

Summary
HD-domain phosphohydrolases have nucleotidase and phosphodiesterase activities and play important roles in the metabolism of nucleotides and in signaling. We present three 2.1 Å resolution crystal structures (one in the free state and two complexed with natural substrates) of a HD-domain phosphohydrolase, the E. coli 5′-nucleotidase YfbR. The free-state structure of YfbR contains a large cavity accommodating the metal-coordinating HD motif (H33, H68, D69, and D137) and other conserved residues (R18, E72, and D77). Alanine scanning mutagenesis confirms that these residues are important for activity. Two structures of the catalytically inactive mutant E72A complexed with Co2+ and either TMP or dAMP disclose the novel binding mode of deoxyribonucleotides in the active site. Residue R18 stabilizes the phosphate on the Co2+, and residue D77 forms a strong hydrogen bond critical for binding the ribose. The indole side chain of W19 is located close to the 2′-carbon atom of the deoxyribose moiety and is proposed to act as the selectivity switch for deoxyribonucleotide, which is supported by comparison to YfdR, another 5′-nucleotidase in E. coli. The nucleotide bases of both dAMP and TMP make no specific hydrogen bonds with the protein, explaining the lack of nucleotide base selectivity. The YfbR E72A substrate complex structures also suggest a plausible single-step nucleophilic substitution mechanism. This is the first proposed molecular mechanism for a HD-domain phosphohydrolase based directly on substrate-bound crystal structures.
Keywords: HD-domain phosphohydrolase, 5′-Nucleotidase, Alanine scanning mutagenesis, Enzyme mechanism, Macromolecular crystallography
Introduction
The HD-domain, characterized by a motif with a doublet of predicted divalent-cation-coordinating His and Asp residues (H…HD…D), is a widely conserved catalytic domain found in almost 5,000 proteins in bacteria, archaea, and eukaryotes. Enzymes containing HD-domains act as phosphohydrolases that can catalyze both metal-dependent and -independent phosphomonoesterase and phosphodiesterase reactions for a broad range of substrates1. In each sequenced genome, there are seven to twenty HD-domain-containing proteins, either encoded as stand-alone proteins or fused to nucleotidyltransferase or helicase domains1. They have diverse functions associated with nucleic acid and nucleotide metabolism and signal transduction1; 2; 3. They are also related to the well-characterized eukaryotic 3′,5′-cyclic phosphodiesterases, which contain the core H…HD…D motif, but are distinct from the HD domain superfamily in that they contain additional conserved regions.
To date, five HD-domain proteins have been characterized biochemically: dGTPase2, RelA/SpoT4, E. coli tRNA nucleotidyltransferase3, Thermus thermophilus dNTPase5, and E. coli YfbR. The substrates of all characterized HD-domain enzymes are nucleotides, and two enzymes have nucleotidase activity: tRNA nucleotidyl transferase3 and YfbR6. Crystal structures have been solved for only two of these enzymes: the catalytic fragment of the RelA/SpoT homolog from Streptococcus equisimilis (PDB code 1vj7)7 and T. thermophilus dNTPase (2dqb)8. The bifunctional RelA/SpoT catalyzes both the synthesis and hydrolysis of (p)ppGpp, an intracellular signaling alarmone, which triggers the stringent response in bacteria9. Microbial dNTPases control the intracellular pool of dNTPs and hydrolyze them to deoxynucleoside and inorganic triphosphate2; 5. However, the great majority of the HD domain proteins remain uncharacterized.
E. coli YfbR is a conserved HD-domain-containing protein (199 amino acids) with over 100 orthologs found in bacteria, archaea, and eukaryotes. YfbR is a 5′-nucleotidase (5′-NT) strictly specific to 2′-deoxyribonucleotide-5′-monophosphates (dNMPs) and does not discriminate among particular nucleotide bases; it is the first 5′-NT discovered to exhibit this pattern of specificity6. YfbR has been established as an essential component of the deoxycytidine pathway for de novo synthesis of thymidylate in E. coli10. The human genome encodes one YfbR ortholog, the uncharacterized HD-domain-containing protein HDDC2 (28% sequence identity), which might represent a novel intracellular 5′-nucleotidase in humans. In addition to YfbR, the E. coli genome encodes two more stand-alone HD-domain proteins, YfdR (178 amino acids) and YedJ (231 amino acids). These uncharacterized proteins show low sequence similarity to YfbR (18.8% and 15.3% sequence identity, respectively). Sequence alignment of several stand-alone HD domain proteins (Fig. 1) reveals the conservation of the predicted metal-coordinating motif H…HD…D.
Figure 1.

Sequence alignment of YfbR with HD-domain proteins of known structure (AF1432, ATU1042, PF0395, PH0347, T. thermophilus dNTPase), paralogs (E. coli YfdR, YedJ) and orthologs from humans (HDDC2) and S. pombe. The alignment was generated with 3DCOFFEE53, which utilizes both sequence and structural information to align the given sequences. Residues identical to the consensus are in blue and similar residues are shown in purple. The secondary structure of YfbR is shown above the alignment. The signature HD motif is boxed in red, and alanine mutation sites that reduced or abolished catalytic activity (see text) are marked with arrows.
5′-NTs (EC 3.1.3.5 and 3.1.3.6) play an important role in the regulation of the intracellular pool of deoxyribo- and ribonucleotides, cell signaling and nucleotide scavenging pathways in eukaryotes, by catalyzing the dephosphorylation of nucleoside 5′-monophosphates to nucleosides and inorganic phosphate11; 12; 13. Six of the seven mammalian 5′-NTs belong to the haloalkanoate dehalogenase-like (HAD) hydrolase superfamily, and structures have been solved for cN-II (PDB id 2j2c), cN-III (2bdu)14, and mdN (dNT-2; 1mh9)15. The cN-II nucleotidase is most active on GMP and IMP, cN-III on CMP and pyrimidine ribonucleotides, and mitochondrial mdN (dNT-2) on dUMP and d™P16. These well-characterized enzymes are thought to share a common two-step catalytic mechanism forming a phosphoenzyme intermediate14; 17; 18. The seventh class of mammalian 5′-nucleotidases (eN) belongs to the calcineurin superfamily of binuclear metallophosphatases. eNs have broad substrate specificity with AMP as the major physiological substrate12; 16, and the enzymes are proposed act via a single-step catalytic mechanism19.
In contrast, little is known about prokaryotic 5′-NTs. A number of membrane-associated, periplasmic and extracellular bacterial 5′-NTs have been identified and purified, but few have been extensively characterized, such as UshA from E. coli, NucA from Haemophilus influenzae20, and HppA from Helicobacter pylori21. Recently, using a systematic general enzymatic screen against a large set of purified bacterial proteins22, we identified three of the first intracellular bacterial 5′-NTs from E. coli: SurE, YjjG, and YfbR. SurE is a 5′(3′)-nucleotidase of the SurE family and shows catalytic activity against both purine and pyrimidine ribonucleotides and deoxyribonucleotides6. YjjG (like the eukaryotic intracellular 5′-NTs) is a member of the HAD superfamily and dephosphorylates UMP, dUMP, and ™P, with much lower activity for a variety of other mono- and diphosphate nucleotides6.
