxylase (ToMOH), affording more exposed diiron sites.6 We postulated that buffer components might quench the reactive intermediates in ToMOH and that mutations to block access to the diiron centers would facilitate the observation of intermediates. Following this strategy, we generated an I100W mutant in the α-subunit of the ToMOH by site-directed mutagenesis and discovered an oxygenated diiron intermediate that oxidizes the newly supplied W100 residue.
The reaction of the chemically-reduced diiron(II) form of I100W ToMOH in the presence of the coupling protein, ToMOD, with O2-saturated buffer was monitored by stopped-flow UV/visible spectroscopy. A transient species with an absorption band centered at 500 nm appeared with formation and decay rate constants kform = 0.804 ± 0.001 s-1 and kdecay = 0.054 ± 0.002 s-1 and an extinction coefficient, ε500, of 1.5 ± 0.2 × 103 M-1cm-1 (Figure S1). Based on these optical properties we tentatively assigned this species as a neutral tryptophan radical, W•.7
To investigate further the reaction of I100W ToMOH with O2, rapid-freeze quench (RFQ) samples were prepared at various aging times and characterized by EPR and Mössbauer spectroscopy. A transient g = 2.00 EPR signal with fine structure (Figure 1) was observed, as expected for a W• species. The intensity of this signal maximized at ∼3.5 s, consistent with the kinetics of formation of the putative W• radical from the UV/visible stopped-flow study. The EPR signal is easily saturated at 10 K and can be detected up to 50 K without any broadening, properties typical of an organic radical. When 57Fe-enriched I100W was used in the reaction, however, hyperfine broadening was observed (Figure 1). This result unambiguously establishes that the diiron cluster contributes to this EPR signal. The observed hyperfine broadening is similar to that observed for the g = 2.00 EPR signals of the spin-coupled diiron(III,IV) X-WH+• in the ribonucleotide reductase R2 subunit (R2)2g and of X-Y• in the R2-W48F variant.2h The optical absorption and EPR spectral data therefore suggest a spin-coupled intermediate comprising W• and a paramagnetic diiron cluster. Mössbauer data (Figure 2) confirmed the spin-coupled nature of this intermediate and revealed the oxidation state of the paramagnetic diiron center to be mixed-valent diiron(III,IV).
Figure 1.
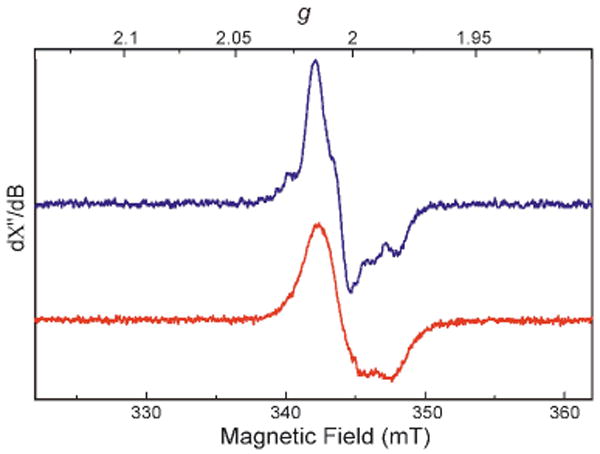
EPR spectra of the diiron(III,IV)-W• intermediate generated in the reaction of reduced ToMOH-I100W with O2. The reaction was quenched at 4 s using natural-abundant diiron(II) protein (blue) or at 3.5 s using 57Fe-enriched protein (red).
Figure 2.
