

Abstract
Development of a safe and effective vaccine for induction of mucosal immunity to the human immunodeficiency virus (HIV) envelope glycoprotein (Env, gp160) represents the best hope for containing the spread of an HIV epidemic worldwide. The highly attenuated modified vaccinia virus Ankara (MVA) is a laboratory virus well suited as a safe vaccine vector. However, the presence of pre-existing immunity to Vaccinia virus in the adult population represents a hindrance that limits the application of the MVA vector for inducing immunity to HIV antigens. Here, cationic liposomes were covalently attached to the surface of recombinant MVA expressing the HIV-1 strain IIIB Env glycoprotein and β-galactosidase (MVAIIIB/β-gal) using tresylmonomethoxypolyethylene glycol (TMPEG) grafted into a lipid membrane without compromising viral infectivity in vitro and in vivo. The orally administered MVAIIIB/β-gal–TMPEG/liposome complexes were capable of delivering the transgenes to mucosal tissues in mice with pre-existing poxvirus immunity based on β-galactosidase gene expression in intestinal tissues measured 18 h after infection. Importantly, the MVAIIIB/β-gal–TMPEG/liposome complexes enhanced Env-specific cellular and humoral immune responses in the mucosal and systemic tissues after repeated oral immunization of BALB/c mice. This approach may prove useful for induction of protective immunity against infectious diseases and cancer in populations with pre-existing immunity to vaccinia from smallpox vaccination.
INTRODUCTION
Induction of a human immunodeficiency virus (HIV) immune response in mucosal as well as systemic compartments of the immune system is a critical feature of an effective AIDS vaccine. As over 90% of HIV-seropositive individuals acquire the infection via the mucosal surface (Kresina & Mathieson, 1999), this implies that HIV-specific immunity at mucosal sites is critical for the control of infection in many individuals exposed to the virus. Induction of mucosal immunity in the gut has been demonstrated with orally delivered DNA vaccines encapsulated in microparticles made of biologically erodible polymer (Hedley et al., 1998; Jones et al., 1997; Kaneko et al., 2000). Encapsulation not only protects the plasmid DNA from the gastric environment, but, due to the propensity of polymeric spheres for antigen-presenting cell uptake, also increases intracellular delivery of DNA designated to activate immune responses. Separate studies performed with orally administered recombinant viral vaccines have demonstrated that recombinant vaccinia virus (rVV) administered by the oral route generates mucosal and systemic immune responses to the recombinant products as well as antigens of the viral vector (Bender et al., 1996; Gherardi & Esteban, 1999). Independent experiments performed by Belyakov et al. (1998) have demonstrate induction of a mucosal cytotoxic T-lymphocyte response by intrarectal immunization with MVA expressing the HIV Env glycoprotein and indicated that the non-replicating recombinant MVA may be at least as effective for mucosal immunization as replicating rVV.
A number of more-attenuated VV-derived strains, including MVA (Mayr et al., 1978; Sutter & Moss, 1992, 1995), and strains attenuated by deletion of some non-essential genes such as the NYVAC strain (Tartaglia et al., 1992), have been shown to be safe in healthy as well as in immunocompromised animals. However, there are data suggesting that pre-existing vaccinia immunity, such as that occurring in a large proportion of the adult population because of smallpox vaccination, limits the effectiveness of rVV as a vaccine (Belyakov et al., 1999). A possible solution to this dilemma has been suggested by studies showing that mucosal vaccination could overcome the barrier to rVV immunization caused by pre-existing poxvirus immunity due to the independent compartmentalization of the mucosal versus the systemic immune systems and the asymmetric trafficking of lymphocytes between them (Belyakov et al., 1999). However, this approach has been challenged by studies showing that systemic immunization of mice and monkeys induced both systemic and mucosal immunity in the vaccinated animals (Belyakov et al., 2004; Fuller et al., 2002; Pal et al., 2006). Additional strategies to overcome the barrier of pre-existing immunity to viral vectors including a heterologous prime/boost immunization strategy with DNA vectors or adenoviruses used for priming followed by a booster with MVA (Gurunathan et al., 2000; Schneider et al., 1998) showed some efficacy in animal models. However, each of these approaches would eventually face limitations related to the generation of neutralizing antibodies to multiple vectors in the exposed individuals.
In an effort to overcome the barrier to poxvirus immunization caused by pre-existing immunity to rVV, we have been developing modifications of the recombinant MVA vector expressing the HIV Env glycoprotein and β-galactosidase (β-gal) genes. One such approach involves a covalent attachment of tresylmonomethoxypolyethylene glycol (TMPEG) to the viral capsid through the epsilon-terminal lysine residue (O’Riordan et al., 1999). Previous studies have shown that therapeutic peptides and proteins can be modified with polyethylene glycol (PEG) (Senior et al., 1991) and the half-life of stealth liposomes that carry exterior PEG chains is extended in the human bloodstream (t1/2 of up to 48 h; Zalipsky, 1993). Although the primary advantages involving PEG modification for proteins and peptides include reduced toxicity and enhanced delivery of encapsulated anti-cancer drugs at tumour sites (Lasic, 1998), one of the inherent problems is the potential for reduced bioactivity of the PEG conjugates. Recently, however, it has been shown that many of these limitations can be overcome by biological optimization using TMPEG (O’Riordan et al., 1999).
The present study explores whether cationic liposomes modified with TMPEG are capable of shielding MVAIIIB/β-gal from poxvirus-neutralizing antibodies in vitro and in vivo. We selected the oral route of immunization, as this method of vaccine delivery has been shown to protect against infection acquired via mucosal surfaces (Chen et al., 1998; Childers et al., 1994; Czerkinsky et al., 1999; Kaneko et al., 2000; McGhee & Mestecky, 1990; Mestecky & Eldridge, 1991). We showed that, after modification using the optimized PEGylation method with cationic liposome complexes, the MVAIIIB/β-gal–TMPEG/liposome complexes retained infectivity in vitro. Furthermore, the same complexes provided an appreciable level of protection in vivo and elicited Env-specific immune responses in mucosal and systemic tissues of BALB/c mice after repeated oral delivery. These results suggest that the MVAIIIB/β-gal vector can be modified for effective shielding against pre-existing poxvirus neutralizing antibodies for multiple deliveries.
METHODS
Reagents
TMPEG glycol and PEG were obtained from Sigma. 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC) were purchased from Avanti Polar Lipids. Lentinan (~500 kDa) was provided by Ajinomoto Central Laboratories.
Recombinant MVAIIIB/β-gal
Recombinant MVA expressing the HIV-1 IIIB Env glycoprotein and β-gal was generated using the pSC11 shuttle vector (a gift from Dr L. Eisenlohr, Thomas Jefferson University, Philadelphia, PA, USA). The gene encoding the HIV-1 IIIB Env glycoprotein was cloned into the SalI and NotI restriction sites of the pSC11 shuttle vector under the control of the VV early/late promoter P7.5 and the β-gal gene was driven by the strong VV late promoter, P11 (Kiszka et al., 2002). The MVAIIIB/β-gal vector was generated by homologous recombination using plasmid pSC11-env/β-gal and wild-type MVA (a gift from Drs B. Moss and L. Wyatt, Laboratories of Viral Diseases, National Institute of Allergy and Infectious Diseases, Bethesda, MD, USA) as described previously (Wyatt et al., 1995, 1996). The virus was selected by β-Gal screening in the presence of X-Gal or by immunostaining with the Env-specific mAb Chessie 8 (Abacioglu et al., 1994). After five consecutive rounds of plaque purification, recombinant virus was amplified in chicken embryo fibroblasts and purified by centrifugation on 24–40% sucrose gradients for 50 min at 26 000 g at 4 °C.