In this work we used X-ray crystallography to explore the molecular mechanisms of substrate selectivity and catalytic activity of E. coli YfbR. We report three 2.1 Å resolution crystal structures of this protein, one in a free state and two with bound natural substrates and metal co-factors, and correlate the structures to kinetic studies of mutants of the protein. Our results explain the structural basis of the substrate specificity of this nucleotidase and suggest a possible catalytic mechanism for HD-domain phosphohydrolases. This work represents the first detailed study of the substrate selectivity and catalytic mechanism of a HD-domain phosphohydrolase based directly on substrate-bound crystal structures.
Results
Purified E. coli HD-domain proteins were screened for crystallization and the structure of SeMet-substituted YfbR was solved by SAD (Fig 2), and the wild-type structure of YfbR was deposited to the PDB with id 2paq. The crystallographic data collection and refinement statistics are shown in Table 1. The globular structure consists of 8 α-helices connected by extended loops. Two protomers are found in the asymmetric unit. In both chains, residues 82–90 in the loop between helices α3 and α4 and residues 188–199 of the C-terminus of the protein were disordered in the structure. In addition, the side chains of several residues, particularly in helix α4, could not be modeled. Analysis of the crystal packing as well as size exclusion chromatography results (data not shown) suggested that the biological unit is a dimer, where the interface surface area is approximately 2100 Å2. The architecture of the four residues (H33, H68, D69, and D137) that compose the cation binding site is very similar to that found in the structure of the catalytic N-terminal fragment of S. equisimilis RelA7, save that the imidazole side chain of H68 is oriented pointing away from the cation binding site. No density for a metal in the divalent cation binding site was observed.
Figure 2.
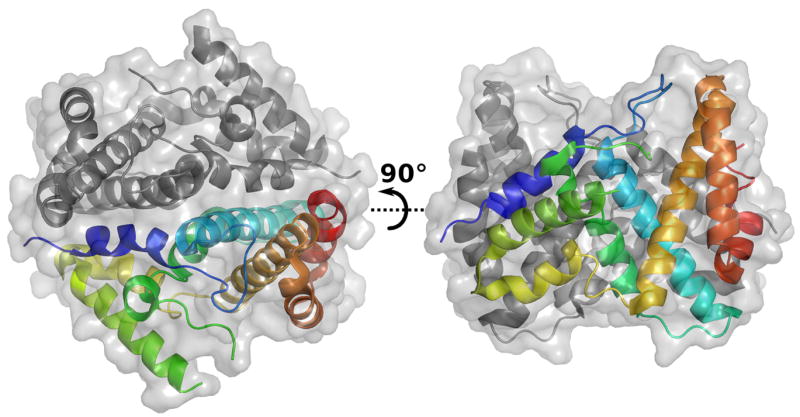
Structure of the wild-type YfbR dimer. One of the two polypeptide chains is colored in rainbow representation (N-terminus in blue to C-terminus in red).
Table 1.
Data collection and refinement statistics for the crystal structures of YfbR described in this study. Statistics for the highest resolution shell for each structure are shown in parentheses.
| Data collection | wild-type YfbR | E72A-Co-TMP | E72A-Co-dAMP |
|---|---|---|---|
| Space group | R3 | R3 | R3 |
| Unit cell parameters (Å) | A=b=137.02
c=56.29 |
a=b=136.14
c=55.42 |
a=b=135.60
c=54.94 |
| Wavelength (Å) | 0.979 | 0.979 | 0.979 |
| Resolution range (Å) | 50–1.95 (2.02–1.95) | 50–2.1 (2.14–2.10) | 50–2.1 (2.18–2.10) |
| I/σI | 24.9 (1.4) | 22.9 (2.8) | 28.2 (3.2) |
| Rmerge | 0.099 | 0.081 | 0.063 |
| Unique reflections | 28633 | 22238 | 21959 |
| Completeness (%) | 98.2 (87.8) | 100.0 (100.0) | 99.9 (100.0) |
| Redundancy | 5.0 (3.6) | 5.1 (5.1) | 4.5 (4.4) |
| Solvent content (%) | 44 | 43 | 42 |
| Solved by | Se-Met SAD | MR | MR |
| Refinement | |||
| Resolution range (Å) | 50–2.1 | 40.4–2.1 | 40.1–2.1 |
| No. protein atoms/AU | 2793 | 2783 | 2777 |
| No. waters/AU | 126 | 42 | 53 |
| No. substrate atoms/AU | 0 | 51 | 53 |
| R/Rfree (%) | 18.7/23.7 | 19.6/24.7 | 19.6/26.0 |
| Mean B-factor (Å2) | 26.6 | 35.0 | 38.3 |
| RMSD bond length (Å) | 0.022 | 0.020 | 0.022 |
| RMSD angles (°) | 1.81 | 1.84 | 1.91 |
| Ramachandran favored (%) | 97.7 | 97.4 | 98.8 |
| Ramachandran allowed (%) | 2.3 | 2.6 | 1.2 |
| Ramachandran outliers (%) | 0.0 | 0.0 | 0.0 |
| PDB id | 2PAQ | 2PAR | 2PAU |
A Dali search for structural homologs of YfbR identified four stand-alone HD-domain protein structures in the PDB: AF1432 from Archaeglobus fulgidus (PDB id 1ynb), PF0395 from Pyrococcus furiosus (1xx7), PH0347 from Pyrococcus horikoshii (2cqz), and ATU1052 from Agrobacterium tumefaciens (2gz4). Although these proteins share relatively low sequence similarity with YfbR (26% to 32% sequence similarity), their crystal structures indicate strong structural similarity (Z-scores of 10.7 to 18.4, r.m.s.d. 2.3 to 2.7 Å). The structures of AF1432, PF0395, and PH0347 were most similar to YfbR in the regions corresponding to helices α2, α3, and α6 (r.m.s.d. 2.3 to 2.5 Å).
Alanine scanning mutagenesis was performed to determine the role of eleven conserved residues located near the predicted active site. The nucleotidase activity (Fig. 3) and substrate affinity (Table 2) of the mutant enzymes was compared to wild type YfbR using dAMP as a substrate. The W19A mutant protein pelleted in the insoluble fraction and could not be purified, while the other mutants were soluble. The V37A mutant showed wild-type activity and substrate affinity, another valine mutant (V30A) and E122A exhibited reduced activity and affinity, and seven mutants (R18A, H33A, H68A, D69A, E72A, D77A, and D137A) had greatly reduced or negligible enzymatic activity (Fig. 3, Table 2). Four of these residues (H33, H68, D69, and D137) comprise the HD metal-binding motif.
Figure 3.
