

Abstract
Passive and active immunization against outer surface protein A (OspA) has been successful in protecting laboratory animals against subsequent infection with Borrelia burgdorferi. Antibodies (Abs) to OspA convey full protection, but only when they are present at the time of infection. Abs inactivate spirochetes within the tick and block their transmission to mammals, but do not affect established infection because of the loss of OspA in the vertebrate host. Our initial finding that the presence of high serum titers of anti-OspC Abs (5 to 10 μg/ml) correlates with spontaneous resolution of disease and infection in experimentally challenged immunocompetent mice suggested that therapeutic vaccination with OspC may be feasible. We now show that polyclonal and monospecific mouse immune sera to recombinant OspC, but not to OspA, of B. burgdorferi resolve chronic arthritis and carditis and clear disseminated spirochetes in experimentally infected C.B.-17 severe combined immunodeficient mice in a dose-dependent manner. This was verified by macroscopical and microscopical examination of affected tissues and recultivation of spirochetes from ear biopsies. Complete resolution of disease and infection was achieved, independent of whether OspC-specific immune sera (10 μg OspC-specific Abs) were repeatedly given (4× in 3- to 4-day intervals) before the onset (day 10 postinfection) or at the time of fully established arthritis and carditis (days 19 or 60 postinfection). The results indicate that in mice spirochetes constitutively express OspC and are readily susceptible to protective OspC-specific Abs throughout the infection. Thus, an OspC-based vaccine appears to be a candidate for therapy of Lyme disease.
Lyme borreliosis is a tick-borne infectious disease, caused by the spirochete Borrelia burgdorferi. The associated clinical manifestations are variable early during infection and involve skin, joint, heart, and nervous system, and they may become chronic at later stages (1). Symptoms such as acute arthritis and neuroborreliosis may undergo spontaneous resolution, however, with episodic recurrence (1). Spirochetes have been isolated repeatedly from untreated patients (1–5), and ample evidence exists for persistent infections, even after extensive antibiotic therapy (6–8). Although most patients develop protective antibodies (Abs) in the course of infection (9), the Abs are not always able to control disease and/or eradicate spirochetes.
Outer surface protein A (OspA) of B. burgdorferi is the most promising vaccine candidate for prophylaxis of Lyme disease, as revealed by the recent phase 3 clinical trial with more than 11,000 individuals in the United States (ref. 10 and Y. Lobet, personal communication). Studies in mice previously have shown that full protection against disease and infection mainly is conveyed by OspA-specific Abs (11, 12). These Abs are effective only when present at the time of infection (11), most probably caused by the fact that OspA is preferentially expressed on spirochetes within ticks, but lost in the mammalian host (13, 14). Thus, OspA-specific immunity may be used for preventive, but not therapeutic, vaccination.
Lyme disease patients and naturally or experimentally (low dose: ≤103 spirochetes) infected mice produce Abs to OspC, but not to OspA (15–20). This suggests that OspC is expressed and immunogenic in mammals. Up-regulation of OspC, but not OspA, on spirochetes after infection of mice has been documented (21). OspC also can serve in protective immunity. Recent studies showed that gerbils and mice were completely protected against experimental challenge and/or tick-borne infection with homologous B. burgdorferi isolates after active immunization with glutathione S-transferase-OspC fusion protein (rOspC) (22, 23). The fact that this immunization protocol does not lead to the elimination of infectious spirochetes from the vector, as was shown with OspA-specific Abs (24), indicates that OspC-based protective immunity is preferentially linked to inactivation of spirochetes in the mammalian host (23).
Passive transfer of immune sera from B. burgdorferi-infected mice resolved, at least temporarily, clinical arthritis in naive immunocompetent (25, 26) or C3H severe combined immunodeficient (SCID) (H-2k) mice (27), but did not cure carditis and infection. However, in none of the studies were the specificities or the amount of the protective Abs identified. By using various mouse strains, we found that disease resolution and/or elimination of spirochetes closely correlated with high titers of OspC-specific serum Abs. Here we analyze the potential of immune serum against rOspC to control persistent infection in C.B.-17 SCID mice.
MATERIALS AND METHODS
Mice and Infection with B. burgdorferi.
Female AKR/N (H-2k), C57BL/6 (H-2b), BALB/c (H-2d), and C.B.-17 SCID (H-2d) mice between 6 and 8 weeks of age were bred under specific pathogen-free conditions at the Max-Planck-Institut für Immunbiologie, Freiburg, Germany, and inoculated s.c. with 1 × 103 low-passage (2–4 in vitro passages) organisms from B. burgdorferi, strain ZS7 (11) into the base of the tail.
Recombinant Antigens.
A full-length recombinant lipidated OspA (rLip-OspA) from B. burgdorferi, strain ZS7, was generated as described (28) and was kindly provided by SmithKline Beecham. rOspC from B. burgdorferi, strain ZS7, was generated by using established protocols (29).
Polyclonal Immune Sera.
BALB/c mice were inoculated with either 10 μg of rLip-OspA or rOspC in 100 μl of ABM2 adjuvant (Sebak, Aldenbach, Germany) into the base of the tail and boosted twice with the same antigen preparation at 10-day intervals. Immune serum (IS) was pooled during 2 months after the last boost and contained the following concentration of OspA- or OspC-specific Abs as determined with ELISA by using rLip-OspA or recombinant OspC (without GST fusion) as substrate: anti-OspA IS, 3.2 mg/ml; anti-OspC IS, 300 μg/ml. Normal mouse serum (NMS) was collected from naive BALB/c mice. The specificities of the IS and NMS were verified by Western blot. As shown in Fig. 1 (lanes 3 and 4), polyclonal IS from either OspA- or OspC-immunized mice reacted selectively with proteins of 31 kDa (OspA) and 24 kDa (OspC), respectively, from a whole-cell lysate of ZS7 spirochetes. No reactivity was found with NMS (Fig. 1, lane 2).