Liposome preparation
MVAIIIB/β-gal-associated liposomes were prepared by dissolving DOTAP, DOPE, DLPC and TMPEG in chloroform at various molar ratios as indicated in Table 1. The lipid monolayer that formed in glass pear-bottomed vials connected to a rotary evaporator (Labconco) was wetted with PBS and liposomes were prepared by intense vortex dispersion and 4 min of sonication. The liposome preparation (800 µl) was mixed with 1×107 p.f.u. MVAIIIB/β-gal in 200 µl PBS, incubated for 30 min at room temperature and sonicated for 1 min. The liposome–MVAIIIB/β-gal complexes were stabilized by the addition of 100 µg lentinan ml−1 prepared in PBS.
Table 1.
Effect of different liposomal and TMPEG formulations on MVAIIIB/β-gal infectivity of BHK-21 cells and protection from neutralizing antibodies
| Compounds* | Molar ratio | Infection (%)† | Protection (%)‡ |
|---|---|---|---|
| None | NA | 100 | 0 |
| Liposomes | |||
| DOTAP : DOPE : DLPC | 0.3 : 0.03 : 0.06 | 71±2 | 17±3 |
| Liposomes/PEG | |||
| DOTAP : DOPE : DLPC : PEG | 0.3 : 0.03 : 0.06 : 0.2 | 115±4 | 25±4 |
| TMPEG | NA | 153±2 | 15±12 |
| Liposomes/TMPEG | |||
| DOTAP : DOPE : DLPC :TMPEG | 0.3 : 0.03 : 0.06 : 0.1 | 96±1 | 65±13 |
| DOTAP : DOPE : DLPC :TMPEG | 0.3 : 0.03 : 0.06 : 0.2 | 130±6 | 88±11 |
| DOTAP : DOPE : DLPC :TMPEG | 0.3 : 0.03 : 0.06 : 0.3 | 115±11 | 86±12 |
MVAIIIB/β-gal-associated liposomes were prepared by dissolving DOTAP, DOPE and DLPC with or without TMPEG in chloroform at various molar ratios. The liposome preparation (800 µl) was mixed with 1×107 p.f.u. MVAIIIB/β-gal in 200 µl PBS, incubated for 30 min at room temperature and sonicated prior to infection studies.
For infection, free MVAIIIB/β-gal or liposome-complexed MVAIIIB/β-gal (10 p.f.u. ml−1) was incubated with a control serum (1 : 10 dilution of pooled sera from naïve mice) for 1 h. The mixtures were then added to a monolayer of BHK cells and incubated for an additional 24 h. The cells were lysed and the level of β-gal was measured using a β-gal reporter gene activity detection kit. The level of β-gal expression, defined as µU β-gal (mg protein)−1, in cells infected with free MVAIIIB/β-gal in the presence of the control serum (diluted 1 : 10) was taken as 100 %. The percentage of liposome-mediated inhibition of infection with MVAIIIB/β-gal complexed with liposomes or TMPEG was calculated as: (β-gal expression in cells infected with complexed MVAIIIB/β-gal/β-gal expression in cells infected with free MVAIIIB/β-gal)×100. Results are presented as the means±SD of at least three independent experiments.
Neutralization assays were performed using BHK-21 cells and pooled sera (diluted 1 : 10) from mice immunized at least three times with the WR strain of non-recombinant VV exhibiting an end-point antibody titre against the VV higher than 1 : 10 000. The level of infection was determined by measuring β-gal expression in cell lysates 24 h after infection. The percentage of liposome-mediated protection of the complexed MVAIIIB/β-galvirus from neutralizing antibodies was determined as: (β-gal expression in cells infected with complexed MVAIIIB/β-gal in the presence of neutralizing serum/β-gal expression in cells infected with complexed MVAIIIB/β-gal in the presence of control serum)×100. Results are presented as the means±SD of three independent experiments.
NA, Not applicable.
In vitro neutralization assays
Neutralization assays were performed using baby hamster kidney cells (BHK-21; ATCC) and pooled sera from mice immunized four times with the WR strain of non-recombinant VV with an end-point antibody titre against VV of ≥1 : 10 000. For infection, free MVAIIIB/β-gal or liposome-complexed MVAIIIB/β-gal (10 p.f.u. ml−1) was combined with pooled immune or control sera (diluted 1 : 10) for 1 h. The mixtures were then added to a monolayer of BHK cells and incubated for an additional 24 h. The cells were lysed and the level of β-gal was measured using a β-gal reporter gene activity detection kit (Sigma) according to the manufacturer’s instructions. All experiments were performed in triplicate.
Analyses of fluorescently labelled MVAIIIB/β-gal–TMPEG/liposome complexes in infected cells
MVAIIIB/β-gal (107 p.f.u.) was labelled with 4 µl DiI (Vybrant Cell-labelling Solutions; Molecular Probes), washed twice with PBS by centrifugation at 30 000 g for 50 min and used for infection of BHK-21 cells in the presence or absence of neutralizing antibodies. In parallel, a liposome film was prepared containing 4 µl DiO dye (Molecular Probes). The DiO-labelled liposomes were washed twice with PBS using a stirred ultrafiltration cell (Millipore) and combined with DiI-labelled MVAIIIB/β-gal for infection of BHK-21 cells. After 15 or 30 min of infection, the cells were washed three times with PBS, fixed with 3.7% paraformaldehyde and mounted for immunofluorescence analyses. The functional integrity of DiI- and DiO-labelled MVAIIIB/βgal–TMPEG/liposome complexes was analysed by confocal microscopy 30 min after infection, as described previously (Gaidarov et al., 1999). Images were collected using a Zeiss Axiovert S100TV microscope with a Zeiss-Neofluar (1.25 NA) 63× objective and a Quantix cooled charge-coupled-device camera containing a Kodak 1400 chip (Photometrics), yielding a size of 0.108 mm in all images. Images were acquired and analysed using IPLab Scientific Imaging software (Scanalytics) and Adobe Photoshop.