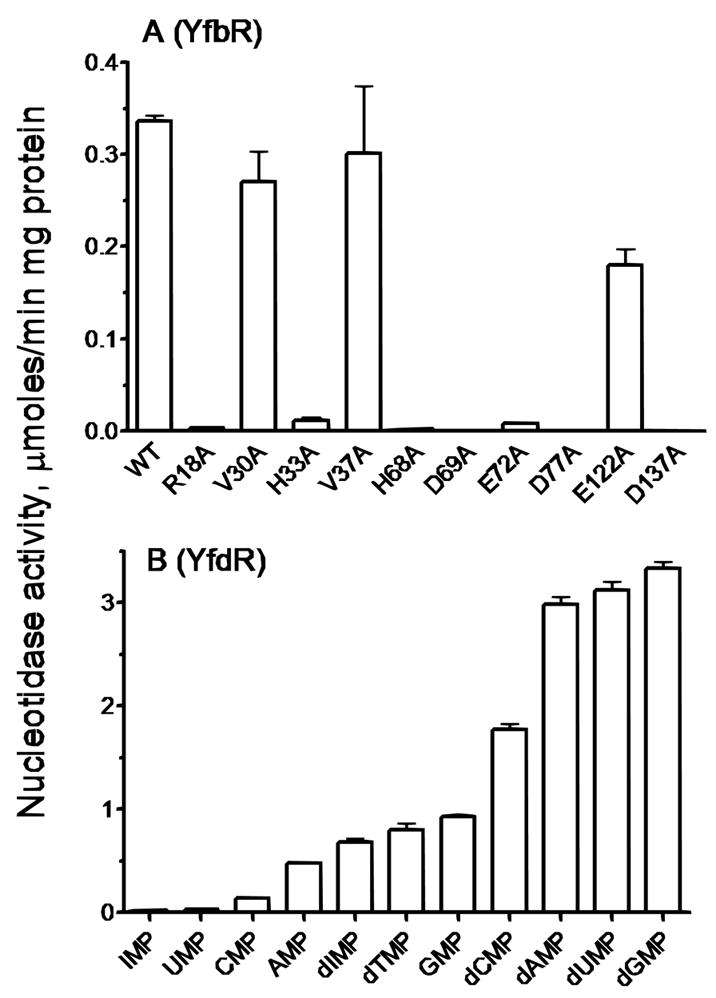
Nucleotidase activity of wild-type (WT) and mutants of YfbR, in the presence of CoCl2 and dAMP, above, and nucleotidase activity of YfdR for selected natural substrates in the presence of CoCl2, below.
Table 2.
Kinetic parameters of the E. coli YfbR (wild type and mutants) and wild type YfdR with various substrates.
| Protein | Variable substrate | Km, μM | Vmax, U/mga | kcat, s−1 | kcat/Km, M−1s−1 |
|---|---|---|---|---|---|
| YfbR (w.t.) | dAMP | 17.0 ± 3.0 | 0.36 ± 0.01 | 0.14 ± 0.004 | 8.09 × 103 |
| YfbR (V30A) | dAMP | 42.0 ± 5.0 | 0.22 ± 0.01 | 0.08 ± 0.004 | 1.98 × 103 |
| YfbR (V37A) | dAMP | 16.0 ± 2.0 | 0.35 ± 0.01 | 0.13 ± 0.004 | 8.41 × 103 |
| YfbR (E122A) | dAMP | 34.0 ± 3.0 | 0.16 ± 0.01 | 0.06 ± 0.004 | 1.72 × 103 |
| YfdR (w.t.) | dAMP | 118.9 ± 19.0 | 5.88 ± 0.35 | 2.28 ± 0.14 | 1.90 × 104 |
| dGMP | 64.7 ± 2.3 | 4.13 ± 0.05 | 1.60 ± 0.02 | 2.46 × 104 | |
| dUMP | 310.5 ± 37.6 | 3.51 ± 0.18 | 1.36 ± 0.07 | 0.44 × 104 | |
| dCMP | 78.5 ± 4.1 | 2.17 ± 0.04 | 0.84 ± 0.02 | 1.06 × 104 | |
| d™P | 129.4 ± 11.3 | 3.08 ± 0.10 | 1.19 ± 0.04 | 0.92 × 104 | |
| dIMP | 47.5 ± 4.2 | 1.56 ± 0.04 | 0.61 ± 0.02 | 1.27 × 104 | |
| GMP | 379.8 ± 23.6 | 1.78 ± 0.05 | 0.69 ± 0.02 | 1.82 × 102 | |
| Co2+ b | 340.0 ± 50.0 | 3.13 ± 0.19 | 1.21 ± 0.07 | 3.56 × 103 |
U/mg, μmoles/min mg protein
determined using 0.5 mM dAMP as substrate
The other stand-alone HD-domain containing proteins found in E. coli, YfdR and YedJ, were over-expressed, purified and tested for phosphohydrolase activity against a range of phosphatase and phosphodiesterase substrates22. In these assays, YedJ exhibited no significant activity, and YfdR exhibited 5′-NT activity with a substrate range broader than that of YfbR. In addition to dNMPs, YfdR also demonstrated non-negligible 5′-nucleotidase activity for GMP, AMP, and CMP (Fig. 3). When compared to YfbR, YfdR demonstrated higher catalytic activity, but lower substrate affinity (Table 2).
The seven YfbR alanine mutants with greatly reduced catalytic activity were subjected to crystallization trials, and the E72A protein produced diffraction-quality crystals in a crystal form similar to that of wild-type (Table 1). Co-crystallizations of YfbR E72A with CoCl2 and dNMPs failed, likely due to the chelating effect of the acetate molecules in the crystallization solution. However, by soaking crystals of the mutant for long periods of time (10–18 hours) with the substrates in a buffer where citrate was replaced by acetate, two well-diffracting datasets were obtained with relatively high occupancy for both metal and nucleotide in the electron density (Table 1). One crystal (E72A-Co-TMP) was soaked with CoCl2 and TMP, and the other (E72A-Co-dAMP) was soaked with CoCl2 and dAMP. Both structures of YfbR E72A were solved by molecular replacement (using wild type YfbR as the search model).
The conformations of the substrate-soaked structures of YfbR E72A are very similar to wild type YfbR, with an r.m.s.d. of 0.45 Å and 0.48 Å for E72A-Co-dAMP and E72A-Co-TMP, respectively. In both structures, the metal-binding HD motif in each monomer contained a single strong peak of density in the 2Fo-Fc maps (~13.5 σ for E72A-C-TMP, ~12 σ for E72A-Co-dAMP), and residue H68 is reoriented to a position coordinating the metal cation in the binding site. An anomalous difference map calculated for a similar crystal soaked with CoCl2 and diffracted at a wavelength of 1.28 Å confirmed there is a single metal ion per monomer, which is presumed to be Co2+ (data not shown). The metal ion is coordinated in a distorted octahedral configuration by the four residues of the HD motif (H33, H68, D69, and D137), a water molecule, and a phosphate oxygen of the bound nucleotide (Fig. 4).
Figure 4.

Structure and 2Fo-Fc density of YfbR E72A mutants with substrate and metal cofactor bound. A) Stereo view of the TMP (blue) and Co2+ (pink sphere) binding site (light gray). dAMP from the equivalent position in the other structure is superimposed (pink). B) σA-weighted 2Fo-Fc electron density maps for nucleotide substrate in the E72A-Co-TMP (blue) and E72A-Co-dAMP (red) structures. Maps are contoured at 1σ.
