

Abstract
Alpha-synuclein (alpha-syn) is implicated in the pathogenesis of Parkinson’s disease (PD). Mutations in alpha-syn gene or alpha-syn locus (SNCA) triplication are associated with mitochondrial abnormalities and early onset of familial PD. The goals of the present study were to examine whether alpha-syn is localized in the mitochondria of alpha-syn overexpressing cells (HEK-syn cells); and whether alpha-syn overexpression causes cells to be more vulnerable to mitochondrial toxin, rotenone. Western blotting and confocal microscopy techniques were employed to assess localization of alpha-syn in the mitochondria of HEK-293 cells that were stably transfected with human wild-type alpha-syn. The results demonstrated that the mitochondrial fractions that were isolated from HEK-syn cells showed the presence of alpha-syn, whereas, no alpha-syn was detected in the mitochondrial fractions of control HEK cells. The mitochondria of HEK-syn cells were found to be more susceptible to rotenone-induced toxicity when compared to control HEK cells. The intracellular ATP levels were significantly decreased in HEK-syn cells in response to sub toxic concentrations of rotenone. These results suggest that under overexpression conditions, alpha-syn may translocate to mitochondria and cause enhanced toxicity in response to sub toxic concentrations of mitochondrial toxins. This study has implications to the pathogenesis of familial PD where alpha-syn overexpression is mainly involved.
Keywords: Parkinson’s disease, neurodegeneration, alpha-synuclein, mitochondria
INTRODUCTION
Parkinson’s disease (PD) is an age-associated, progressive neurodegenerative disease that is characterized by gradual loss of dopamine producing neurons in the substantia nigra pars compacta. The mechanisms by which these neurons die in PD are not well understood, however, oxidative stress and mitochondrial complex I deficiency were consistently observed in degenerating dopaminergic neurons [13,14]. Alpha-synuclein (alpha-syn) is a pre-synaptic protein which is also associated with the pathogenesis of PD. It is a major component of protein aggregates termed as Lewy bodies that appear in the brains of PD patients [18]. Mutations in alpha-syn gene or alpha-syn locus (SNCA) triplication are associated with early onset of familial PD [7,12,15]. Individuals with SNCA triplication were found to have increased alpha-syn protein in their blood and brains [10]. However, how this increased alpha-syn dosage is related to the pathogenesis of PD remains unknown. Interestingly, mitochondrial defects were consistently observed in the cells that were overexpressed with the alpha-syn gene and such defects were also found in alpha-syn transgenic mice [4,9]. It has been demonstrated that alpha-syn knock-out mice were resistant to the parkinsonism-inducing neurotoxin, MPTP. On the contrary, enhanced mitochondrial pathology has been observed in the substantia nigra of alpha-syn transgenic mice [1,17]. Moreover, abnormal mitochondria with swollen and rounded structures were also commonly found in the brains of PD patients [20]. These observations lead us to hypothesize that alpha-syn overexpression causes accumulation of alpha-syn in mitochondria and alter mitochondrial function. The goals of the present study were to investigate whether alpha-syn colocalizes to mitochondria; and whether alpha-syn overexpressing cells show increased susceptibility to parkinsonism-inducing mitochondrial toxin, rotenone. We have used the cell line, HEK-293 (Human embryonic kidney-293; HEK) which was stably transfected with human wildtype alpha-syn gene (HEK-syn). For control HEK cells, we used cells that were transfected with an empty vector.