Oral administration of liposome-complexed MVAIIIB/β-gal virus to mice with pre-existing poxvirus immunity
BALB/c (H-2d) mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and maintained in a specific pathogen-free microisolator environment. Mice (n=6) were immunized orally with 1×107 p.f.u. of the WR strain of non-recombinant VV using a 24-gauge feeding needle (Popper & Sons). The immunization was repeated every 3 weeks until antibody titres against VV were higher than 1 : 10 000 and neutralizing antibody titres exceeded 1 : 50. WR VV-immunized BALB/c mice with pre-existing poxvirus immunity were then orally administered free or TMPEG/liposome-complexed MVAIIIB/β-gal (3×107 p.f.u.) virus. Naive BALB/c mice (n=6) treated with the same preparations of MVAIIIB/β-gal served as controls. The mice were sacrificed 18 h after infection to analyse β-gal gene expression in intestinal tissues using the β-gal reporter gene activity kit (Sigma). The protein concentration in individual samples was determined using a DC reagent kit (Bio-Rad) according to the manufacturer’s protocol and the results were expressed as µU β-gal (mg protein)−1.
Oral vaccination
Groups of BALB/c mice (n=4) were immunized orally with 3×107 p.f.u. MVAIIIB/β-gal, MVAIIIB/β-gal–TMPEG/liposome complex or MVAIIIB/β-gal and TMPEG/liposomes delivered separately using a 24-gauge feeding needle. Control mice were immunized with wild-type MVA and TMPEG/liposomes. Booster immunizations were repeated three times at 4 week intervals. The immunized and control mice were sacrificed 4 weeks after the last immunization to analyse the induction of Env-specific cellular and humoral responses.
Cell purification
Induction of Env-specific cell-mediated immune responses in Peyer’s patches and spleen was analysed 4 weeks after specific immunization. After Peyer’s patches were identified and isolated from the intestinal wall, dead cells and tissue debris were removed by passing the cells through a cotton/glass wool column (Fisher Scientific) and then layering them on to discontinuous gradients of 75 and 40% Percoll (Sigma). After centrifugation for 20 min (600 g) at 4°C, the interface between the two Percoll layers was carefully removed and washed with medium. This procedure resulted in over 95% viable lymphocytes. Spleen cells were aseptically removed and a single-cell suspension was prepared by gently teasing the cells through sterile screens.
ELISPOT assay
The number of CD8+ T lymphocytes secreting gamma interferon (IFN-γ) in the spleen and Peyer’s patches stimulated with a CD8+ T-cell peptide epitope of the HIV Env glycoprotein,I10 (RGPGRAFVTI, aa 315–329; Takahashi et al., 1996) was determined using an ELISPOT assay. Briefly, 96-well nitrocellulose plates (Multiscreen-MAIP; Millipore) were coated with 15 µg rat anti-mouse mAb ml−1 directed against IFN-γ (mAb 785; R&D Systems) in 0.05 M carbonate/bicarbonate buffer (pH 9.6). After overnight incubation at 4 °C, the wells were washed with PBS containing 0.05% Tween 20 (PBS/Tween 20) and non-specific binding was blocked for 1 h with RPMI 1640 containing 10% FCS. Cells from immunized mice were resuspended at a concentration of 107 cells ml−1 and added at various densities to antibody-coated wells in the presence of the Env-specific I10 peptide. After 24 h incubation at 37 °C, the plates were washed six times with PBS containing 0.05% Tween 20 and incubated for 2 h with 50 µl biotinylated mAb(1 µg ml−1) directed against mouse IFN-γ (mAb 485; R&D Systems). The plates were washed and incubated for 1 h with 50 µl streptavidin-conjugated alkaline phosphatase (SA-5100; Vector Laboratories), diluted 1 : 1000. After a final wash with PBS, spots were developed with the alkaline phosphatase substrate NBT/BCIP (SK-5400; Vector Laboratories) and counted under a stereomicroscope.
Anti-Env antibody ELISA
A direct ELISA assay was used to determine the presence of Env-specific antibodies in the sera of the immunized mice. Ninety-six-well ELISA plates (Nunc) were coated overnight at 4 °C with 100 µl recombinant Env (3 µg ml−1; Immunodiagnostics) as described previously (Kaneko et al., 2000). Following a wash with PBS/Tween 20, the wells were blocked for 2 h with 2% BSA (Sigma) in PBS/Tween 20 at room temperature. Sera were prepared from murine blood samples, serially diluted in PBS/Tween 20 and added to wells. After incubation at room temperature for 1 h, the plates were washed three times and then incubated with a 1 : 2000 dilution of a peroxidase-conjugated goat anti-mouse Ig (IgG, IgM and IgA; Sigma) in PBS/Tween 20. The plates were washed three times and developed with O-phenylenediamine (0.4 mg ml−1; Sigma) in 0.05 M phosphate/citrate buffer containing 0.03% sodium perborate (Sigma), stopped with 0.2 M sulfuric acid and analysed at 450 nm with an ELISA plate reader (Dynatech MRX).
Statistical analyses
The statistical significance of the difference in Env-specific immune responses between the immunized groups of mice was performed using a two-tailed Student’s t-test assuming equal variance. Data were presented as arithmetic mean±SD and analysed using the JMP program (SAS Institute) on a Windows-based platform.
RESULTS
Protection of MVAIIIB/β-gal from neutralization with poxvirus-specific antibodies by TMPEG/liposome complexes
In our initial experiments, we explored approaches for testing the hypothesis that coating of MVAIIIB/β-gal with liposomes would shield the virus from neutralizing antibodies. We first examined the effect of different formulations of cationic liposomes on the biological activity of MVAIIIB/β-gal by measuring the ability of the virus to infect BHK-21 cells in vitro. Infection efficiency was quantified by measuring the level of β-gal gene expression in the infected cultures. The level of infection with free MVAIIIB/β-gal in the presence of sera from unimmunized mice was taken as 100 %. As shown in Table 1, retention of infectivity was largely dependent on the composition of liposomes and the type of liposome conjugation. For example, if MVAIIIB/β-gal was conjugated with cationic liposomes that consisted of DOTAP, DOPE and DLPC (0.3 : 0.03 : 0.06), its infectivity was reduced by approximately 30% compared with that achieved using unconjugated MVAIIIB/β-gal for infection. However, the same liposomes prepared with PEG at a molar ratio of 0.2 preserved infectivity to a greater extent than that observed with the unconjugated virus. The efficiency of infection was further augmented when MVAIIIB/β-gal was complexed with TMPEG or TMPEG/liposomes.
We then evaluated the effectiveness of shielding of the MVAIIIB/β-gal virus from neutralizing antibodies by the different liposomal formulations. Neutralizing assays were performed using BHK-21 cells and pooled sera (diluted 1 : 10) from mice immunized at least three times with the WR strain of non-recombinant VV with an end-point antibody titre against VV of ≥1 : 10 000. The level of infection was determined by measuring β-gal expression in the cell lysates 24 h after infection. Table 1 shows that liposomes alone or associated with PEG provided only a moderate level of protection (17±3 and 25±4%, respectively). TMPEG had the lowest shielding effect with MVAIIIB/β-gal, which is in contrast to results obtained with an adenoviral vector (O’Riordan et al., 1999). However, attaching TMPEG to liposomes provided the highest protection from neutralization, eliciting 88±11% of the level of infection measured with the untreated MVAIIIB/β-gal and control sera. Protection was also influenced by the concentration of TMPEG, with the highest levels achieved at a molar ratio of 0.2 (Table 1).