In both E72A-Co-dAMP and E72A-Co-TMP, the nucleotide substrates bind in similar configurations (Fig. 4). The mode of binding for TMP is shown schematically in Fig. 5. The occupancies of the nucleotide and Co2+ in chains A and B of E72A-Co-dAMP are 0.8 and 0.9, respectively, while there is complete substrate occupancy in E72A-Co-TMP. The side chain of residue R18 forms a strong hydrogen bond with the nucleotide phosphate (Fig. 5). The 2′-deoxyribofuranose rings of the substrates in E72A-Co-TMP and E72A-Co-dAMP adopt C2′-endo configurations, the conformation preferred in B-DNA. Three hydrogen bonds are formed with the deoxyribose: a strong H-bond from the 3′ hydroxyl to the carboxyl group of D77 and two weaker H-bonds from backbone nitrogens to the ribose: W19 N-H … O3′ and T80 N-H … O4′. Significant van der Waals contacts are also made with residues W19, P79, T80, P81, A141, and L163 (Fig. 4). No hydrogen bonds or other specific contacts are formed with the residues on the edge of the bases of either nucleotide in either structure, which is exposed to bulk solvent.
Figure 5.
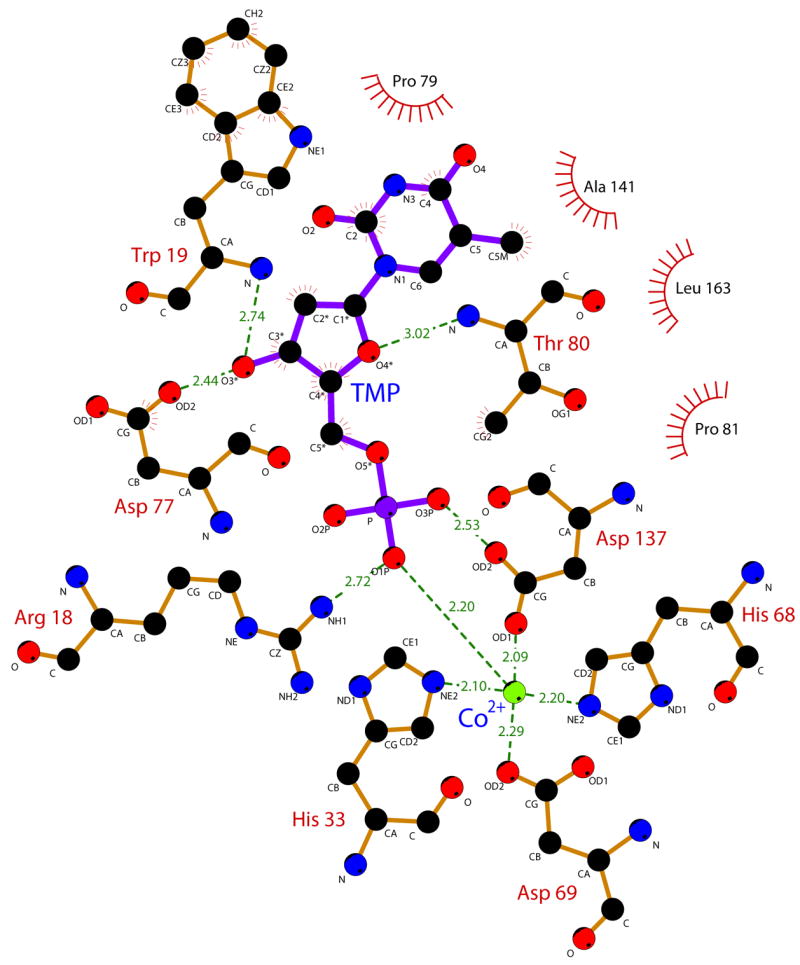
Schematic representation of the binding mode of TMP to YfbR E72A. Protein residues are shown with brown bonds, TMP is shown with purple bonds, and Co2+ is shown as a green sphere. Atoms and residues involved in van der Waals contacts are marked with red lines. Substrate-protein hydrogen bonds and cation contacts are shown as green lines, with bond distances in Å. The figure was generated using LIGPLOT56.
Discussion
The binding of TMP and dAMP in the E72A-Co-TMP and E72A-Co-dAMP structures demonstrate a novel architecture for substrate recognition for a 5′-nucleotidase. As in the mammalian phosphodiesterase 423, the metal atom bound to the HD motif makes a direct contact with the phosphate oxygen atom of the substrate in the E. coli YfbR (Figs. 4, 5). In contrast, the metal cofactor does not coordinate a phosphate of the bound natural inhibitor guanosine-5′-diphosphate-2′,3′-cyclic monophosphate found in the structure of RelA from S. equisimilis7. The binding sites of S. equisimilis RelA and of T. thermophilus dNTPase8 are superimposed on the structure of E72A-Co-TMP in Fig. 6. In all three structures, the four residues of the HD motif, as well as the three α-helical fragments to which they belong, share a conserved structural architecture and bind divalent cations in equivalent positions. However, the architecture of the residues involved in substrate recognition are not conserved, either by sequence or in structure. Although all of the ligands seem to bind in the same general position relative to the metal cation, the ribose rings and nitrogenous bases all adopt significantly different orientations, and bind to non-conserved motifs. There is no substrate in the structure of dNTPase, but the residues predicted to be involved in substrate binding are not conserved8.
Figure 6.
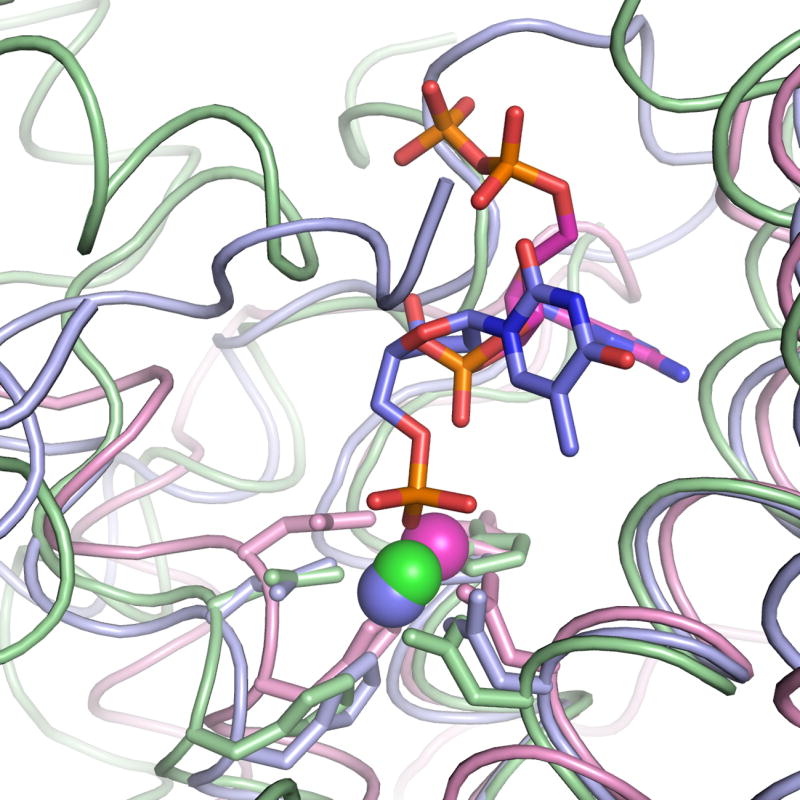
Superposition of three structures of HD-domain proteins with divalent cations and substrates (or natural inhibitors) bound. YfbR E72A complexed with Co2+ and TMP (this work) is shown in blue. S. equisimilis RelA complexed with Mn2+ and guanosine-5′-diphosphate-2′,3′-cyclic monophosphate is shown in magenta7. T. thermophilus dNTPase complexed with Mg2+ is shown in green8.