MATERIALS AND METHODS
Control HEK and HEK-syn cells that were stably transfected with human wild-type alpha-syn or blank vector (pc DNA 3.1), using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) were obtained from Dr. Nussbaum’s Laboratory, National Institutes of Health, Bethesda, MD. Cells were grown in Dulbecco’s modified eagle’s medium (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum and antibiotics (penicillin and streptomycin) and placed in a humidified incubator (Thermo Forma, Minneapolis, MN) at 37°C with 5% CO2 in air. Alpha-syn levels were determined in whole cell lysates and also in mitochondrial and cytosolic fractions of both HEK and HEK-syn cells by western blot analysis. Briefly, the cell lysates were prepared by lysing cells in a RIPA buffer containing protease inhibitors (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM PMSF, 1 mM EDTA, 5 μg/ml Aprotinin, 5 μg/ml Leupeptin, 1% Triton X-100, 1% Sodium deoxycholate and 0.1% SDS). The lysates were centrifuged and the protein content in the supernatants was estimated using Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA). The mitochondrial and cytosolic fractions were separated from HEK and HEK-syn cells according to the method of Vander Heiden et al., [21]. Cells (1×107) were placed in 0.8 ml of ice-cold buffer A (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM dithiothreitol, and protease inhibitors cocktail) and homogenized with glass Dounce homogenizer with 40 strokes of the B pestle in cold. Unlysed cells, cell debris, and nuclei were pelleted by a 10 min, 1,000 × g centrifugation. The supernatant fluids were then centrifuged at 10,000 × g for 25 min, and the pellets were washed twice, resuspended in buffer A and represented as the mitochondrial fractions. Supernatants were used as cytosolic fractions. Proteins obtained from whole cell lysates and also from both fractions (mitochondrial and cytosolic) were loaded (ten microgram per lane) separately onto 15% polyacrylamide gel, separated by electrophoresis and transferred onto nitrocellulose membrane. The membranes were blocked with 3% bovine albumin and probed with anti-alpha-syn antibodies (R & D systems, Minneapolis, MN). After incubation with HRP-labeled anti-goat secondary antibodies, the membranes were developed using an ECL chemiluminescence method (Amersham Pharmacia Biotech, Piscataway, NJ). To check the purity of mitochondrial fractions, the membranes were stripped with stripping buffer and reprobed with antibodies to complex I enzyme (Molecular Probes, Inc., Eugene, OR), which is a specific marker for mitochondria. Equal loading of protein in whole cell lysates was confirmed by probing with antibodies to β-actin.
The localization of alpha-syn to mitochondria in HEK-syn cells was further confirmed by using double labeling confocal microscopy. Briefly, HEK-syn cells were grown on sterile glass cover slips (BD Biosciences, Bedford, MA) and fixed with 4% paraformaldehyde solution. After permeabilization in 0.2% Triton X-100 in PBS, cells were blocked with 10% goat serum in PBS at RT for 30 min and incubated with anti alpha-syn (R & D Sytems, MN) and anti-COX (cytochrome oxidase; Molecular Probes, Eugene, OR) antibodies for 2 hrs. We used COX antibodies to localize mitochondria. After washing with the washing buffer, the cells were incubated with rhodamine-conjugated anti-goat IgG (red) and FITC-labelled anti-mouse IgG (green) for 1 hr at RT. The cells were washed and the fluorescent signals were examined with an Olympus FluoView 300 Confocal laser scanning microscope.
To study the differential susceptibility of HEK and HEK-syn cells to rotenone toxicity, we treated cells with various concentrations of rotenone (5 nM to 50 nM) and ATP levels were measured using an ATP Lite kit (Perkin-Elmer, Wellesley, MA) after 12 hrs of incubation. Briefly, ten micro liters of cell lysates were added to 50 μl of ATP-substrate solution in a 96-well plate (black-colored). The plate was shaken for 5 min, dark adapted for 10 min, and the luminescence was measured with a luminescence counter (Top Count NXT; Packard, Downers Grove, IL, USA). ATP values were normalized with their respective protein values.
STATISTICS
Results were expressed as Mean ± SEM. Statistical comparisons were made using unpaired student’s t test and one way ANOVA.
RESULTS
Alpha-syn protein levels were found several folds higher in alpha-syn overexpressing HEK-syn cells when compared to control HEK cells (Fig 1A). Alpha-syn was found both in cytosolic and mitochondrial fractions of HEK-syn cells but not detected in HEK control cells of both fractions (Fig 1C). To confirm that the mitochondrial fractions were indeed enriched with mitochondria, we probed these fractions with antibodies to complex I enzyme, which is a specific marker for mitochondria. The complex I enzyme was detected only in the mitochondrial fractions of both HEK and HEK-syn cells, but not in the cytosolic fractions (Fig 1B). This indicated that the mitochondrial fractions isolated were, indeed, highly enriched with mitochondria. However, to rule out completely the possibility of cytosolic contamination of mitochondrial fractions, we conducted double labeling confocal microscopic studies using antibodies to alpha-syn and a mitochondrial respiratory enzyme, cytochrome c-oxidase (COX). As shown in Fig 2C, the merged image (yellow-color) indicated colocalization of alpha-syn with COX enzyme in HEK-syn cells. To our knowledge, this is the first report to indicate that alpha-syn is localized in the mitochondria of alpha-syn overexpressing HEK cells. We further investigated the sensitivity of HEK-syn cells to mitochondrial toxin, rotenone, by analyzing intracellular ATP levels in both control HEK and HEK-syn cells. No differences were observed in basal ATP levels between HEK-control and HEK-syn cells, however, lower concentrations of rotenone (5 nM and 10 nM) caused a significant reduction in ATP levels in HEK-syn cells (p<0.05, p<0.01; Fig 3). At higher concentration of rotenone (50 nM), both cells became susceptible to rotenone-induced ATP depletion. However, more ATP depletion was observed in HEK-syn cells compared to control HEK cells (Fig 3). These results suggest that mitochondria of HEK-syn cells are more vulnerable to rotenone-induced toxicity compared to mitochondria of control HEK cells.