Analysis of fluorescently labelled MVAIIIB/β-gal–TMPEG/liposome complexes during infection
Considering the possibility that the TMPEG/cationic lipids might have disrupted the virus and that viral DNA was simply being transfected rather than actively infecting the cells, we analysed the infection process with MVAIIIB/β-gal–TMPEG/liposome complexes in BHK-21 cells. The MVAIIIB/β-gal virion and TMPEG/liposomes were labelled separately with the lipophilic dyes DiI and DiO, respectively. BHK-21 cells were then infected with the dual-labelled MVAIIIB/β-gal–TMPEG/liposome complexes in the presence of neutralizing antibodies and the status of infection was analysed by fluorescence microscopy. The results showed that most BHK-21 cells expressing DiI-labelled MVAIIIB/β-gal (red fluorescence; Fig. 1a) on the cell surface were also positive for DiO staining (green fluorescence; Fig. 1b) 15 min after infection, indicating the presence of both virus and liposomal complexes on the surface of infected cells (Fig. 1c). In contrast, infection was almost completely eliminated if free DiI-labelled MVAIIIB/β-gal was incubated with neutralizing antibodies (Fig. 1d). These results demonstrated that the TMPEG/liposome complex did not compromise the integrity of MVAIIIB/β-gal as the virus retained its infectivity, but it shielded the virus from neutralizing antibodies.
Fig. 1.
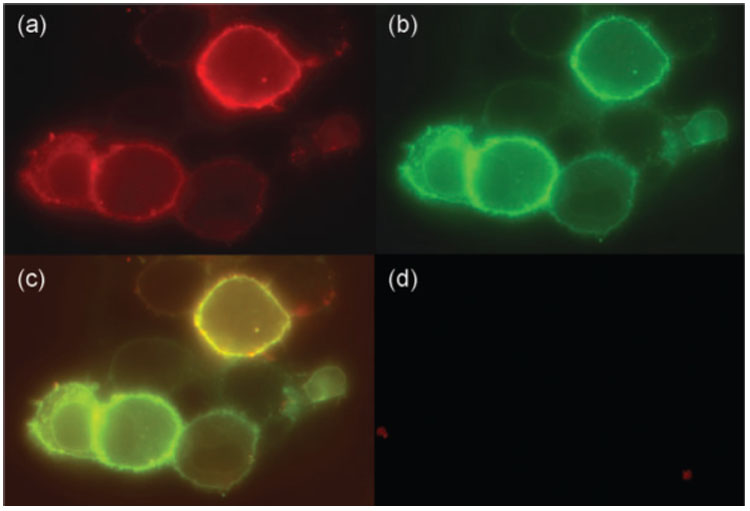
Fluorescence analyses of BHK-21 cells infected with Dil-labelled MVAIIIB/β-gal/DiO-labelled TMPEG/liposome complexes. BHK-21 cells were infected with the Dil/DiO-labelled MVAIIIB/β-gal/TMPEG/liposomes (a–c) or free Dil-labelled MVAIIIB/β-gal virus (d) in the presence of neutralizing antibodies (serum dilution of 1 : 10) and analysed by fluorescence microscopy after 15 min. Red fluorescence denotes cells positive for Dil-labelled MVAIIIB/β-gal (a); green fluorescence denotes cells positive for DiO-labelled TMPEG/liposomes (b); orange staining denotes double-positive cells (c).
Using confocal microscopy, we next examined the co-localization of DiI-labelled MVAIIIB/β-gal and DiO-labelled TMPEG/liposomes in BHK-21 cells 30 min after infection in the presence of VV-specific antibodies. Fig. 2(a–c) shows that DiI-labelled MVAIIIB/β-gal and DiO-labelled liposomes were similarly distributed within the infected cells. Fluorescence was intense in the trans-Golgi network region, in some endosomal structures and in the cytoplasm near the plasma membrane. These findings indicated that the fluorescently labelled MVAIIIB/β-gal–TMPEG/liposome complexes were capable of infecting BHK-21 cells in the presence of neutralizing antibodies.
Fig. 2.
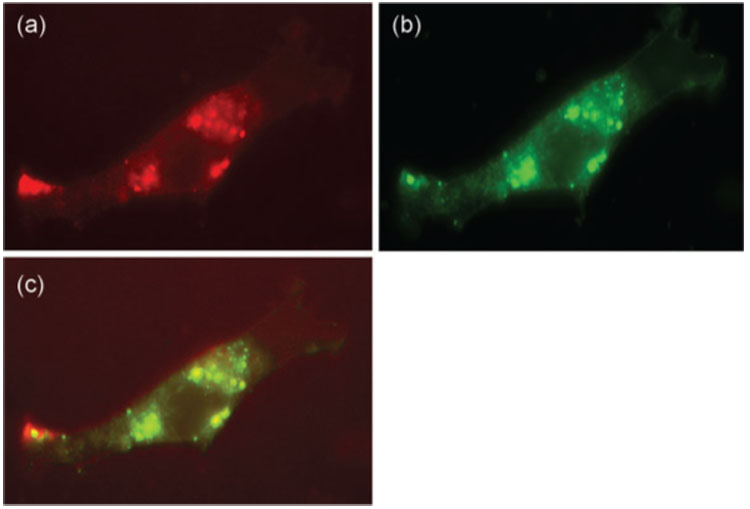
Confocal microscopy images of fluorescently labelled MVAIIIB/β-gal–TMPEG/liposome complexes in infected cells. BHK-21 cells were infected with Dil-labelled MVAIIIB/β-gal/DiO-labelled TMPEG/liposome complexes in the presence of neutralizing antibodies (serum dilution of 1 : 10) for 30 min. The cells were then fixed with 3.7% paraformaldehyde and mounted for analysis by confocal microscopy. Excitation wavelengths alternating between 488 and 554 nm produced a 1.7 s interval between successive frames of individual channels. Images were produced using Adobe Photoshop. Red fluorescence denotes cells positive for Dil-labelled MVAIIIB/β-gal (a); green fluorescence denotes cells positive for DiO-labelled TMPEG/liposomes (b); orange staining denotes overlaid images (c).
Administration of MVAIIIB/β-gal complexed with TMPEG/liposomes to mice with pre-existing poxvirus immunity
We investigated whether the TMPEG/liposomes complexed with MVAIIIB/β-gal would be able to deliver the transgenes to mucosal tissues in animals with pre-existing poxvirus immunity. For these experiments, BALB/c mice were first immunized orally with the WR strain of VV (107 p.f.u. per mouse) for generation of neutralizing antibodies. The titres of neutralizing antibodies were determined in vitro by measuring the ability of the serially diluted sera from the WR VV-immunized mice to inhibit MVAIIIB/β-gal infection of BHK-21 cells. Immunized mice (n=6) with serum neutralizing titres ranging from 1 : 50 to over 1 : 100, usually detected after the third immunization, received 3×107 p.f.u. of MVAIIIB/β-gal orally either as a free virus or complexed with the TMPEG/liposomes. Naïve mice infected with the free or TMPEG/liposome-complexed MVAIIIB/β-gal served as controls. Infection was determined by measuring expression of the β-gal gene in intestinal tissues 18 h after virus delivery. Fig. 3 shows that the level of β-gal expression delivered by free MVAIIIB/β-gal to mice with the pre-existing poxvirus immunity represented approximately 35% of that measured in the naïve mice. In contrast, the MVAIIIB/β-gal vector complexed with TMPEG/liposomes infected the VV-pre-immunized or naïve mice with similar efficiencies. Collectively, these results indicated that the TMPEG/liposome complexes were attached to MVAIIIB/β-gal without compromising the infectivity of the viral vector in vivo.