The phosphate is stabilized in position near the Co2+ cation by the side chain of R18. Residue R18 appears to be important in orienting the phosphate for catalysis, as the R18A mutant had negligible catalytic activity. The deoxyribose is bound by several specific hydrogen bonds, including a strong H-bond from the side chain of residue D77 to the 3′-OH of the deoxyribose. This H-bond is also critical for proper substrate binding, as the D77A mutant also had negligible catalytic activity. In the structure of the human mitochondrial deoxyribonucleotidase dNT-2 (which is not a member of the HD-domain superfamily), the 3′-OH group of the deoxyribose is coordinated to an aspartate residue (D43)15. In contrast to dNT-2, YfbR does not hydrolyze deoxyribonucleoside 3′-monophosphates6, which agrees with the observation that the 3′-OH group is located far away from the metal cofactor and thus any putative metal-activated nucleophiles. In addition, in both E72A-substrate complex structures, the 3′-O of ribose is located close to the side chain of P20 (3.8 Å – 4.0 Å) suggesting that YfbR will not bind these nucleotides. It is also apparent that the binding site can only accommodate one phosphate group of the substrate, explaining the lack of activity for deoxyribonucleoside di- or triphosphates. In both nucleotide-bound structures (one with a purine and one with a pyridimine), no hydrogen bonds or other specific contacts are made with the polar atoms on the edge of the nucleotide base, explaining the lack of specificity for particular nucleotides observed in YfbR.
The sterical hindrance between W19 side chain and 2′-OH of ribonucleotide is likely to determine the selectivity of YfbR for 2′-deoxyribonucleotides. The indole plane of the side chain of W19 is located approximately 3.5 Å from the 2′ carbon atom of the deoxyribose (Fig. 4A). When an 2′-OH moiety is modeled on the ribose, its oxygen atom would be located approximately 2.5 Å from the indole plane, too close even to form an aromatic hydrogen bond. Human mitochondrial (mdN) and cytosolic (cdN) deoxyribonucleotidases are the only other known 5′-nucleotidases that prefer the deoxyribo-form over ribo-form of nucleoside 5′-monophosphates16; 18. Structural studies of mdN revealed that its selectivity for deoxyribonucleotides is due to the presence of a hydrophobic pocket formed by F49, F102, and I133 near the 2′-carbon of the sugar18. E. coli YfdR, an ortholog of YfbR where Trp is replaced by Phe (Fig. 1), demonstrated high nucleotidase activity against both deoxyribo- and ribonucleoside monophosphates and substrate selectivity similar to that of human deoxyribonucleotidases mdN and cdN (Fig. 3). When compared to YfbR, YfdR demonstrated higher catalytic activity, but lower substrate affinity (Table 2), suggesting that this protein is likely to play a different role in E. coli cells. The presence of a smaller Phe side chain in YfdR is the likely reason for the reduced selectivity of this protein toward deoxyribonucleotides. In the structures of the uncharacterized YfbR orthologs PF0395 and PH0347, the conserved Trp is positioned 3 Å further away from the substrate binding site due to the insertion of two amino acid residues after the conserved Arg (Fig. 1), suggesting that these proteins might have even lower selectivity for deoxyribonucleotides.
The structures of the complexes of YfbR with the natural substrates suggest a catalytic mechanism. Virtually all enzymatic substitution reactions of phosphates proceed via an inline attack by a nucleophile, where the attacking and leaving groups are located on opposite sides of the phosphorus atom24. In the case of the E72A-Co-TMP and E72A-Co-dAMP structures, the most likely candidate for the attacking nucleophile is the water moiety coordinated to the Co2+, which we propose is in hydroxide form. This model is supported by the strict metal dependence of the YfbR nucleotidase activity demonstrated previously6. While a hydroxide ion and a water molecule cannot be directly distinguished by electron density at 2.1 Å resolution, the shortened Co2+—O bond distance of 1.9 Å suggests that this moiety is a hydroxide ion.
The source of the proton for the leaving 5′-O atom of the deoxynucleoside product is not apparent based on the crystal structures reported here. No residue (conserved or otherwise) capable of donating a proton to the product is located close enough to do so in the E72A structures. When the wild-type structure is superimposed on the E72A-Co-TMP structure, the E72 residue is too far away to donate this proton. However, as substituting Glu with Ala is a drastic mutation, the conformation of the protein and/or the substrate in the complex of wild-type enzyme with nucleotide may differ significantly from that seen in the crystal structures. One possibility is that E72, which is important for YfbR activity as determined by mutagenic studies, donates a proton mediated through a water bridge. The water in the wild-type structure labelled HOH1101 in Fig. 7A is in the approximate position of this possible water bridge (Fig. 7A), though the position of this water is not positioned ideally for this role. (A schematic of this speculated mechanism is shown in Fig. 7B). However, it is unclear if E72 is energetically capable of donating a proton, though the residue’s pKa may be shifted upward by interaction with acidic residue D77. A water is not universally present in this position in the nucleotide-bound structures. A more conclusive answer as to the role of E72 and/or waters in the catalytic mechanism awaits structures of substrate or substrate-analogs bound to either wild-type enzyme or a more conservative mutant such as E72Q.
Figure 7.

Proposed mechanism for catalytic activity of YfbR. A) Stereo view of the binding site of YfbR E72A (light gray) superimposed on WT YfbR (yellow). Waters from YfbR E72A and from wild-type YfbR are shown as gray and yellow spheres, respectively. B) Schematic of a possible catalytic mechanism.
Since the phosphate conformation is rotated slightly away from the optimal position for nucleophilic attack by a Co2+-bound hydroxide ion, the possibility that another nucleophile is involved in the reaction cannot be excluded. For example, the oxygen of the carboxylate group of D137 not coordinated to the metal could serve as the nucleophile, forming a covalent phosphoaspartyl intermediate. The phosphate then would be liberated by a second substitution reaction with water. Such a two-step mechanism has been proposed for human deoxynucleotidase17; 19. Alternatively, another water bound to an acidic residue could serve as the nucleophile, as was suggested for the conserved E81 in S. equisimilis RelA7. However, in YfbR this mechanism would require a kinetically slow pseudorotation of the hypervalent phosphorus intermediate25, which is rarely observed in phosphohydrolases or phosphotransferases. Additional functional and structural experiments with transition state analogs, such as AlF3, are necessary to determine the mechanism conclusively.