Fig 1.
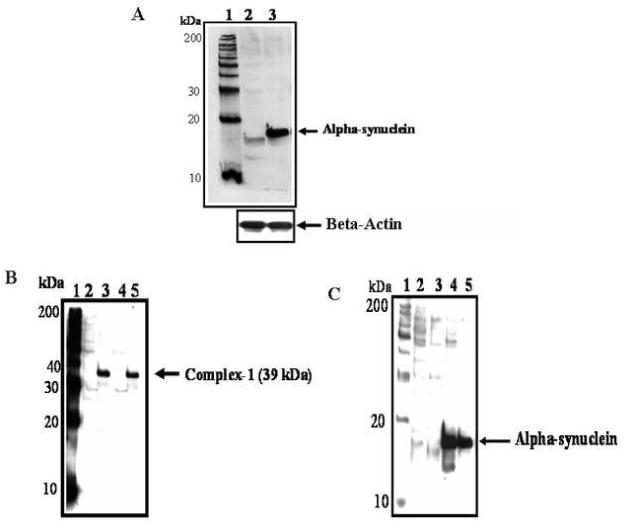
A Alpha-syn protein in HEK control and HEK-syn cells. Alpha-syn was detected by western blot analysis. Ten micrograms of protein from HEK control and HEK-syn cells was resolved separately by polyacrylamide gel-electrophoresis, transferred onto nitrocellulose membrane and probed with antibodies to alpha-syn. Lanes 1. Protein marker; 2. HEK-Control cells; 3. HEK cells overexpressing alpha-syn (HEK-syn cells). The blot is representative of three independent experiments.
B Complex I enzyme in mitochondrial fractions of HEK control and HEK-syn cells. Mitochondrial and cytosolic fractions were separated and complex I enzyme was detected by western blot analysis using antibodies to complex I. Lanes 1. Standard protein marker; 2. Control HEK cells (cytosolic fraction); 3. Control HEK cells (mitochondrial fraction); 4. HEK-syn cells (cytosolic fraction); 5. HEK-syn cells (mitochondrial fraction). Complex I enzyme was identified only in mitochondrial fractions of both HEK control (lane 3) and HEK-syn cells (lane 5) but not in the cytosolic fractions of both cells. The blot is representative of three independent experiments.
C Alpha-syn in cytosolic and mitochondrial fractions of HEK-syn cells. Mitochondrial and cytosolic fractions were separated from cell lysates of HEK control and HEK-syn cells. Alpha-syn was detected by western blot analysis using antibodies to alpha-syn. Lanes 1. Standard protein marker; 2. Control HEK cells (cytosolic fraction); 3. Control HEK cells (mitochondrial fraction); 4. HEK-syn cells (cytosolic fraction); 5. HEK-syn cells (mitochondrial fraction). Alpha-syn was found in the cytosolic as well as mitochondrial fractions of HEK-syn cells (lanes 4 & 5) but not found in either fractions of control HEK cells (lanes 2 & 3).
Fig 2.
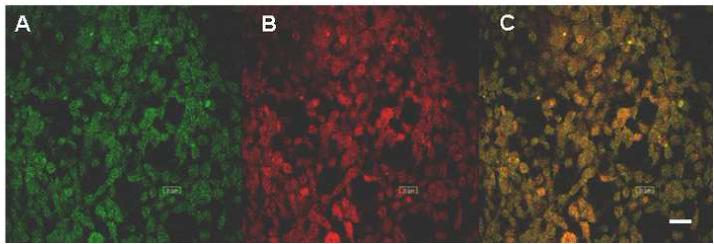
Colocalization of alpha-syn with COX enzyme in mitochondria of HEK-syn cells. HEK-syn cells were stained with both anti-COX and anti-alpha-syn antibodies (primary) and then with FITC-conjugated (Green; Panel A), and rhodamine-conjugated (Red; Panel B) secondary antibodies, respectively. Alpha-syn and COX immunoreactivity overlaps with each other producing yellow color (Panel, C). The bar in panel C indicates 20 μm.