Fig. 3.

Expression of β-gal in intestinal tissues of mice after oral delivery of free MVAIIIB/β-gal (vertically hatched bars) or MVAIIIB/β-gal–TMPEG/liposome complexes (diagonally hatched bars). Naïve BALB/c mice or mice with pre-existing poxvirus immunity (n=6) received 3×107 p.f.u. MVAIIIB/β-gal delivered orally either as a free virus or as a complex with the TMPEG/liposomes. The expression of β-gal in intestinal tissues was determined 18 h after virus delivery. Results are presented as the means±SD of four independent experiments. **, P<0.001.
Induction of HIV Env-specific cellular and humoral immune responses after repeated immunizations with free or TMPEG/liposome-associated MVAIIIB/β-gal
The expression of β-gal in intestinal tissues after oral administration of MVAIIIB/β-gal associated with TMPEG/liposomes in mice with pre-existing poxvirus immunity suggested that the MVAIIIB/β-gal virus was protected from neutralizing antibodies in the gastric environment. We next analysed whether the MVAIIIB/β-gal vector complexed with TMPEG/liposomes was capable of inducing HIV Env glycoprotein-specific immune responses in systemic and mucosal tissues of the immunized mice and whether the responses could be increased after multiple vaccinations. BALB/c mice (n=4) were vaccinated four times with free or TMPEG/liposome complexed-MVAIIIB/β-gal (3×107 p.f.u. per mouse) at 4 week intervals. An additional group of mice received TMPEG/liposomes and MVAIIIB/β-gal separately to determine whether the virus needed to be complexed with the TMPEG/liposomes prior to in vivo delivery to elicit optimal levels of immune responses. The induction of Env-specific IFN-γ responses in CD8+ T lymphocytes in spleen and Peyer’s patches was analysed after each immunization by ELISPOT using a CD8+ T-cell peptide epitope of the Env glycoprotein, I10.
As shown in Fig. 4(a), Env-specific IFN-γ responses in spleen were more than twofold higher after the second immunization in all groups of immunized mice, consistent with the undetectable level of neutralizing antibody responses to MVAIIIB/β-gal after the first immunization.Mice that were immunized with the TMPEG and MVAIIIB/β-gal preparations, delivered separately or as MVAIIIB/β-gal–TMPEG/liposome complexes, had similar responses to those induced by the MVAIIIB/β-gal virus, suggesting that this preparation of TMPEG/liposomes had no adjuvant effect on the level of Env-specific IFN-γ production or the neutralizing antibody response to the viral vector. The profile of the IFN-γ response in Peyer’s patches was similar to that measured in the spleens of immunized mice, although the frequencies of IFN-γ-secreting CD8+ T cells were lower after the first and second immunizations compared with those in splenocytes of the immunized mice. The level of l10 peptide-specific IFN-γ responses in spleen and Peyer’s patches decreased after the third vaccination in mice immunized with the free MVAIIIB/β-gal virus or MVAIIIB/β-gal and TMPEG/liposomes delivered separately. This could be related to the loss of some Env-specific CD8+effector T cells induced by the first and second immunizations and the ineffective delivery of the env gene by MVAIIIB/β-gal due to generation of neutralizing antibodies that interfered with the infection process. Although we were unable to demonstrate any neutralizing antibodies in faecal washes from immunized mice due to the faecal material being diluted extensively, neutralizing antibodies were detectable after the second immunization at serumdilutions lower than 1 : 10. In contrast, the numbers of IFN-γ producing CD8+ T lymphocytes in mice immunized with the MVAIIIB/β-gal–TMPEG/liposome complexes remained relatively stable after the third and fourth vaccinations and higher than those measured in the remaining groups of mice (P<0.05).
Fig. 4.

Induction of IFN-γ-producing CD8+ T-cell responses by repeated immunization with free or TMPEG/liposome-complexed MVAIIIB/β-gal in BALB/c mice. BALB/c mice (n=4) were immunized with an unconjugated MVAIIIB/β-gal viral vector (vertically hatched bars), TMPEG/liposome-complexed MVAIIIB/β-gal (diagonally hatched bars) or TMPEG/liposomes and MVAIIIB/β-gal delivered separately (filled bars). Control mice (empty bars) were immunized with the wild-type MVA and TMPEG/liposomes. Booster immunizations were carried out every 4 weeks. The frequencies of CD8+ T cells secreting IFN-γ in cultures established from spleen (a) and Peyer’s patches (b) were determined by ELISPOT assay with the Env-specific peptide l10. Results are shown as the means±SD of at least three independent experiments. The frequencies of cells secreting IFN-γ were determined by regression analysis from curves generated by plotting numbers of spots versus effector cells. *, P<0.05; **, P<0.001.
An ELISA assay used to analyse humoral responses to the Env glycoprotein revealed the presence of Env-specific antibody responses in sera from all groups of MVAIIIB/β-gal-immunized mice after the second immunization, with end-point titres ranging from 1 : 1300 to 1 : 1600 (Fig. 5). Consistent with the profile of cellular responses, Env-specific antibody titres increased after the third and fourth immunizations in mice vaccinated with the MVAIIIB/β-gal–TMPEG/liposome complexes and were significantly higher compared with animals vaccinated with free MVAIIIB/β-gal virus or MVAIIIB/β-gal and TMPEG/liposomes delivered separately (P<0.01).
Fig. 5.

Induction of Env-specific antibody responses by repeated immunization with free MVAIIIB/β-gal or MVAIIIB/β-gal–TMPEG/liposome complexes. BALB/c mice (n=4) were immunized with the unconjugated MVAIIIB/β-gal viral vector (vertically hatched bars), TMPEG/liposome-complexed MVAIIIB/β-gal (diagonally hatched bars) or TMPEG/liposomes and MVAIIIB/β-gal delivered separately (filled bars). Blood samples were collected 4 weeks after each immunization and ELISA was used to analyse the titre of Env-specific antibody responses in the sera of immunized mice. Titres are shown as the highest serum dilutions with significantly different absorbance values compared with sera from control mice. *, P<0.01; **, P<0.001.