The crystal structures of YfbR provide a molecular explanation for the novel pattern of substrate selectivity of the 5′-nucleotidase. The structures also suggest the mechanism of catalysis, which is based on a single-step substitution mechanism (Fig. 7B). These results provide further insight into the function of YfbR, an essential component of the deoxycytidine nucleotide scavenging pathway for thymidylate synthesis in E. coli10. The substrate binding mode demonstrated here differs significantly from those demonstrated and proposed for other members of the HD-domain phosphohydrolase superfamily. The known substrates of the HD-domain phosphohydrolases include such various nucleotides as 5′- and 2′-deoxyribo- and ribonucleoside monophosphates, (p)ppGpp, and (d)NMPs2; 5; 6; 7. Thus, the members of the HD domain superfamily have developed different modes of substrate binding while retaining a similar structural fold and using the same metal binding motif to catalyze a phosphohydrolase reaction.
Materials and Methods
Cloning and Protein Purification
Genes yfbR, yfdR, and yedJ from E. coli were cloned into a modified pET15b (Novagen) vector (p11) that introduced a hexahistidine affinity tag and a tobacco etch virus (TEV) protease site to the N-terminus of the translated proteins, and native protein was over-expressed as previously described26. To produce the selenomethionine (SeMet)-containing YfbR protein for structural studies, the vector was transformed into the E. coli methionine-auxotrophic strain B834(DE3)pLysS (Novagen) and the cells were grown in 1L flasks of M9 minimal media with 8 mg/L methionine. When the cultures reached stationary phase, SeMet was added to a final concentration of 50 mg/L. Both native and SeMet-incorporated cultures were then induced, harvested, and purified on nickel nitrilotriacetic acid (Ni-NTA) affinity resin (Qiagen). Soluble cell lysates from 1 L cultures were loaded onto 5 mL (packed volume) columns of Ni-NTA resin equilibrated with binding buffer (5% v/v glycerol, 0.5 M NaCl, 50 mM HEPES pH 7.5, and 5 mM imidazole). The columns were washed with 10 column volumes of binding buffer containing 30 mM imidazole, then purified protein was eluted with binding buffer containing 250 mM imidazole. To prepare for crystallization of YfbR, the polyhistidine affinity tag was cleaved by digestion with recombinant TEV protease27 at 4 °C for 2–4 days, and the cleavage was confirmed by SDS-PAGE and mass spectroscopy. Cleaved protein was dialyzed back into binding buffer and applied to a second Ni-NTA column prepared as before, but unexpectedly bound again to the affinity resin. The protein was eluted and further purified by size-exclusion chromatography on a HiPrep 16/60 Superdex 200 column using an AKFA FPLC system (GE Healthcare).
Alanine Mutagenesis
Site-directed mutagenesis was performed using the QuikChange™ site-directed mutagenesis kit (Stratagene) according to the manufacturer’s protocol. Eleven conserved residues of YfbR located in the putative catalytic site (R18, W19, V30, H33, V37, H68, D69, E72, D77, E122, and D137) were mutated to alanine. The yfbR open reading frame cloned on the expression vector p11 was used as a template. The experimental conditions were as previously described28. Plasmids were purified using Qiaprep Mini Prep kit (Qiagen), and all mutations were verified by DNA sequencing. Mutated proteins were then over-expressed and purified as described above.
Phosphatase Screening and Enzymatic Assays
General phosphatase and natural phosphatase substrate screening was performed essentially as previously described22. Briefly, YfdR was screened for phosphatase activity using the general phosphatase substrate p-nitrophenyl phosphate (pNPP) in 200 μL reactions. The reactions contained 50 mM HEPES-K pH 7.5, 5 mM MgCl2, 1 mM MnCl2, 0.5 mM NiCl2, 20 mM pNPP and 10μg of purified protein. Reactions were incubated for one hour at 37°C and then activity was measured by absorbance at 410 nm. Natural phosphatase substrates were identified by screening a set of 90+ endogenous phosphate-containing compounds (nucleotides and phosphorylated sugars, amino acids, and organic acids) in 96-well microplates in 160 μL reactions. Reaction wells contained 50 mM HEPES-K pH 7.5, 0.5 mM CoCl2, 0.08–0.25 mM substrate and 3 μg of purified protein. After one hour of incubation at 37°C, the reactions were halted by the addition of 40 μL of the Malachite Green reagent29 and the level of Pi released was measured by absorbance at 630 nm. To establish kinetic constants, saturation curves were prepared for numerous phosphorylated nucleotides. The 160 μL reactions contained 50 mM HEPES-K pH 7.5, 0.5 mM CoCl2, 3 μM–4 mM substrate, and 0.07 – 0.15 μg of YfdR (depending on activity level with the specific substrate). The reactions were incubated at 37°C for 25 minutes and stopped with 40 μL Malachite Green reagent and the free Pi was measured at 630 nm. The resultant curves were plotted and analyzed using GraphPad Prism software (www.graphpad.com). The YfbR mutant profile was studied by testing the mutants for activity against dAMP under saturating conditions for the wild-type protein. The 160 μL reaction mixtures contained 50 mM HEPES-K pH 8.0, 0.5 mM CoCl2, 0.1 mM dAMP, and 1 μg of a YfbR mutant (or wild-type). The reactions were incubated for 20 minutes, then 40 μL of the Malachite Green reagent was added and the free Pi was measured at 630 nm. The dAMP saturation curves for the YfbR mutants that showed activity were done with 160 μL reaction mixtures containing 50 mM HEPES-K pH 8.0, 0.5 mM CoCl2, 3 μM – 1.25 mM dAMP, and 0.6 – 1.2 μg of a YfbR mutant or wild-type protein.
Crystallization and Crystal Harvesting
Purified protein was dialyzed into 10 mM HEPES pH 7.5 and 0.5 M NaCl, and concentrated to 11–20 mg/mL. Protein was screened for crystallization in a series of commercial screens, and promising hits were optimized. Crystallization experiments were designed and tracked using the Xtaldb crystallization expert system (Zimmerman & Minor, manuscript in preparation). Crystals of SeMet-substituted wild-type YfbR were crystallized by hanging-drop vapor diffusion, where the well contained a solution of 10% w/v PEG 3350, 0.3 M NH4 citrate, and 5 mM β-mercaptoethanol, and the drop contained 2 μL of protein solution mixed with 2 μL of well solution. Crystals were harvested, briefly soaked in a 4:1 mixture of well solution and glycerol, and flash-frozen in liquid N2.