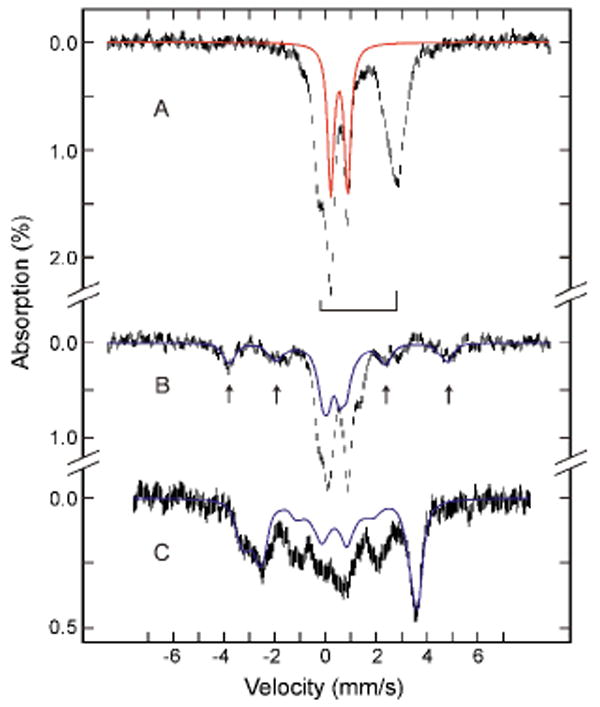
Mössbauer spectra of samples freeze-quenched at 70 ms (A) and 3.5 s (B and C) in the reaction of diiron(II) I100W ToMOH with O2. The data were recorded at 4.2 K in a parallel applied field of 50 mT (A and B) or 8 T (C). For the spectra in B and C, the diiron(II) contributions (50% of the total Fe absorption) were removed from the raw data for clarity. Simulated spectra of the peroxo intermediate (A, red line) and of the X-Y• species in R2-W48F (B and C, blue lines) are superimposed and scaled to 30% of the total absorption of that sample. The bracket (A) indicates the diiron(II) protein. Raw data are denoted as vertical bars.
Figure 2B displays the 4.2-K Mössbauer spectrum of the 3.5 s RFQ sample recorded with a 50-mT magnetic field applied parallel to the γ-beam. A paramagnetic spectral component with features (arrows) similar to the diiron(III,IV) intermediate, X, in R22g,h was observed. Analysis of the spectrum indicates that this component accounts for 15 ± 2 % of the total Fe absorption. The spectrum of this sample recorded with a strong applied field of 8 T (Figure 2C) showed a two-fold increase in the intensity of the paramagnetic component to 30 ± 3 % of total Fe absorption. This field-dependent increase of the paramagnetic absorption indicates that the S = 1/2 diiron(III,IV) cluster is weakly spin-coupled to a nearby S = 1/2 paramagnet.2h For a weakly-coupled system (J < 1 cm-1) in a weak applied field of 50 mT, the energy separations of the four electronic levels are small, whereas in a strong applied field of 8 T, the Zeeman interaction increases the energy separations between these levels. At 4.2 K and 50 mT, all four levels are nearly equally populated and the spectrum contains two equal-intensity components: a paramagnetic component (two MS = ±1 sublevels of the triplet state) superposed on a “diamagnetic” component (the singlet state and the MS = 0 sublevel of the triplet state). In contrast, at 4.2 K and 8 T, only the lowest MS = − 1 sublevel is populated and the “diamagnetic” component disappears, doubling the intensity of the paramagnetic component.2h We compared a simulated spectrum of the X-Y• species in R2-W48F (Figure 2) to the spectrum of the 3.5 s sample and the agreement is remarkable and consistent with our assignment. The observed discrepancy in the central region of the spectrum indicates the presence of another intermediate (vide infra) and a diiron(III) product; in addition, the Mössbauer parameters for the I100W diiron(III,IV) may be slightly different than those of X in R2-W48F.