DISCUSSION
The primary goal of an effective HIV vaccine is to identify an immunogen that can be formulated in such a way that the vaccine induces a sustained level of anti-HIV immune responses in mucosal and systemic tissues. In this study, we have described a novel approach for delivery of MVAIIIB/β-gal to mice with pre-existing poxvirus immunity. It is becoming increasingly evident that the main concerns that hinder the application of VV-based vectors in AIDS vaccines are related to safety in a potentially immunodeficient human population (Fenner, 1989), as well as the efficacy of the vectors in people previously immunized with a VV vaccine for smallpox (Cooney et al., 1991). These concerns have encouraged the use of avian poxvirus vectors as an alternative delivery system. Although these vectors are considered safer because they do not replicate in mammalian cells and are not affected by previous VV immunity, they appear to be less potent than VV vectors (Hanke et al., 1998; Konishi et al., 1997). The problem of safety has recently been overcome with the introduction of highly attenuated VV strains such as NYVAC (Tartaglia et al., 1992) and MVA (Mayr et al., 1978; Meyer et al., 1991; Sutter & Moss, 1992) as vectors. MVA is replication incompetent in most mammalian cells, but recombinants are as or more immunogenic than replication-competent strains (Belyakov et al., 1998; Hanke et al., 1998). Thus, replication-defective VV vectors appear to have the safety of avian poxvirus vectors without a decrease in immunogenicity. In addition, some recent studies have demonstrated shared modes of protection against poxvirus infection by replication-deficient VVs and conventional smallpox vaccine viruses (Belyakov et al., 2003). These properties, together with the reported protection against lethal VV challenge by a CD8+ T-cell peptide epitope of vaccinia and variola viruses (Snyder et al., 2004), provide a basis for further evaluation of these replication-deficient VVs as safer vaccines against smallpox or complications from VV.
Concern over the use of attenuated VVs as vaccine vectors in the setting of pre-existing immunity to poxvirus has not been resolved fully. Our results show that repeated oral vaccination with MVAIIIB/β-gal covalently attached to TMPEG-modified cationic liposomes induces Env-specific immune responses in VV-immunized mice, suggesting that this strategy may be applicable to individuals with pre-existing immunity to poxvirus and may also be used with other recombinant viral vectors. Although liposomes have been used extensively as a delivery system for a variety of antigens including proteins and peptides, their use in association with viral vectors for oral vaccination remains to be elucidated. Our results indicate that MVAIIIB/β-gal complexed with TMPEG/liposomes improves the ability of the recombinant poxviral vector to deliver transgenes repeatedly to mice and also increases the efficacy of gene transfer both in vitro and in vivo. The TMPEG/liposome association with MVAIIIB/β-gal preserved the activity of the virus more effectively than viral complexes formed with cationic liposomes or with TMPEG alone, although the infectivity and protection of the modified virus was affected by the concentration of TMPEG, as well as by the lipid component of the liposomes. The modification procedure with TMPEG described here has several advantages over other methods (Francis et al., 1998), as the stable, secondary amine linkage that attaches TMPEG leaves no chemical linker groups on the target molecule (Delgado et al., 1990; Malik et al., 1992). Moreover, in contrast to methods where the linkers compromise the surface charge, the nitrogen atom to which the TMPEG chains is attached results in better conservation of the surface charge than many indirect linkages (O’Riordan et al., 1999). The major additional advantages of the TMPEG method are that protein coupling occurs under physiological conditions (Delgado et al., 1990)and that the presence of TMPEG on the surface of liposomes inhibits aggregation (Harasym et al., 1995). TMPEG has also been coupled to proteins and liposomes to prolong their circulation, as it acts as a surface barrier to plasma factors that otherwise bind to liposomes in the blood and accelerate vesicle removal (Fuertges & Abuchowski, 1990; Senior et al., 1991). Similarly, incorporating hydrophilic TMPEG molecules in liposomes may provide a steric barrier that inhibits the association of serum proteins, including neutralizing antibodies, with the virion. Furthermore, the direct association of cationic liposomes with the recombinant viral vector may add an additional adjuvant property to the complex, as cationic liposomes have been widely applied to enhance immune responses (Bangham, 1963; Gregoriadis & Ryman, 1972; Ishii et al., 1997; Wierzbicki et al., 2002). In our studies, however, we were unable to detect any adjuvant effect of the TMPEG/liposomal complex on the induction of Env-specific immune responses in MVAIIIB/β-gal-immunized mice. As the adjuvant properties of liposomes may be dependent on their lipid compositions, additional experiments are required to promote beneficial immune responses induced by the MVAIIIB/β-gal–TMPEG/liposome vaccine. This strategy may also be useful for the delivery of replication-attenuated recombinant viruses, as their association with liposomes may improve viral dissemination in vivo or protect the viral vector from the acidic environment of the gastrointestinal tract.
The important feature of our approach is versatility, as all viral vectors could be susceptible to TMPEG modification alone or in association with liposomes, regardless of the transgene contained within the vector. Although the condition of TMPEG modification and the composition of liposomes may differ for each viral vector, our approach offers a relatively safe solution to the issue of vector neutralization. An additional advantage of this delivery system is that liposomes can be used as carriers for adjuvant delivery, concomitant with the immunogen at the time of vaccination. In summary, our study describes the potential of oral vaccines with replication-attenuated viral vectors for repeated induction and augmentation of HIV Env-specific cellular and humoral responses. Although a successful HIV vaccine is likely to require the incorporation of other HIV antigens together with co-stimulatory molecules or immuno-modulatory cytokines that amplify antigen presentation, our results offer a strategy that can facilitate the development of prophylactic and therapeutic AIDS vaccines.
ACKNOWLEDGEMENTS
We are grateful to Drs Bernard Moss and Patricia Earl for reagents. We acknowledge the assistance of the Confocal Microscopy Facility at Thomas Jefferson University in Philadelphia, PA, USA. This study was supported by research grants from the National Institute of Allergy & Infectious Diseases to Dr Danuta Kozbor (R21AI060375), the Japan Health Sciences Foundation and funds to commemorate Dr Goro Chihara’s research activity. T. N. was a Research Scholar of the Japan Health Sciences Foundation.