Figure 1.
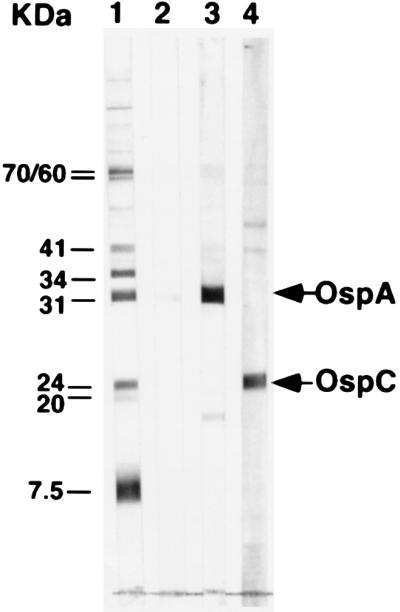
Western blot analysis of NMS and IS used for passive immunization of C.B.-17 SCID mice. Lane 1, standard consisting of mouse mAbs to Hsp-70 (70 kDa), Hsp-60 (60 kDa), flagellin (41 kDa), OspB (34 kDa), OspA (31 kDa), OspC (24 kDa), pLA7 (20 kDa), and p7.5 (7.5 kDa). Lane 2, NMS, pooled from naive BALB/c mice. Lane 3, polyclonal IS generated in BALB/c mice immunized with rLip-OspA in ABM2 adjuvant. Lane 4, polyclonal IS generated in BALB/c mice immunized with rOspC in ABM2 adjuvant, as described in Materials and Methods.
Analysis of Serum Abs by ELISA and Western Blot.
Serum Abs to B. burgdorferi OspA, or OspC were quantified by a solid-phase ELISA as described (30) by using 1 μg/ml of either whole-cell lysate of B. burgdorferi strain ZS7, rLip-OspA (ZS7), or rOspC (ZS7) as substrates, respectively. Western blot analysis, by using whole-cell lysate of B. burgdorferi strain ZS7 as antigen preparation, was done as described (31).
Passive Transfer of Immune Serum.
For passive protection, C.B.-17 SCID mice were injected i.p. with either OspA- or OspC-reactive polyclonal IS 1 hr before infection. Control mice received 100 μl of NMS. Alternatively, for passive treatment of established infection, SCID mice first were infected s.c. and subsequently given repeatedly (four times at 3- to 4-day intervals) various amounts of polyclonal IS specific for either OspA or OspC (i.p.), starting at either day 10, 19, or 60 postinfection (p.i.). Animals were monitored for the development of clinical arthritis in the tibiotarsal joints under double-blind conditions. The severity of arthritis was scored in the right and left tibiotarsal joints.
At indicated time points, mice were investigated for the presence of spirochetes by cultivation of ear biopsies, as described (32).
For histopathological examination, mice were sacrificed at indicated time points p.i. Tibiotarsal joint, heart, liver, and muscle adjacent to the tibiotarsal joint were fixed in 10% formaldehyde, embedded in paraffin, and stained with hematoxylin and eosin. Samples were examined under double-blind conditions.
RESULTS
AKR/N, BALB/c, and C57BL/6 mice with differential susceptibility to B. burgdorferi-induced disease (33) were inoculated with 103 spirochetes. The kinetics of specific Ab responses, development of arthritis, and persistence of spirochetes were monitored up to 90 days p.i. All animals seroconverted (Fig. 2A and data not shown) and produced similar amounts of B. burgdorferi-specific IgG Abs, which first were detectable 14 days p.i. and increased steadily during the entire observation period. Furthermore, all mice developed significant, but variable, amounts of OspC-specific IgM and IgG Abs (Fig. 2A). OspC-specific IgM Abs first were detected 4 days p.i., peaked 14 days p.i., and declined to baseline levels 23 days p.i. OspC-specific IgG Abs first were seen at 14 days p.i., peaked 23 days p.i. (peak values for AKR/N, C57BL/6: ≈8 μg/ml; BALB/c: ≈3 μg/ml). Ab titers declined in AKR/N and C57BL/6 mice over time, but remained at detectable levels (≈3 μg/ml) up to 90 days p.i. In contrast, BALB/c mice produced lower amounts of OspC-specific IgG Abs in the early phase of infection, but serum titers increased with time p.i. As expected from previous studies (18, 19), antibodies to OspA were not detectable in any of the sera from infected mice during the entire 90-day observation period by ELISA or Western-blot analysis that used spirochetal lysate or rLip OspA (data not shown).
Figure 2.

Kinetics of appearance of B. burgdorferi-specific (IgG) or OspC-specific (IgM/IgG) Abs (A) and correlation analysis of serum levels of either total B. burgdorferi-specific or OspC-specific Abs (IgG) and elimination of spirochetes from infected mice (B). AKR/N, C57BL/6, and BALB/c mice (6–8 weeks old) were infected by syringe injection with 103 spirochetes into the base of the tail (s.c.). The amounts of B. burgdorferi-specific (IgG) and OspC-specific Abs (IgM and IgG) in sera from individual mice were tested with ELISA by using either whole-cell lysates (B. burgdorferi strain ZS7) or rOspC (ZS7) as substrates. The data represent the mean of individual serum samples tested (AKR/N and C57BL/c: 10 mice per group; BALB/c: seven mice per group). (A) Correlation between serum levels of total B. burgdorferi-specific IgG Abs, OspC-specific IgG Abs (day 23 p.i.), and the ability to recultivate spirochetes from ear biopsies (day 90 p.i.) was analyzed by ranking correlation assay (B).