YfbR alanine mutants were screened both in commercial screens and in conditions that produced crystals for the wild-type protein. Well-diffracting crystals of the E72A mutant were produced by hanging-drop vapor diffusion, as described above, in 0.1 M NH4 citrate, 5% w/v PEG 3350, 0.2–0.8% w/v NDSB 256, and 1% v/v glycerol. Crystals were transferred into a soaking buffer containing 5 mM CoCl2, 0.15 M Na acetate pH 4.5, 5% w/v PEG 3350, and either 8.3 mM 2′-deoxyriboadenosine-5′-monophosphate (dAMP) or 7.5 mM thymidine-5′-monophosphate (™P), and incubated for 10–18 hours. The crystals were then briefly transferred into a 3:1 mixture of soaking buffer and PEG 400 and flash-frozen in liquid N2.
Diffraction, Structure Solution, and Refinement
Diffraction data on a SeMet-substituted wild-type YfbR crystal were collected at the Se peak wavelength at beamline 19-ID of the Structural Biology Center (SBC-CAT) at the Advanced Photon Source (APS)30. The data were indexed, integrated, and scaled with HKL-200031. The structure was solved by SAD phasing and an initial model was generated using an early version of HKL-300032, integrated with MLPHARE33, DM34, SHELXD35, SHELXE36, CCP437, SOLVE38 and RESOLVE39. Independently, the structure was solved manually, by finding Se sites using SHELXD, calculating a map using SHARP/autoSHARP40, and building an initial model with ARP/wARP41. The HKL-3000 model and the ARP/wARP model were then manually combined. The combined model was refined by cycles of simulated annealing and restrained maximum-likelihood refinement in CNS 1.142 and manual rebuilding in O43. This model was deposited to the PDB with id 1wph. The wild-type model was further improved by maximum-likelihood restrained refinement in REFMAC544 from the CCP4 package37, and manual rebuilding with COOT45. TLS restraints46 were used in refinement. The TLS model consisted of 12 groups (6 segments per chain) as identified by the TLS Motion Determination server47, and a corrected model was deposited with PDB id 2paq.
Diffraction data on substrate-soaked YfbR E72A mutant crystals were also collected at APS beamline 19-ID. Reflection data were collected, indexed, integrated, and scaled using HKL-3000. Data for the dAMP-soaked crystal were partially merohedrally twinned by the operation (h, -h-k, -l), with a twin fraction of 0.3548. Corrected structure factors produced by the DETWIN program from the CCP4 distribution37 were used for structure solution and refinement. Structures were solved by molecular replacement with MOLREP49, using the molecular replacement mode of HKL-3000. The wild-type structure, with waters removed and SeMet replaced by methionine, was used as a search model. The structures were iteratively improved by manual rebuilding with COOT, followed by maximum-likelihood restrained refinement with REFMAC5. 2-fold non-crystallography symmetry (NCS) and TLS restraints were used in refinement. The TLS model consisted of 14 groups, 7 per chain, identified as described above.
Alignments and figures
The structures of YfbR, AF1432, PF0395, PH0347, and ATU1052 were aligned and all structural similarity statistics were calculated using DaliLite50. Other structural alignments were generated by secondary-structure matching (SSM) superposition51, as implemented in COOT. The sequence alignment was created by inputting the set of HD-domain proteins along with the 31 sequences of the COG1896 domain52 to the 3DCOFFEE web server53, which generates alignments using both sequence and structural information. The sequence alignment figure was created with TEXSHADE54, and structure and electron density figures were created with PyMOL55.
Acknowledgments
We thank A. Joachimiak, M. Chruszcz, D. Cooper and SBC and MCSG members for help and discussion. We also thank A. Savchenko and M. Kudritska for providing clones. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by U. Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. Work was supported by NIH PSI grants GM62414 and GM074942 and by Genome Canada.
Footnotes
ACCESSION CODE: Structure factor has been deposited in the Protein Data Bank with accession number 2PAQ
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aravind L, Koonin EV. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem Sci. 1998;23:469–72. doi: 10.1016/s0968-0004(98)01293-6. [DOI] [PubMed] [Google Scholar]
- 2.Seto D, Bhatnagar SK, Bessman MJ. The purification and properties of deoxyguanosine triphosphate triphosphohydrolase from Escherichia coli. J Biol Chem. 1988;263:1494–9. [PubMed] [Google Scholar]
- 3.Yakunin AF, Proudfoot M, Kuznetsova E, Savchenko A, Brown G, Arrowsmith CH, Edwards AM. The HD domain of the Escherichia coli tRNA nucleotidyltransferase has 2′,3′-cyclic phosphodiesterase, 2′-nucleotidase, and phosphatase activities. J Biol Chem. 2004;279:36819–27. doi: 10.1074/jbc.M405120200. [DOI] [PubMed] [Google Scholar]
- 4.An G, Justesen J, Watson RJ, Friesen JD. Cloning the spoT gene of Escherichia coli: identification of the spoT gene product. J Bacteriol. 1979;137:1100–10. doi: 10.1128/jb.137.3.1100-1110.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kondo N, Kuramitsu S, Masui R. Biochemical characterization of TT1383 from Thermus thermophilus identifies a novel dNTP triphosphohydrolase activity stimulated by dATP and dTTP. J Biochem (Tokyo) 2004;136:221–31. doi: 10.1093/jb/mvh115. [DOI] [PubMed] [Google Scholar]
- 6.Proudfoot M, Kuznetsova E, Brown G, Rao NN, Kitagawa M, Mori H, Savchenko A, Yakunin AF. General enzymatic screens identify three new nucleotidases in Escherichia coli. Biochemical characterization of SurE, YfbR, and YjjG. J Biol Chem. 2004;279:54687–94. doi: 10.1074/jbc.M411023200. [DOI] [PubMed] [Google Scholar]
- 7.Hogg T, Mechold U, Malke H, Cashel M, Hilgenfeld R. Conformational antagonism between opposing active sites in a bifunctional RelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response [corrected] Cell. 2004;117:57–68. doi: 10.1016/s0092-8674(04)00260-0. [DOI] [PubMed] [Google Scholar]
- 8.Kondo N, Nakagawa N, Ebihara A, Chen L, Liu ZJ, Wang BC, Yokoyama S, Kuramitsu S, Masui R. Structure of dNTP-inducible dNTP triphosphohydrolase: insight into broad specificity for dNTPs and triphosphohydrolase-type hydrolysis. Acta Cryst D. 2007;63:230–9. doi: 10.1107/S0907444906049262. [DOI] [PubMed] [Google Scholar]
- 9.Chatterji D, Ojha AK. Revisiting the stringent response, ppGpp and starvation signaling. Curr Opin Microbiol. 2001;4:160–5. doi: 10.1016/s1369-5274(00)00182-x. [DOI] [PubMed] [Google Scholar]
- 10.Weiss B. The deoxycytidine pathway for thymidylate synthesis in Escherichia coli. J Bacteriol. 2007;189:7922–6. doi: 10.1128/JB.00461-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bianchi V, Pontis E, Reichard P. Interrelations between substrate cycles and de novo synthesis of pyrimidine deoxyribonucleoside triphosphates in 3T6 cells. Proc Natl Acad Sci U S A. 1986;83:986–90. doi: 10.1073/pnas.83.4.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmermann H. 5′-Nucleotidase: molecular structure and functional aspects. Biochem J. 1992;285(Pt 2):345–65. doi: 10.1042/bj2850345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunsucker SA, Mitchell BS, Spychala J. The 5′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol Ther. 2005;107:1–30. doi: 10.1016/j.pharmthera.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Bitto E, Bingman CA, Wesenberg GE, McCoy JG, Phillips GN., Jr Structure of pyrimidine 5′-nucleotidase type 1. Insight into mechanism of action and inhibition during lead poisoning. J Biol Chem. 2006;281:20521–9. doi: 10.1074/jbc.M602000200. [DOI] [PubMed] [Google Scholar]