REFERENCES
- Abacioglu YH, Fouts TR, Laman JD, Claassen E, Pincus SH, Moore JP, Roby CA, Kamin-Lewis R, Lewis GK. Epitope mapping and topology of baculovirus-expressed HIV-1 gp160 determined with a panel of murine monoclonal antibodies. AIDS Res Hum Retroviruses. 1994;10:371–381. doi: 10.1089/aid.1994.10.371. [DOI] [PubMed] [Google Scholar]
- Bangham AD. Physical structure and behavior of lipids and lipid enzymes. Adv Lipid Res. 1963;64:65–104. doi: 10.1016/b978-1-4831-9937-5.50008-9. [DOI] [PubMed] [Google Scholar]
- Belyakov IM, Wyatt LS, Ahlers JD, Earl P, Pendleton CD, Kelsall BL, Strober W, Moss B, Berzofsky JA. Induction of a mucosal cytotoxic T-lymphocyte response by intrarectal immunization with a replication-deficient recombinant vaccinia virus expressing human immunodeficiency virus 89.6 envelope protein. J Virol. 1998;72:8264–8272. doi: 10.1128/jvi.72.10.8264-8272.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyakov IM, Moss B, Strober W, Berzofsky JA. Mucosal vaccination overcomes the barrier to recombinant vaccinia immunization caused by preexisting poxvirus immunity. Proc Natl Acad Sci U S A. 1999;96:4512–4517. doi: 10.1073/pnas.96.8.4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyakov IM, Earl P, Dzutsev A, Kuznetsov VA, Lemon M, Wyatt LS, Snyder JT, Ahlers JD, Franchini G, et al. Shared modes of protection against poxvirus infection by attenuated and conventional smallpox vaccine viruses. Proc Natl Acad Sci U S A. 2003;100:9458–9463. doi: 10.1073/pnas.1233578100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyakov IM, Hammond SA, Ahlers JD, Glenn GM, Berzofsky JA. Transcutaneous immunization induces mucosal CTLs and protective immunity by migration of primed skin dendritic cells. J Clin Invest. 2004;113:998–1007. doi: 10.1172/JCI20261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender BS, Rowe CA, Taylor SF, Wyatt LS, Moss B, Small PA., Jr Oral immunization with a replication-deficient recombinant vaccinia virus protects mice against influenza. J Virol. 1996;70:6418–6424. doi: 10.1128/jvi.70.9.6418-6424.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SC, Jones DH, Fynan EF, Farrar GH, Clegg JCS, Greenberg HB, Herrmann JE. Protective immunity induced by oral immunization with a rotavirus DNA vaccine encapsulated in microparticles. J Virol. 1998;72:5757–5761. doi: 10.1128/jvi.72.7.5757-5761.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childers NK, Zhang SS, Michalek SM. Oral immunization of humans with dehydrated liposomes containing Streptococcus mutans glucosyltransferase induces salivary immunoglobulin A2 antibody responses. Oral Microbiol Immunol. 1994;9:146–153. doi: 10.1111/j.1399-302x.1994.tb00051.x. [DOI] [PubMed] [Google Scholar]
- Cooney EL, Collier AC, Greenberg PD, Coombs RW, Zarling J, Arditti DE, Hoffman MC, Hu S-L, Corey L. Safety of and immunological response to a recombinant vaccinia virus vaccine expressing HIV envelope glycoprotein. Lancet. 1991;337:567–572. doi: 10.1016/0140-6736(91)91636-9. [DOI] [PubMed] [Google Scholar]
- Czerkinsky C, Anjuere F, McGhee JR, George-Chandy A, Holmgren J, Kieny MP, Fujiyashi K, Mestecky JF, Pierrefite-Carle V, et al. Mucosal immunity and tolerance: relevance to vaccine development. Immunol Rev. 1999;170:197–222. doi: 10.1111/j.1600-065X.1999.tb01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado C, Patel JN, Francis GE, Fisher D. Coupling of poly(ethylene glycol) to albumin under very mild conditions by activation with tresyl chloride: characterization of the conjugate by partitioning in aqueous two-phase systems. Biotechnol Appl Biochem. 1990;12:119–128. [PubMed] [Google Scholar]
- Fenner F. Risks and benefits of vaccinia vaccine use in the worldwide smallpox eradication campaign. Res Virol. 1989;140:465–466. doi: 10.1016/s0923-2516(89)80126-8. [DOI] [PubMed] [Google Scholar]
- Francis GE, Fisher D, Delgado C, Malik F, Gardiner A, Neale D. PEGylation of cytokines and other therapeutic proteins and peptides: the importance of biological optimisation of coupling techniques. Int J Hematol. 1998;68:1–18. doi: 10.1016/s0925-5710(98)00039-5. [DOI] [PubMed] [Google Scholar]
- Fuertges F, Abuchowski A. The clinical efficacy of poly(ethylene glycol)-modified proteins. J Control Release. 1990;11:139–148. [Google Scholar]
- Fuller DH, Rajakumar PA, Wilson LA, Trichel AM, Fuller JT, Shipley T, Wu MS, Weis K, Rinaldo CR, et al. Induction of mucosal protection against primary, hetero-logous simian immunodeficiency virus by a DNA vaccine. J Virol. 2002;76:3309–3317. doi: 10.1128/JVI.76.7.3309-3317.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidarov I, Santini F, Warren RA, Keen JH. Spatial control of coated-pit dynamics in living cells. Nat Cell Biol. 1999;1:1–7. doi: 10.1038/8971. [DOI] [PubMed] [Google Scholar]
- Gherardi MM, Esteban M. Mucosal and systemic immune responses induced after oral delivery of vaccinia virus recombinants. Vaccine. 1999;17:1074–1083. doi: 10.1016/s0264-410x(98)00324-7. [DOI] [PubMed] [Google Scholar]
- Gregoriadis G, Ryman BE. Fate of protein-containing liposomes injected into rats. An approach to the treatment of storage diseases. Eur J Biochem. 1972;24:485–491. doi: 10.1111/j.1432-1033.1972.tb19710.x. [DOI] [PubMed] [Google Scholar]
- Gurunathan S, Wu C-Y, Freidag BL, Seder RA. DNA vaccines: a key for inducing long-term cellular immunity. Curr Opin Immunol. 2000;12:442–447. doi: 10.1016/s0952-7915(00)00118-7. [DOI] [PubMed] [Google Scholar]
- Hanke T, Blanchard TJ, Schneider J, Hannan CM, Becker M, Gilbert SC, Hill AVS, Smith GL, McMichael A. Enhancement of MHC class I-restricted peptide-specific T cell induction by a DNA prime/MVA boost vaccination regime. Vaccine. 1998;16:439–445. doi: 10.1016/s0264-410x(97)00226-0. [DOI] [PubMed] [Google Scholar]
- Harasym TO, Tardi P, Longman SA, Ansell SM, Bally MB, Cullis PR, Choi LSL. Poly(ethylene glycol)-modified phospholipids prevent aggregation during covalent conjugation of proteins to liposomes. Bioconjug Chem. 1995;6:187–194. doi: 10.1021/bc00032a006. [DOI] [PubMed] [Google Scholar]
- Hedley ML, Curley J, Urban R. Microspheres containing plasmid-encoded antigens elicit cytotoxic T-cell responses. Nat Med. 1998;4:365–368. doi: 10.1038/nm0398-365. [DOI] [PubMed] [Google Scholar]
- Ishii N, Fukushima J, Kaneko T, Okada E, Tani K, Tanaka SI, Hamajima K, Xin K-Q, Kawamoto S, et al. Cationic liposomes are a strong adjuvant for a DNA vaccine of human immunodeficiency virus type 1. AIDS Res Hum Retroviruses. 1997;13:1421–1428. doi: 10.1089/aid.1997.13.1421. [DOI] [PubMed] [Google Scholar]