Nine of 10 disease-susceptible AKR/N mice developed mild clinical arthritis to B. burgdorferi, which was apparent on day 40 p.i. Swelling of tibiotarsal joints increased with time in six AKR/N mice, but completely disappeared in the other three mice 110 days p.i. One infected AKR/N mouse did not show any signs of clinical arthritis during the entire 90-day observation time. In contrast, none of the disease-resistant C57BL/6 (10) or BALB/c (seven) mice developed clinical arthritis at any time p.i. However, spirochetes could be reisolated from 12 of 20 ear biopsies taken from individual mice when tested 90 days p.i. (two ear samples were contaminated). Most importantly, a close correlation was found between serum titers of anti-OspC Abs and spontaneous resolution of infection irrespective of the mouse strains tested. As shown in Fig. 2B, the amounts of OspC-specific IgG Abs produced were significantly higher in those eight mice from which no spirochetes could be cultivated, as compared with those from which positive cultures were obtained (10 ± 5.0 μg/ml vs. 4.8 ± 2.8 μg/ml). On the other hand, no correlation was found between total B. burgdorferi-specific IgG Abs and elimination of spirochetes. This indicated that OspC-specific Abs are able to inactivate spirochetes in the vertebrate host and to control infection.
To confirm this hypothesis, we used passive transfer experiments to examine the ability of polyclonal IS specific for rOspC to cure an established B. burgdorferi infection in SCID mice. Polyclonal immune serum specific for rLip-OspA served as control. It was found initially that different doses of OspC-specific Abs were required for prevention and treatment of infection. Passive transfer of 3 μg of OspC-specific Abs into SCID mice 1 hr before infection led to complete protection against disease and infection in all five mice tested (Table 1), whereas only one of three animals tested was protected with 0.3 μg Abs (data not shown). As shown before, full protection also was observed with 3 μg OspA-specific Abs, but not with normal mouse serum under similar conditions (Table 1 and ref. 31). In contrast, repeated administration of 3 μg/mouse of OspC-specific Abs (4× in 3- to 4-day intervals), starting at day 10 p.i., a time point when spirochetes have disseminated (34) and inflammation of joints and heart starts to evolve, only partially prevented development of clinical arthritis. However, no effect was seen on clearance of spirochetes; 0.3 μg/mouse of the same IS had no effect on either disease or infection (data not shown). Moreover, when the same protocol (3 μg/mouse) was applied to mice 20 days p.i., a time point at which arthritis and carditis has fully developed, only a marginal intermittent attenuation of the severity of clinical arthritis was observed, with no effect at all on infection (data not shown).
Table 1.
Differential therapeutic effect of immune sera against OspA and OspC on established B. burgdorferi infection of C.B.-17 SCID mice
| Intervals of serum transfer, days p.i. | Serum transferred, dose | Mouse no. | Clinical arthritis, days p.i.
|
Ear culture, days p.i.
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 19 | 30 | 40 | 50 | 70 | 80 | 40 | |||
| minus 1 hr | NMS, 100 μl/mouse | 1 | −/± | ±/± | ++/++ | ++/++ | + | |||
| 2 | (±)/− | ±/++ | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 3 | −/− | ±/± | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 4 | −/± | ++/+ | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 5 | −/− | ++/+ | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| anti-OspA I.S., 3 μg/mouse | 1 | −/− | ±/− | (±)/− | (±)/− | (±)/− | ±/− | ±/− | − | |
| 2 | −/− | −/− | −/− | −/− | − | |||||
| 3 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | − | ||
| 4 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | − | ||
| 5 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | − | ||
| anti-OspC I.S., 3 μg/mouse | 1 | −/− | −/− | −/− | −/− | − | ||||
| 2 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | − | ||
| 3 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | − | ||
| 4 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | − | ||
| 5 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | − | ||
| 10, 14, 19, 22 | NMS, 100 μl/mouse | 1 | −/− | +/++ | ++/++ | ++/++ | + | |||
| 2 | −/− | (±)/− | +/+ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 3 | (±)/− | ±/+ | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 4 | (±)/(±) | ±/+ | +/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 5 | −/(±) | +/+ | +/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| anti-OspA I.S., 10 μg/mouse | 1 | −/(±) | ±/± | ++/++ | ++/++ | + | ||||
| 2 | −/− | +/± | ++/+ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 3 | −/(±) | +/++ | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 4 | (±)/(±) | ±/± | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 5 | −/− | ±/++ | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| anti-OspC I.S., 10 μg/mouse | 1 | (±)/± | −/− | (±)/− | (±/− | ±/− | ±/− | ++/± | + | |
| 2 | −/(±) | −/− | −/− | −/− | − | |||||
| 3 | −/(±) | −/− | −/− | −/− | −/− | −/− | −/− | − | ||
| 4 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | − | ||
| 5 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | − | ||
| 19, 22, 26, 30 | NMS, 100 μl/mouse | 1 | −/− | −/− | ±/± | ±/± | ±/± | ±/± | ±/± | + |
| 2 | −/− | ±/± | ++/++ | ++/++ | + | |||||
| 3 | −/− | ±/+ | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 4 | −/− | ++/+ | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 5 | −/(±) | (±)/− | −/− | −/− | −/− | −/− | −/− | − | ||
| anti-OspA I.S., 10 μg/mouse | 1 | −/− | ±/+ | ++/++ | ++/++ | + | ||||
| 2 | (±)/− | +/+ | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 3 | −/− | ±/+ | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| 4 | −/− | −/− | −/− | −/− | −/− | −/− | −/− | − | ||
| 5 | −/(±) | ++/+ | ++/++ | ++/++ | ++/++ | ++/++ | ++/++ | + | ||
| anti-OspC I.S., 10 μg/mouse | 1 | −/− | ±/± | ±/(±) | (±)/− | − | ||||
| 2 | −/+ | ++/++ | ±/+ | ±/+ | −/± | −/± | −/± | − | ||
| 3 | −/− | ±/+ | ±/(±) | −/(±) | −/− | −/− | −/− | − | ||
| 4 | −/± | +/± | ±/(±) | ±/− | −/− | −/− | −/− | − | ||
| 5 | −/(±) | ++/++ | ±/+ | ±/+ | −/(±) | −/(±) | −/(±) | − | ||
C.B.-17 SCID mice (6–8 weeks old) were inoculated with 103 B. burgdorferi strain ZS7 into the base of the tail (s.c.). Normal mouse serum (NMS, pooled from naive BALB/c) as well as monospecific immune serum against either OspA or OspC (prepared by immunizing BALB/c mice with recombinant OspA and OspC, respectively) were passively transferred into SCID mice (i.p.) at intervals and doses indicated.