Time-dependent EPR experiments indicate that the diiron(II) g = 16 signal decays more rapidly (∼ 60 s-1) than the formation rate of the diiron(III,IV)-W• g = 2 signal (data not shown). An EPR-silent precursor to the diiron(III,IV)-W• species therefore must exist, the concentration of which would maximize at a reaction time of ∼100 ms. Consistent with this conclusion, the Mössbauer spectrum of a sample quenched at 70 ms (Figure 2A) shows a sharp quadrupole doublet accounting for approximately 30% of the total Fe absorption, as well as the broad quadrupole doublet of the initial diiron(II) protein. The parameters of the sharp quadrupole doublet (δ = 0.54 ± 0.02 mm/s and ΔEQ = 0.66 ± 0.03 mm/s) are typical of high-spin FeIII species. Spectra recorded with strong applied fields and the EPR data indicate that this doublet arises from an antiferromagnetically coupled diiron(III) cluster. In the 3.5 s sample, the intensity of this doublet is diminished substantially and accounts for less than 7% of the Fe absorption, establishing that this diiron(III) intermediate decays during formation of the spin-coupled diiron(III,IV)-W• species. This diiron(III) species is the first observable intermediate after reaction with O2 and, based on work with other non-heme diiron proteins,2 we tentatively assign this species as a peroxodiiron(III) unit. The Mössbauer parameters for this species in I100W ToMOH, however, differ from those reported for the peroxodiiron(III) intermediates in other carboxylate-bridged diiron proteins, for which δ > 0.6 mm/s and ΔEQ > 1.00 mm/s.8 Furthermore, the peroxo species in these proteins exhibit optical absorptions with λmax greater than 650 nm2d-g and no such features are observed for I100W ToMOH. Peroxodiiron(III) synthetic complexes with a planar μ-1,2-peroxo coordination mode and a second bridging ligand9 have δ and ΔEQ values similar to those reported here for the putative peroxodiiron(III) intermediate. The mechanism by which the diiron(III,IV) center and W• radical form in I100W ToMOH may be similar to that proposed for the generation of X and Y• in R2.2h Preliminary evidence indicates that decay of the diiron(III,IV) and W• species may proceed via hydroxylation of W100 (unpublished results).
The detection of an early transient for I100W ToMOH with Mössbauer parameters atypical of those for peroxodiiron(III) species and no obvious visible absorption band led us to examine the time-dependent Mössbauer spectra of the reaction of the wild-type enzyme with O2. The results indicate unambiguously the accumulation of a transient with a spectrum identical to that observed for the diiron(III) intermediate in the I100W ToMOH variant (unpublished results). Detailed spectroscopic investigations of this intermediate are in progress. Peroxo3c,10 and hydroperoxo11 species are proposed to be reactive intermediates in metalloenzymes, and this putative peroxo adduct may be the active oxidant for aromatic hydroxylation in the ToMO systems.
Supplementary Material
Supporting Information Available. Experimental details for sample preparation, stopped-flow UV/visible data, and EPR acquisition parameters. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by NIH Grants GM32134 (to SJL) and GM 47295 (to BHH). LJM was supported in part by a fellowship from the Martin Society.
References
- 1.Kurtz DM., Jr J Biol Inorg Chem. 1997;2:159–167. [Google Scholar]