Fig 3.
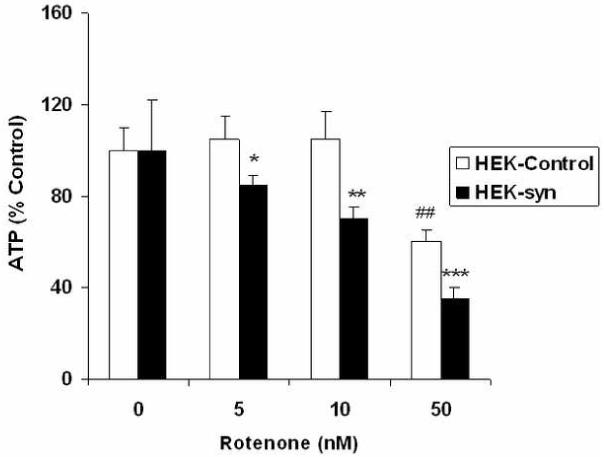
Intracellular ATP levels in HEK control and HEK-syn cells after treatment with various concentrations of rotenone. Cells were treated with rotenone for 12 h and ATP levels were measured by ATP Lite kit. Each value represents Mean +/- SEM of six separate determinations. The symbols *, **, *** represent significance between vehicle-treated HEK-syn and rotenone-treated HEK-syn cells at p<0.05, p<0.01 and p<0.001, respectively. The symbol ## represents significance between vehicle-treated control HEK-cells and HEK-cells that were treated with 50 nM of rotenone at p<0.01.
DISCUSSION
Previous studies have indicated that alpha-syn is widely expressed in presynaptic vesicles and also, as an aggregated protein in the Lewy bodies of PD patients. The functional role of alpha-syn in neurons is still not completely understood. In this study, we provide the first evidence that alpha-syn protein is localized in the mitochondria of alpha-syn overexpressing cells. In addition, we also found that alpha-syn overexpressing cells are increasingly vulnerable to subtoxic concentrations of rotenone, a mitochondrial toxin, that is linked to the pathogenesis of PD. Our study has implications to familial PD where alpha-syn overexpression is mainly involved [15]. Increased alpha-syn levels were found in the blood and brains of individuals that carried extra copies of the alpha-syn gene [10]. It is also demonstrated that increased alpha-syn levels can cause mitochondrial defects and degeneration of dopamine neurons, however, the precise mechanisms behind it were not well understood. The present results demonstrate that alpha-syn is localized to the mitochondria of alpha-syn overexprssing HEK cells, but not in the mitochondria of control cells that have very low expression. These results indicate that alpha-syn may translocate into mitochondria under overexpressed conditions. Although the precise colocalization of alpha-syn in the mitochondria is currently unknown, the double labeling confocal microscopy results with alpha-syn and COX antibodies suggested that it may be localized in the inner membrane of mitochondria. Based upon these results, we presume that alpha-syn may have certain regulatory functions in the mitochondria, which remains to be elucidated. Recent studies have suggested that alpha-syn is also present in the mitochondria of other neuronal cells [8,11].
The second goal of the study was to determine the mitochondrial function by measuring ATP levels in HEK control and HEK-syn cells after treatment with various concentrations of rotenone. Alpha-syn overexpressing cells showed increased susceptibility to subtoxic concentrations of rotenone in terms of cell viability and ATP levels. Low concentrations of rotenone caused a significant decrease in ATP levels in HEK-syn cells, suggesting that mitochondria of HEK-syn cells are more vulnerable to rotenone toxicity. It is interesting to note that alpha-syn knock-out mice were found resistant to MPTP (mitochondrial toxin) neurotoxicity [1], whereas, enhanced mitochondrial pathology was found in human alpha-syn transgenic mice after treatment with the same toxin [17]. On the other hand, knockdown of alpha-syn by RNA interferance protected the cells from MPP+ toxicity in the cell culture model of PD [2]. Our results also suggest that mitochondrial functions were significantly impaired in HEK-syn cells by subtoxic concentrations of rotenone. Overall, these observations indicate that in the presence of excess alpha-syn, the mitochondrial toxins such as MPTP or rotenone may exert strong inhibitory actions on complex I enzyme in the mitochondria of alpha-syn overexpressing cells. Future studies should reveal whether alpha-syn regulates the mitochondrial complex I enzyme activity in dopaminergic neurons.