Scoring of findings for the right and left tibiotarsal joints: ++, severe; +, moderately severe; ±, mild swelling; (±), redding; −, no clinical signs.
One animal of each group was sacrificed on day 45 p.i. for histopathological examination.
Based on these findings, IS containing 10 μg of anti-OspC Abs was used in all further passive transfer experiments. As shown in Table 1, IS repeatedly applied (10 μg, 4×, 3- to 4-day intervals) either 10 or 19 days p.i., completely prevented the appearance of and cured established clinical arthritis in 5 of 5 infected SCID mice. With one exception, spirochetes could not be recultivated from ear biopsies. In contrast and as expected from previous studies (11), repeated passive transfer of similar amounts of anti-OspA-specific Abs on either day 10 or day 19 had no effect on clinical arthritis and infection. Most notably, anti-OspC-specific Abs also were able to resolve chronic disease and infection in SCID mice. This is indicated by the fact that passive transfer of the respective IS, starting at 60 days p.i. led to substantial reduction of clinical arthritis within 10 days post-treatment and to nearly complete resolution in the following 30 days (Table 2). Whereas the pathogen could be cultivated from ear biopsies of 3 of 3 infected SCID mice before treatment (day 38 p.i.), none of the specimens taken from the same animals at day 20 after antibody transfer (day 80 p.i.) contained any detectable spirochetes. No relapse of clinical arthritis was observed up to 40 days post-treatment, independent on whether IS (10 μg/mouse anti-OspC specific Abs) was transferred at day 10, 19 or 60 p.i. No effect on progressive clinical arthritis and infection was seen in SCID mice repeatedly treated with only 0.3 μg of anti-OspC Abs at day 60 p.i. (Table 2, and data not shown).
Table 2.
Therapeutic effect of immune serum against OspC 60 days after infection of C.B.-17 SCID mice with 103 B. burgdorferi ZS7
| Anti-OspC serum, μg/mouse | Intervals of serum transfer, days p.i. | Mouse | Clinical arthritis (days p.i.)
|
Ear culture of spirochetes
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 | 19 | 60 | 70 | 80 | 100 | 1st (d38) | 2nd (d80) | |||
| 0.3 | 10, 14, 19, 22 | 1 | −/− | +/++ | ++/++ | ++/++ | ++/++ | n.d. | n.d. | + |
| 2 | −/− | ++/++ | ++/++ | ++/++ | ++/++ | n.d. | n.d. | + | ||
| 3 | −/− | ++/++ | ++/++ | ++/++ | ++/++ | n.d. | n.d. | + | ||
| 10 | 60, 63, 66, 70 | 1 | −/− | +/+ | ++/++ | ±/+ | −/± | −/± | + | − |
| 2 | −/− | ++/+ | ++/++ | ±/± | −/− | * | + | − | ||
| 3 | (±)/− | ++/++ | ++/++ | +/++ | −/(±) | −/(±) | + | − | ||
The same protocol for infection of mice and the same scoring system for clinical arthritis as described in Table 1 was used. n.d., not determined.
The mouse was sacrificed for histopathological examination.
The therapeutic effect of polyclonal IS reactive to OspC on established disease and infection of SCID mice was confirmed further by histopathological examination of affected organs. As shown in Table 3, significant histopathological changes were detected in joints, heart, liver, and muscle of infected, but otherwise untreated, SCID mice or those receiving only NMS. As shown before (35), chronic, progressive inflammatory changes were most pronounced in tibiotarsal joints, heart and liver, and persisted throughout the entire period of observation (45 days p.i.). Mice that had received IS specific for either OspC or OspA (3 μg specific Abs per mouse) 1 hr before infection did not show pathological alterations in any of the four organs when examined at day 45 p.i. Furthermore, mice that had received IS reactive to OspC (10 μg specific Abs per mouse) starting at either day 10 or day 19 after inoculation showed, if at all, only marginal inflammatory lesions in joints, heart, liver, and muscle. In contrast, IS reactive with OspA (10 μg specific Ab/mouse) had no effect on the development or progression of inflammation in the affected organs (day 19 p.i.).
Table 3.