- Jones DH, Corris S, McDonald S, Clegg JCS, Farrar GH. Poly(dl-lactide-co-glycolide)-encapsulated plasmid DNA elicits systemic and mucosal antibody responses to encoded protein after oral administration. Vaccine. 1997;15:814–817. doi: 10.1016/s0264-410x(96)00266-6. [DOI] [PubMed] [Google Scholar]
- Kaneko H, Bednarek I, Wierzbicki A, Kiszka I, Dmochowski M, Wasik TJ, Kaneko Y, Kozbor D. Oral DNA vaccination promotes mucosal and systemic immune responses to HIV envelope glycoprotein. Virology. 2000;267:8–16. doi: 10.1006/viro.1999.0093. [DOI] [PubMed] [Google Scholar]
- Kiszka I, Kmieciak D, Gzyl J, Naito T, Bolesta E, Sieron A, Singh SP, Srinivasan A, Trinchieri G, et al. Effect of the V3 loop deletion of envelope glycoprotein on cellular responses and protection against challenge with recombinant vaccinia virus expressing gp160 of primary human immunodeficiency virus type 1 isolates. J Virol. 2002;76:4222–4232. doi: 10.1128/JVI.76.9.4222-4232.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi E, Kurane I, Mason PW, Shope RE, Ennis FA. Poxvirus-based Japanese encephalitis vaccine candidates induce JE virus-specific CD8+ cytotoxic T lymphocytes in mice. Virology. 1997;227:353–360. doi: 10.1006/viro.1996.8331. [DOI] [PubMed] [Google Scholar]
- Kresina TF, Mathieson B. Human immunodeficiency virus type 1 infection, mucosal immunity, and pathogenesis and extramural research programs at the National Institutes of Health. J Infect Dis. 1999;179 Suppl. 3:S392–S396. doi: 10.1086/314815. [DOI] [PubMed] [Google Scholar]
- Lasic DD. Novel applications of liposomes. Trends Biotechnol. 1998;16:307–321. doi: 10.1016/s0167-7799(98)01220-7. [DOI] [PubMed] [Google Scholar]
- Malik F, Delgado C, Knusli C, Irvine AE, Fisher D, Francis GE. Polyethylene glycol (PEG)-modified granulocyte-macrophage colony-stimulating factor (GM-CSF) with conserved biological activity. Exp Hematol. 1992;20:1028–1035. [PubMed] [Google Scholar]
- Mayr A, Stickl H, Muller HK, Danner K, Singer H. The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behaviour in organisms with a debilitated defence mechanism. Zentralbl Bakteriol. 1978;167:375–390. in German. [PubMed] [Google Scholar]
- McGhee JR, Mestecky J. In defense of mucosal surfaces. Development of novel vaccines for IgA responses protective at the portals of entry of microbial pathogens. Infect Dis Clin North Am. 1990;4:315–341. [PubMed] [Google Scholar]
- Mestecky J, Eldridge JH. Targeting and controlled release of antigens for the effective induction of secretory antibody responses. Curr Opin Immunol. 1991;3:492–495. doi: 10.1016/0952-7915(91)90009-p. [DOI] [PubMed] [Google Scholar]
- Meyer H, Sutter G, Mayr A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol. 1991;72:1031–1038. doi: 10.1099/0022-1317-72-5-1031. [DOI] [PubMed] [Google Scholar]
- O’Riordan CR, Lachapelle A, Delgado C, Parkes V, Wadsworth SC, Smith AE, Francis GE. PEGylation of adenovirus with retention of infectivity and protection from neutralizing antibody in vitro and in vivo. Hum Gene Ther. 1999;10:1349–1358. doi: 10.1089/10430349950018021. [DOI] [PubMed] [Google Scholar]
- Pal R, Venzon D, Santra S, Kalyanaraman VS, Montefiori DC, Hocker L, Hudacik L, Rose N, Nacsa J, et al. Systemic immunization with an ALVAC-HIV-1/protein boost vaccine strategy protects rhesus macaques from CD4+ T-cell loss and reduces both systemic and mucosal simian-human immunodeficiency virus SHIVKU2 RNA levels. J Virol. 2006;80:3732–3742. doi: 10.1128/JVI.80.8.3732-3742.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J, Gilbert SC, Blanchard TJ, Hanke T, Robson KJ, Hannan CM, Becker M, Sinden R, Smith GL, Hill AV. Enhanced immunogenicity for CD8+ T cell induction and complete protective efficacy of malaria DNA vaccination by boosting with modified vaccinia virus Ankara. Nat Med. 1998;4:397–402. doi: 10.1038/nm0498-397. [DOI] [PubMed] [Google Scholar]
- Senior J, Delgado C, Fisher D, Tilcock C, Gregoriadis G. Influence of surface hydrophilicity of liposomes on their interaction with plasma protein and clearance from the circulation: studies with poly(ethylene glycol)-coated vesicles. Biochim Biophys Acta. 1991;1062:77–82. doi: 10.1016/0005-2736(91)90337-8. [DOI] [PubMed] [Google Scholar]
- Snyder JT, Belyakov IM, Dzutsev A, Lemonnier F, Berzofsky JA. Protection against lethal vaccinia virus challenge in HLA-A2 transgenic mice by immunization with a single CD8+ T-cell peptide epitope of vaccinia and variola viruses. J Virol. 2004;78:7052–7060. doi: 10.1128/JVI.78.13.7052-7060.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter G, Moss B. Nonreplicating vaccinia vector efficiently expresses recombinant genes. Proc Natl Acad Sci U S A. 1992;89:10847–10851. doi: 10.1073/pnas.89.22.10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter G, Moss B. Novel vaccinia vector derived from the host range restricted and highly attenuated MVA strain of vaccinia virus. Dev Biol Stand. 1995;84:195–200. [PubMed] [Google Scholar]
- Takahashi H, Nakagawa Y, Leggatt GR, Ishida Y, Saito T, Yokomuro K, Berzofsky JA. Inactivation of human immunodeficiency virus (HIV)-1 envelope-specific CD8+ cytotoxic T lymphocytes by free antigenic peptide: a self-veto mechanism? J Exp Med. 1996;183:879–889. doi: 10.1084/jem.183.3.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia J, Perkus ME, Taylor J, Norton EK, Audonnet J-C, Cox WI, Davis SW, van der Hoeven J, Meignier B, et al. NYVAC: a highly attenuated strain of vaccinia virus. Virology. 1992;188:217–232. doi: 10.1016/0042-6822(92)90752-b. [DOI] [PubMed] [Google Scholar]
- Wierzbicki A, Kiszka I, Kaneko H, Kmieciak D, Wasik TJ, Gzyl J, Kaneko Y, Kozbor D. Immunization strategies to augment oral vaccination with DNA and viral vectors expressing HIV envelope glycoprotein. Vaccine. 2002;20:1295–1307. doi: 10.1016/s0264-410x(01)00480-7. [DOI] [PubMed] [Google Scholar]
- Wyatt LS, Moss B, Rozenblatt S. Replication-deficient vaccinia virus encoding bacteriophage T7 RNA polymerase for transient gene expression in mammalian cells. Virology. 1995;210:202–205. doi: 10.1006/viro.1995.1332. [DOI] [PubMed] [Google Scholar]
- Wyatt LS, Shors ST, Murphy BR, Moss B. Development of a replication-deficient recombinant vaccinia virus vaccine effective against parainfluenza virus 3 infection in an animal model. Vaccine. 1996;14:1451–1458. doi: 10.1016/s0264-410x(96)00072-2. [DOI] [PubMed] [Google Scholar]
- Zalipsky S. Synthesis of an end-group functionalized polyethylene glycol-lipid conjugate for preparation of polymer-grafted liposomes. Bioconjug Chem. 1993;4:296–299. doi: 10.1021/bc00022a008. [DOI] [PubMed] [Google Scholar]