- 15.Rinaldo-Matthis A, Rampazzo C, Reichard P, Bianchi V, Nordlund P. Crystal structure of a human mitochondrial deoxyribonucleotidase. Nat Struct Biol. 2002;9:779–87. doi: 10.1038/nsb846. [DOI] [PubMed] [Google Scholar]
- 16.Bianchi V, Spychala J. Mammalian 5′-nucleotidases. J Biol Chem. 2003;278:46195–8. doi: 10.1074/jbc.R300032200. [DOI] [PubMed] [Google Scholar]
- 17.Himo F, Guo JD, Rinaldo-Matthis A, Nordlund P. Reaction mechanism of deoxyribonucleotidase: a theoretical study. J Phys Chem B. 2005;109:20004–8. doi: 10.1021/jp0546150. [DOI] [PubMed] [Google Scholar]
- 18.Wallden K, Ruzzenente B, Rinaldo-Matthis A, Bianchi V, Nordlund P. Structural basis for substrate specificity of the human mitochondrial deoxyribonucleotidase. Structure. 2005;13:1081–8. doi: 10.1016/j.str.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 19.Knofel T, Strater N. Mechanism of hydrolysis of phosphate esters by the dimetal center of 5′-nucleotidase based on crystal structures. J Mol Biol. 2001;309:239–54. doi: 10.1006/jmbi.2001.4656. [DOI] [PubMed] [Google Scholar]
- 20.Zagursky RJ, Ooi P, Jones KF, Fiske MJ, Smith RP, Green BA. Identification of a Haemophilus influenzae 5′-nucleotidase protein: cloning of the nucA gene and immunogenicity and characterization of the NucA protein. Infect Immun. 2000;68:2525–34. doi: 10.1128/iai.68.5.2525-2534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reilly TJ, Calcutt MJ. The class C acid phosphatase of Helicobacter pylori is a 5′ nucleotidase. Protein Expr Purif. 2004;33:48–56. doi: 10.1016/j.pep.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 22.Kuznetsova E, Proudfoot M, Sanders SA, Reinking J, Savchenko A, Arrowsmith CH, Edwards AM, Yakunin AF. Enzyme genomics: Application of general enzymatic screens to discover new enzymes. FEMS Microbiol Rev. 2005;29:263–79. doi: 10.1016/j.femsre.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 23.Xu RX, Rocque WJ, Lambert MH, Vanderwall DE, Luther MA, Nolte RT. Crystal structures of the catalytic domain of phosphodiesterase 4B complexed with AMP, 8-Br-AMP, and rolipram. J Mol Biol. 2004;337:355–65. doi: 10.1016/j.jmb.2004.01.040. [DOI] [PubMed] [Google Scholar]
- 24.Frey PA. Stereochemistry of Enzymatic-Reactions of Phosphates. Tetrahedron. 1982;38:1541–1567. [Google Scholar]
- 25.Westheimer FH. Pseudo-Rotation in the Hydrolysis of Phosphate Esters. Acc Chem Res. 1968;1:70–78. [Google Scholar]
- 26.Zhang RG, Skarina T, Katz JE, Beasley S, Khachatryan A, Vyas S, Arrowsmith CH, Clarke S, Edwards A, Joachimiak A, Savchenko A. Structure of Thermotoga maritima Stationary Phase Survival Protein SurE: A Novel Acid Phosphotase. Structure. 2001;9:1095–1106. doi: 10.1016/s0969-2126(01)00675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez CF, Proudfoot M, Brown G, Korniyenko Y, Mori H, Savchenko AV, Yakunin AF. Molecular basis of formaldehyde detoxification. Characterization of two S-formylglutathione hydrolases from Escherichia coli, FrmB and YeiG. J Biol Chem. 2006;281:14514–22. doi: 10.1074/jbc.M600996200. [DOI] [PubMed] [Google Scholar]
- 29.Baykov AA, Evtushenko OA, Avaeva SM. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal Biochem. 1988;171:266–70. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
- 30.Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, Ginell SL, Duke N, Naday I, Lazarz J, Molitsky MJ, Keefe L, Gonczy J, Rock L, Sanishvili R, Walsh MA, Westbrook E, Joachimiak A. The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J Synchrotron Radiat. 2006;13:30–45. doi: 10.1107/S0909049505036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 32.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution - from diffraction images to an initial model in minutes. Acta Cryst D. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z. CCP4. SERC Daresbury Laboratory; Warrington, UK: 1991. [Google Scholar]
- 34.Cowtan K. DM: an automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsletter on Protein. Crystallography. 1994;31:34–38. [Google Scholar]
- 35.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Cryst D. 2002;58:1772–9. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 36.Sheldrick GM. Macromolecular phasing with SHELXE. Z Kristallogr. 2002;217:644–650. [Google Scholar]
- 37.CCP4. The CCP4 suite: programs for protein crystallography. Acta Cryst D. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 38.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Cryst D. 1999;55(Pt 4):849–61. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terwilliger TC. Automated structure solution, density modification and model building. Acta Cryst D. 2002;58:1937–40. doi: 10.1107/s0907444902016438. [DOI] [PubMed] [Google Scholar]
- 40.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Cryst D. 2003;59:2023–30. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 41.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–63. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 42.Brunger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR System (CNS): A New Software System for Macromolecular Structure Determination. Acta Cryst D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 43.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst D. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 44.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst D. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 45.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst D. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotrophics displacements in macromolecular refinement. Acta Cryst D. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 47.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst. 2006;39:109–111. [Google Scholar]
- 48.Yeates TO. Detecting and overcoming crystal twinning. Methods Enzymol. 1997;276:344–58. [PubMed] [Google Scholar]
- 49.Vagin A, Teplyakov A. MOLREP: an Automatic Program for Molecular Replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 50.Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics. 2000;16:566–7. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 51.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst D. 2004;60:2256–68. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 52.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poirot O, Suhre K, Abergel C, O’Toole E, Notredame C. 3DCoffee@igs: a web server for combining sequences and structures into a multiple sequence alignment. Nucleic Acids Res. 2004;32:W37–40. doi: 10.1093/nar/gkh382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beitz E. TEXshade: shading and labeling of multiple sequence alignments using LATEX2ε. Bioinformatics. 2000;16:135–9. doi: 10.1093/bioinformatics/16.2.135. [DOI] [PubMed] [Google Scholar]
- 55.DeLano WL. The PyMOL Molecular Graphics System. 2002 http://www.pymol.org/
- 56.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–34. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]