Histopathological examination of affected organs from individual infected SCID mouse after therapeutic treatment with immune sera
| Intervals of serum transfer, days p.i. | Serum transferred, dose | Day of necropsy p.i. | Clinical arthritis* | Histopathological examination†
|
|||
|---|---|---|---|---|---|---|---|
| Joint | Heart | Liver | Muscle | ||||
| None | None | 10 | −/(±) | + | + | n.t. | n.t. |
| 27 | ++/++ | ++ | ++ | ++ | + | ||
| minus 1 hr | NMS, 100 μl/mouse | 45 | ++/++ | +++ | ++ | − | + |
| anti-OspA I.S., 3 μg/mouse | 45 | −/− | + | − | − | − | |
| anti-OspC I.S., 3 μg/mouse | 45 | −/− | − | − | − | − | |
| 10, 14, 19, 22 | NMS, 100 μl/mouse | 45 | ++/++ | +++ | ++ | − | + |
| anti-OspA I.S., 10 μg/mouse | 45 | ++/++ | +++ | + | − | ± | |
| anti-OspC I.S., 10 μg/mouse | 45 | −/− | − | − | − | − | |
| 19, 22, 26, 30 | NMS, 100 μl/mouse | 45 | ++/++ | +++ | +++ | ± | + |
| anti-OspA I.S., 10 μg/mouse | 45 | ++/++ | +++ | +++ | ± | n.t. | |
| anti-OspC I.S., 10 μg/mouse | 45 | ±/(±) | + | − | − | − | |
Tissues from tibiotarsal joint, heart, liver, and muscle adjacent to the joint of mice from experimental groups described in Table 1 were fixed, stained with hematoxylin and eosin, and subjected to histopathological examination.
The same scoring system for clinical arthritis as described in Table 1.
Scoring: +++, extensive; ++, strong; +, moderate; ±, mild; −, none. n.t., not tested.
DISCUSSION
This study shows that passive transfer of polyclonal OspC-reactive immune serum into B. burgdorferi-infected SCID mice leads to complete resolution of chronic arthritis and carditis as well as the elimination of the pathogen. For an efficient control of infection, a critical threshold level of OspC-specific Abs appears to be of importance. This implies that spirochetes express OspC in the mammalian host and are susceptible to protective Abs within affected tissues throughout infection. Therefore, targeting of OspC is relevant for a successful therapeutic treatment of Lyme disease.
Earlier studies have shown that IS from B. burgdorferi-infected mice conferred complete protection against disease and infection when adoptively transferred into either naive or immunodeficient recipients before, or at the time of, but not after experimental or natural inoculation with the pathogen (11, 25–27). It also was found that IS from infected mice significantly reduced established arthritis, however, with no effect on carditis and persistent infection (27). Although the IS used in these studies consisted of a whole array of B. burgdorferi-specific Abs, including those to OspC, but not to OspA, (27), the specificities and quantities of Abs relevant for the obvious differential control of infection and disease have not been identified. The present finding of a close correlation between high serum titers of anti-OspC-specific (10 μg/ml), but not total B. burgdorferi-specific Abs, and spontaneous resolution of infection demonstrates that Abs to OspC are critical in the inactivation of spirochetes in the vertebrate host. Furthermore, we found that polyclonal OspC-specific IS was able to resolve both arthritis and carditis as well as persistent spirochete infection in C.B.-17 SCID mice, independent of whether the Abs were administered before the onset (day 10 p.i.) or, alternatively, at the time of fully established (day 19 p.i.) or chronic disease (day 60 p.i). This therapeutic effect was strictly dose-dependent. Complete resolution of disease and elimination of the pathogen was achieved with ≥ 10 μg of anti-OspC Abs per mouse, whereas 3 or 0.3 μg of the same serum preparation had only a transient effect on the course of arthritis and failed to clear persistent spirochetes. This value of ≥10 μg of anti-OspC Abs per mouse also compares to titers found in those 2 of 7 experimentally infected immunocompetent BALB/c mice (average 10.2 μg/ml), which showed spontaneous resolution of disease and infection (Fig. 2B). At present it is unclear why infection was not controlled in those AKR/N or C57BL/6 mice in which naturally induced Abs to OspC approached 8–10 μg/ml, at least transiently (Fig. 2A). One explanation may be the differential kinetics of production of anti-OspC Abs in the three mouse strains, which rapidly declined in AKR/N and C57BL/6 mice after peak response at day 23 p.i., but increased with time p.i. in BALB/c mice. It is also possible that the anti-OspC Ab population induced in AKR/N, C57BL/6, and BALB/c mice expressed different specificities to OspC epitopes with distinct protective potential. However, the present results suggest that the induction of protective Abs alone is not sufficient to eliminate persisting spirochetes. Rather, there seems to be a minimal requirement for both the level of OspC-reactive serum Abs and the duration of their production to reach and eliminate spirochetes in affected organs.
The finding that chronic Lyme disease can be cured with high concentrations of homologous OspC-specific Abs indicates that spirochetes express the target structure and are accessible to immune attack throughout infection. One obstacle to the general applicability of this therapeutic treatment appears to be the well documented heterogeneity of OspC among members of the species B. burgdorferi sensu lato (36–38). Evidence indicates that the majority of European and North American B. burgdorferi isolates contain the OspC gene and also express the protein (38, 39). In addition, it has been shown that polyclonal anti-OspC IS generated in response to one strain of B. burgdorferi crossreacted with the majority of heterologous isolates analyzed (refs. 36 and 40 and unpublished data). This suggests the existence of crossreactive B cell epitopes among OspC molecules within the species of B. burgdorferi. Although the protective character of these crossreactive epitopes has not been proven, it is possible that IS to one OspC species also may resolve chronic disease and infection induced by heterologous spirochetes, provided that the threshold level for strain-specific protective Abs is reached.