- 2.(a) Lee SK, Fox BG, Froland WA, Lipscomb JD, Münck E. J Am Chem Soc. 1993;115:6450–6451. [Google Scholar]; (b) Liu KE, Wang D, Huynh BH, Edmonsdon DE, Salifoglou A, Lippard SJ. J Am Chem Soc. 1994;116:7465–7466. [Google Scholar]; (c) Burdi D, Willems JP, Riggs-Gelasco P, Antholine WE, Stubbe J, Hoffman BM. J Am Chem Soc. 1998;120:12910–12919. [Google Scholar]; (d) Moënne-Loccoz P, Baldwin J, Ley BA, Loehr TM, Bollinger JM., Jr Biochemistry. 1998;37:14659–14663. doi: 10.1021/bi981838q. [DOI] [PubMed] [Google Scholar]; (e) Pereira AS, Small W, Krebs C, Tavares P, Edmondson DE, Theil EC, Huynh BH. Biochemistry. 1998;37:9871–9876. doi: 10.1021/bi980847w. [DOI] [PubMed] [Google Scholar]; (f) Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Biochemistry. 1998;37:14664–14671. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]; (g) Baldwin J, Krebs C, Ley BA, Edmondson DE, Huynh BH, Bollinger JM., Jr J Am Chem Soc. 2000;122:12195–12206. [Google Scholar]; (h) Krebs C, Chen S, Baldwin J, Ley BA, Patel U, Edmonsdon DE, Huynh BH, Bollinger JM., Jr J Am Chem Soc. 2000;122:12207–12219. [Google Scholar]
- 3.(a) Nesheim JC, Lipscomb JD. Biochemistry. 1996;35:10240–10247. doi: 10.1021/bi960596w. [DOI] [PubMed] [Google Scholar]; (b) Valentine AM, Stahl SS, Lippard SJ. J Am Chem Soc. 1999;121:3876–3887. [Google Scholar]; (c) Beauvais LG, Lippard SJ. J Am Chem Soc. 2005;127:7370–7378. doi: 10.1021/ja050865i. [DOI] [PubMed] [Google Scholar]
- 4.Leahy JG, Batchelor PJ, Morcomb SM. FEMS Micro Rev. 2003;27:449–479. doi: 10.1016/S0168-6445(03)00023-8. [DOI] [PubMed] [Google Scholar]
- 5.(a) Rosenzweig AC, Brandstetter H, Whittington DA, Nordlund P, Lippard SJ, Frederick CA. Proteins. 1998;29:141–152. [PubMed] [Google Scholar]; (b) Whittington DA, Rosenzweig AC, Frederick CA, Lippard SJ. Biochemistry. 2001;40:3476–3482. doi: 10.1021/bi0022487. [DOI] [PubMed] [Google Scholar]
- 6.Sazinsky MH, Bard J, Di Donato A, Lippard SJ. J Biol Chem. 2004;279:30600–30610. doi: 10.1074/jbc.M400710200. [DOI] [PubMed] [Google Scholar]
- 7.Aubert C, Vos MH, Mathis P, Eker APM, Brettel K. Nature. 2000;405:586–590. doi: 10.1038/35014644. [DOI] [PubMed] [Google Scholar]
- 8.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Angew Chem Int Ed. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 9.(a) Menage S, Brennan BA, Juarez-Garcia C, Münck E, Que L., Jr J Am Chem Soc. 1990;112:6423–6425. [Google Scholar]; (b) Dong Y, Yan S, Young VG, Jr, Que L., Jr Angew Chem Int Ed. 1996;35:618–620. [Google Scholar]; (c) Zhang X, Furutachi H, Fujinami S, Nagatomo S, Maeda Y, Watanabe Y, Kitagawa T, Suzuki M. J Am Chem Soc. 2004;127:826–827. doi: 10.1021/ja045594a. [DOI] [PubMed] [Google Scholar]
- 10.(a) Decker H, Dillinger R, Tuczek F. Angew Chem Int Ed. 2000;39:1591–1594. doi: 10.1002/(sici)1521-3773(20000502)39:9<1591::aid-anie1591>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]; (b) Baldwin J, Voegtli WC, Khidekel N, Moënne-Loccoz P, Krebs C, Pereira AS, Ley BA, Huynh BH, Loehr TM, Riggs-Gelasco PJ, Rosenzweig AC, Bollinger JM., Jr J Am Chem Soc. 2001;123:7017–7030. doi: 10.1021/ja002114g. [DOI] [PubMed] [Google Scholar]; (c) Yamazaki Si, Morioka C, Itoh S. Biochemistry. 2004;43:11546–11553. doi: 10.1021/bi048908f. [DOI] [PubMed] [Google Scholar]
- 11.(a) Vatsis KP, Coon MJ. Arch Biochem Biophys. 2002;397:119–129. doi: 10.1006/abbi.2001.2665. [DOI] [PubMed] [Google Scholar]; (b) Jin S, Bryson TA, Dawson JH. J Biol Inorg Chem. 2004;9:644–653. doi: 10.1007/s00775-004-0575-7. [DOI] [PubMed] [Google Scholar]; (c) Nam W, Ryu YO, Song WJ. J Biol Inorg Chem. 2004;9:654–660. doi: 10.1007/s00775-004-0577-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available. Experimental details for sample preparation, stopped-flow UV/visible data, and EPR acquisition parameters. This material is available free of charge via the Internet at http://pubs.acs.org.