The precise mechanisms by which alpha-syn overexpression causes mitochondrial dysfunction and dopamine neuron death are not well understood. Several mechanisms were proposed for alpha-syn mediated neurotoxicity in dopaminergic neurons. Alpha-syn overexpression is known to increase intracellular reactive oxygen species [6] and activate MAPK pathway [5], which may contribute to neuronal cell death. Further, it has also been demonstrated that increased expression of alpha-syn predisposes dopaminergic cells to proteasomal dysfunction [19]. Previous studies from our laboratory suggested that oxidative stress increases alpha-syn expression, whereas, antioxidants that decrease free radical stress protect from alpha-syn toxicity [16]. As mitochondria are a major source for oxidative stress [3], post-translationally modified alpha-syn by toxins like MPTP and rotenone may induce mitochondrial defects and dysfunction in PD.
In conclusion, our present results provide a novel initial evidence that alpha-syn is localized in the mitochondria of alpha-syn overexpressing cells; and alpha-syn overexpression increases susceptibility to mitochondrial toxins. Future studies with dopaminergic neuronal cells that overexpress alpha-syn should provide insights into the mechanisms by which alpha-syn causes mitochondrial defects and dopamine neuron death in PD.
Acknowledgements
The authors gratefully acknowledge Dr. R.L. Nussbaum, Division of Medical Genetics, University of California School of Medicine, San Francisco for providing the alpha-synuclein overexpressing HEK-293 cells. The authors also thank Ms. Sarah Rolling and Donna Leturnus for confocal microscopy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, Schmitz Y, Yuan CA, Rocha M, Jackson-Lewis V, Hersch S, Sulzer D, Przedborski S, Burke R, Hen R. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fountaine TM, Wade-Martins R. RNA interference-mediated knockdown of alpha-synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. J. Neurosci. Res. 2007;85:351–363. doi: 10.1002/jnr.21125. [DOI] [PubMed] [Google Scholar]
- [3].Ghafourifar P, Sen CK. Mitochondrial nitric oxide synthase. Front Biosci. 2007;12:1072–1078. doi: 10.2741/2127. [DOI] [PubMed] [Google Scholar]
- [4].Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Iwata A, Maruyama M, Kanazawa I, Nukina N. Alpha-Synuclein affects the MAPK pathway and accelerates cell death. J Biol Chem. 2001;276:45320–45329. doi: 10.1074/jbc.M103736200. [DOI] [PubMed] [Google Scholar]
- [6].Junn E, Mouradian MM. Human alpha-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci. Lett. 2002;320:146–150. doi: 10.1016/s0304-3940(02)00016-2. [DOI] [PubMed] [Google Scholar]
- [7].Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- [8].Li WW, Yang R, Guo JC, Ren HM, Zha XL, Cheng JS, Cai DF. Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport. 2007;18:1543–1546. doi: 10.1097/WNR.0b013e3282f03db4. [DOI] [PubMed] [Google Scholar]
- [9].Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, Singleton AB. Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology. 2004;62:1835–1838. doi: 10.1212/01.wnl.0000127517.33208.f4. [DOI] [PubMed] [Google Scholar]
- [11].Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci. 2008;65:1272–1284. doi: 10.1007/s00018-008-7589-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- [13].Przedborski S, Ischiropoulos H. Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson’s disease. Antioxid Redox Signal. 2005;7:685–693. doi: 10.1089/ars.2005.7.685. [DOI] [PubMed] [Google Scholar]
- [14].Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- [15].Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;320:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- [16].Shavali S, Carlson EC, Swinscoe JC, Ebadi M. 1-Benzyl-1,2,3,4-tetrahydroisoquinoline, a Parkinsonism-inducing endogenous toxin, increases alpha-synuclein expression and causes nuclear damage in human dopaminergic cells. J. Neurosci. Res. 2004;76:563–571. doi: 10.1002/jnr.20082. [DOI] [PubMed] [Google Scholar]
- [17].Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp. Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- [18].Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- [19].Sun F, Anantharam V, Latchoumycandane C, Kanthasamy A, Kanthasamy AG. Dieldrin induces ubiquitin-proteasome dysfunction in alpha-synuclein overexpressing dopaminergic neuronal cells and enhances susceptibility to apoptotic cell death. J. Pharmacol Exp. Ther. 2005;315:69–79. doi: 10.1124/jpet.105.084632. [DOI] [PubMed] [Google Scholar]
- [20].Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP, Jr., Miller SW, Davis RE, Parker WD., Jr. Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp. Neurol. 2000;162:37–50. doi: 10.1006/exnr.2000.7333. [DOI] [PubMed] [Google Scholar]
- [21].Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]