Inflammatory lesions of joints and heart of B. burgdorferi-inoculated SCID mice were completely resolved by polyclonal immune sera to OspC, even when applied at the stage of chronic disease, i.e. 60 days p.i. This indicates that the pathological processes seen in infected mice directly depend on the presence of live spirochetes. Indeed, it has been shown that only viable, but not killed, spirochetes are pathogenic in mice (41) and that the severity of organ destruction is related to the number of microorganisms in the affected tissues (42). It currently is believed that in early stages of infection, pathology is elicited mainly through spirochetal activation of cells of the vasculature and recruited inflammatory cells, and through concomitant induction of inflammatory cytokines (43–45), rather than by specific lymphocytes. Although the therapeutic protocol proposed herein is effective in immunodeficient mice, it is not clear whether it is also operative in immunocompetent hosts. In the latter case, chronic stages of disease also may develop independent of spirochetes and may be governed by activated T and/or B cells (46–48).
It is well established that B. burgdorferi can persist for years in mammalian hosts in the presence of high titers of spirochete-specific Abs (49, 50). Although a number of putative mechanisms for their immune evasion have been described, such as antigenic variation (51), binding of host-derived enzymes (52–54), or intracellular survival (55, 56), it is presently unclear if any of these strategies contribute to the persistence of spirochetes. Our finding that chronic B. burgdorferi infection of SCID mice is resolved by Abs to OspC, at least for >80 days p.i., strongly indicates that in vivo the majority of spirochetes are localized extracellularly in affected tissues of the mammalian host. However, the data do not exclude that a few spirochetes escape this immune surveillance and survive intracellularly or in otherwise immunoprivileged sites.
The present and previous studies suggest a further strategy of B. burgdorferi to obtain persistent infection and to maintain the balance between spirochetes and host immunity within the zoonotic cycle. Accordingly, OspA is a prominent antigen on spirochetes within the midgut of ticks, but is lost upon their transmission to the mammalian host (13, 21). Consequently, anti-OspA Abs are not produced, and because of the lack of this specificity, vector-associated spirochetes are not vulnerable to immune serum ingested by feeding ticks. Conversely, OspC is not expressed on spirochetes within ticks, but produced in the mammalian host and may be critical for their infectivity (57). However, because of their slow growth rate or other unknown reasons, the amount of OspC antigen produced is only sufficient to induce a humoral immune response suitable to control continuous growth of spirochetes, and to some extent also disease, but not to eradicate spirochetes (refs. 49 and 50 and shown here).
Effective prophylactic vaccination against B. burgdorferi infection has been readily documented in laboratory animals and in humans using preparations of recombinant OspA and/or OspA Abs. The conclusion of the present study is that OspC is a suitable target structure for therapeutic treatment of established Lyme disease and persistent infection. Most importantly, complete elimination of spirochetes is critically dependent on the quantity of protective Abs. This is reminiscent of past successes with specific antisera for prevention and treatment of a number of bacterial infections (58). It is tempting to speculate that serum Ab titers to OspC may be of prognostic value for the course of Lyme disease in humans. The results do not exclude the possibility that Abs with different specificities and proven protective potential, such as OspB, P35, and P37 also are involved in the control of Lyme disease (31, 59–61). Together, the data suggest that, at present, a combination of OspA and OspC is the most promising vaccine formula for prevention and treatment of Lyme disease.
Acknowledgments
We thank Melanie Witt for excellent technical assistance and Ian Haidl for critically reading the manuscript. This study was supported in part by the Bundesministerium für Forschung und Technologie, Germany (01 KI 9506/4) and by SmithKline Beecham.
ABBREVIATIONS
- Ab
antibody
- OspA
outer surface protein A
- rLip-OspA
recombinant outer surface lipoprotein A
- rOspC
glutathione S-transferase-OspC fusion protein
- NMS
normal mouse serum
- IS
immune serum
- p.i.
postinfection
- SCID
severe combined immunodeficient
References
- 1.Steere A C. N Engl J Med. 1989;321:586–596. doi: 10.1056/NEJM198908313210906. [DOI] [PubMed] [Google Scholar]
- 2.Benach J L, Bosler E M, Hanrahan J P, Coleman J L, Habicht G S, Bast T F, Cameron D J, Ziegler J L, Barbour A G, Burgdorfer W, Edelman R, Kaslow R A. N Engl J Med. 1983;308:740–742. doi: 10.1056/NEJM198303313081302. [DOI] [PubMed] [Google Scholar]
- 3.Asbrink E, Hovmark A. Acta Pathol Microbiol Immunol Scand. 1985;93:161–163. doi: 10.1111/j.1699-0463.1985.tb02870.x. [DOI] [PubMed] [Google Scholar]
- 4.Pfister H W, Einhäupl K, Preac-Mursic V, Wilske B, Schierz G. J Neurol. 1984;231:141–144. doi: 10.1007/BF00313682. [DOI] [PubMed] [Google Scholar]
- 5.de Koning J, Hoogkamp-Korstanje A A, van der Linde M R, Crijna H J G M. J Infect Dis. 1989;1:150–153. doi: 10.1093/infdis/160.1.150. [DOI] [PubMed] [Google Scholar]
- 6.Preac-Mursic V, Weber K, Pfister H W, Wilske B, Gross B, Baumann A, Prokop J. Infection. 1989;17:355–359. doi: 10.1007/BF01645543. [DOI] [PubMed] [Google Scholar]
- 7.Leigner K, Shapiro J, Ramsey D, Halperin A, Hogrefe W, Kong L. J Am Acad Dermatol. 1993;28:312–314. doi: 10.1016/0190-9622(93)70043-s. [DOI] [PubMed] [Google Scholar]
- 8.Strle F, Cheng Y, Cimperman J, Maraspin V, Lotric-Furlan S, Nelson J A, Picken M M, Ruzic-Sabljic E, Picken R N. Clin Infect Dis. 1995;21:380–389. doi: 10.1093/clinids/21.2.380. [DOI] [PubMed] [Google Scholar]
- 9.Fikrig E, Bockenstedt L K, Barthold S W, Chen M, Tao H, Ali-Salaam P, Telford S R, Flavell R A. J Infect Dis. 1994;169:568–574. doi: 10.1093/infdis/169.3.568. [DOI] [PubMed] [Google Scholar]
- 10.Edelman R. Vaccine. 1997;15:463–464. [Google Scholar]
- 11.Schaible U E, Kramer M D, Eichmann K, Modolell M, Museteanu C, Simon M M. Proc Natl Acad Sci USA. 1990;87:3768–3772. doi: 10.1073/pnas.87.10.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fikrig E, Barthold S W, Kantor F S, Flavell R A. Science. 1990;250:553–556. doi: 10.1126/science.2237407. [DOI] [PubMed] [Google Scholar]
- 13.Schwan T G, Piesman J, Golde W T, Dolan M C, Rosa P A. Proc Natl Acad Sci USA. 1995;92:2909–2913. doi: 10.1073/pnas.92.7.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Silva A M, Telford S R, III, Brunet L R, Barthold S W, Fikrig E. J Exp Med. 1996;183:271–275. doi: 10.1084/jem.183.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilske B, Preac-Mursic V, Schierz G, Busch K V. Zentralbl Bakteriol Mikrobiol Hyg Ser A. 1986;263:92–102. doi: 10.1016/s0176-6724(86)80108-0. [DOI] [PubMed] [Google Scholar]
- 16.Fung B P, McHugh G L, Leong J M, Steere A C. Infect Immun. 1994;62:3213–3221. doi: 10.1128/iai.62.8.3213-3221.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hauser U, Kehnert G, Lobentanzer R, Wilske B. J Clin Microbiol. 1997;35:1433–1444. doi: 10.1128/jcm.35.6.1433-1444.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaible U E, Gern L, Wallich R, Kramer M D, Prester M, Simon M M. Immunol Lett. 1993;36:219–226. doi: 10.1016/0165-2478(93)90056-8. [DOI] [PubMed] [Google Scholar]
- 19.Gern L, Schaible U E, Simon M M. J Infect Dis. 1993;167:971–975. doi: 10.1093/infdis/167.4.971. [DOI] [PubMed] [Google Scholar]
- 20.Zhong, W., Gern, L., Kramer, M., Wallich, R. & Simon, M. M. (1997) Eur. J. Immunol., in press. [DOI] [PubMed]
- 21.Montgomery R R, Malawista S E, Feen K J M, Bockenstedt L K. J Exp Med. 1996;183:261–269. doi: 10.1084/jem.183.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Preac-Mursic V, Wilske B, Patsouris E, Jauris S, Will G, Soutschek E, Rainhardt S, Lehnert G, Klockmann U, Mehraein P. Infection. 1992;20:342–349. doi: 10.1007/BF01710681. [DOI] [PubMed] [Google Scholar]
- 23.Gilmore R D, Jr, Kappel K J, Dolan M C, Burkot T R, Johnson B J B. Infect Immun. 1996;64:2234–2239. doi: 10.1128/iai.64.6.2234-2239.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fikrig E, Telford S R, III, Barthold S W, Kantor F S, Spielman A, Flavell R A. Proc Natl Acad Sci USA. 1992;89:5418–5421. doi: 10.1073/pnas.89.12.5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barthold S W, Bockenstedt L K. Infect Immun. 1993;61:4696–4702. doi: 10.1128/iai.61.11.4696-4702.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shih C M, Spielman A, Telford S R., III Am J Trop Med Hyg. 1995;52:72–74. doi: 10.4269/ajtmh.1995.52.72. [DOI] [PubMed] [Google Scholar]
- 27.Barthold S W, deSouza M, Feng S. Lab Invest. 1996;74:67–67. [PubMed] [Google Scholar]
- 28.Gern L, Rais O, Capiau C, Hauser P, Lobet Y, Simoen E, Voet P, Pêtre J. Immunol Lett. 1994;39:249–258. doi: 10.1016/0165-2478(94)90166-x. [DOI] [PubMed] [Google Scholar]
- 29.Wallich R, Brenner C, Kramer M D, Simon M M. Infect Immun. 1995;63:3327–3335. doi: 10.1128/iai.63.9.3327-3335.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kramer M D, Schaible U E, Wallich R, Moter S, Petzoldt D, Simon M M. Immunobiology. 1990;181:357–366. doi: 10.1016/S0171-2985(11)80504-8. [DOI] [PubMed] [Google Scholar]
- 31.Simon M M, Schaible U E, Kramer M D, Müller-Hermelink H K, Wallich R. J Infect Dis. 1991;164:123–132. doi: 10.1093/infdis/164.1.123. [DOI] [PubMed] [Google Scholar]
- 32.Sinsky R J, Piesman J. J Clin Microbiol. 1989;27:1723–1727. doi: 10.1128/jcm.27.8.1723-1727.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaible U E, Kramer M D, Wallich R, Tran T, Simon M M. Eur J Immunol. 1991;21:2397–2405. doi: 10.1002/eji.1830211016. [DOI] [PubMed] [Google Scholar]
- 34.Barthold S W, Persing D H, Armstrong A L, Peebles R A. Am J Pathol. 1991;139:263–273. [PMC free article] [PubMed] [Google Scholar]
- 35.Schaible U E, Gay S, Museteanu C, Kramer M D, Zimmer G, Eichmann K, Museteanu U, Simon M M. Am J Pathol. 1990;137:811–820. [PMC free article] [PubMed] [Google Scholar]
- 36.Theisen M, Frederiksen B, Lebech A-M, Vuust J, Hansen K. J Clin Microbiol. 1993;31:2570–2576. doi: 10.1128/jcm.31.10.2570-2576.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilske B, Busch U, Fingerle V, Jauris-Heipke S, Preac-Mursic V P, Rössler D, Will G. Infection. 1996;24:208–212. doi: 10.1007/BF01713341. [DOI] [PubMed] [Google Scholar]
- 38.Wilske B, Busch U, Eiffert H, Fingerle V, Pfister H-W, Rössler D, Preac-Mursic V. Med Microbiol Immunol. 1996;184:195–201. doi: 10.1007/BF02456135. [DOI] [PubMed] [Google Scholar]
- 39.Schwan T G, Schrumpf M E, Karstens R H, Clover J R, Wong J, Daugherty M, Struthers M, Rosa P A. J Clin Microbiol. 1993;31:3096–3108. doi: 10.1128/jcm.31.12.3096-3108.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Busch U, Will G, Hizoteufel C, Wilske B, Preac-Mursic V. Res Microbiol. 1997;148:109–118. doi: 10.1016/S0923-2508(97)87642-5. [DOI] [PubMed] [Google Scholar]
- 41.Schaible U E, Kramer M D, Museteanu C, Zimmer G, Mossman H, Simon M M. J Exp Med. 1989;170:1427–1432. doi: 10.1084/jem.170.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang L, Weis J H, Eichwald E, Kolbert C P, Persing D H, Weis J J. Infect Immun. 1994;62:492–500. doi: 10.1128/iai.62.2.492-500.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hurtenbach U, Museteanu C, Gasser J, Schaible U E, Simon M M. Int J Pathol. 1995;76:111–123. [PMC free article] [PubMed] [Google Scholar]
- 44.Simon M M, Kramer M D, Wallich R, Schaible U. In: The Immunology of the Connective Tissue Diseases. Johnstone P, editor. Boston: Kluwer; 1994. pp. 205–229. [Google Scholar]
- 45.Seiler K P, Weis J J. Curr Opin Immunol. 1996;8:503–509. doi: 10.1016/s0952-7915(96)80038-0. [DOI] [PubMed] [Google Scholar]
- 46.Kalish R A, Leong J M, Steere A C. Infect Immun. 1993;61:2774–2779. doi: 10.1128/iai.61.7.2774-2779.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lengl-Janssen B, Strauss A F, Steere A C, Kamradt T. J Exp Med. 1994;180:2069–2078. doi: 10.1084/jem.180.6.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamradt T, Lengl-Janssen B, Strauss A F, Bansal G, Steere A C. Infect Immun. 1996;64:1284–1289. doi: 10.1128/iai.64.4.1284-1289.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwan T G, Kime K K, Schrumpf M E, Coe J E, Simpson W J. Infect Immun. 1989;57:3445–3451. doi: 10.1128/iai.57.11.3445-3451.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barthold S W, de Souza M S, Janotka J L, Persing D H. Am J Pathol. 1993;143:959–972. [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J R, Hardham J M, Barbour A G, Norris S J. Cell. 1997;89:275–285. doi: 10.1016/s0092-8674(00)80206-8. [DOI] [PubMed] [Google Scholar]
- 52.Fuchs H, Wallich R, Simon M M, Kramer M D. Proc Natl Acad Sci USA. 1994;91:12549–12598. doi: 10.1073/pnas.91.26.12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klempner M S, Noring R, Epstein M P, McCloud B, Hu R, Limentani S A, Rogers R A. J Infect Dis. 1995;171:1258–1265. doi: 10.1093/infdis/171.5.1258. [DOI] [PubMed] [Google Scholar]
- 54.Coleman J L, Sellatti T J, Testa J E, Kew R R, Furie M B, Benach J L. Infect Immun. 1995;63:2478–2484. doi: 10.1128/iai.63.7.2478-2484.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma Y, Sturrock A, Weis J J. Infect Immun. 1991;59:671–678. doi: 10.1128/iai.59.2.671-678.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Girschick H J, Huppertz H I, Rüssman H, Krenn V, Karch H. Rheumatol Int. 1996;16:125–132. doi: 10.1007/BF01409985. [DOI] [PubMed] [Google Scholar]
- 57.Masuzawa T, Kurita T, Kawabata H, Yanagihara Y. FEMS Microbiol Lett. 1994;123:319–324. doi: 10.1111/j.1574-6968.1994.tb07242.x. [DOI] [PubMed] [Google Scholar]
- 58.Krause R M, Dimmock N J, Morens D M. J Infect Dis. 1997;176:549–559. doi: 10.1086/514074. [DOI] [PubMed] [Google Scholar]
- 59.Fikrig E, Barthold S W, Marcantonio N, Deponte K, Kantor F S, Flavell R A. Infect Immun. 1992;60:657–661. doi: 10.1128/iai.60.2.657-661.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Probert W S, Crawford M, LeFebvre R B. Vaccine. 1997;15:15–19. doi: 10.1016/s0264-410x(96)00123-5. [DOI] [PubMed] [Google Scholar]
- 61.Fikrig E, Barthold S W, Sun W, Feng W, Telford S R, Flavell R A. Immunity. 1997;6:531–539. doi: 10.1016/s1074-7613(00)80341-6. [DOI] [PubMed] [Google Scholar]