

Abstract
The methylotrophic yeast, Pichia pastoris, is widely used as a host organism for the expression of heterologous proteins. Currently, the Zeocin and blasticidin resistance genes are the only dominant selectable markers that can be used for primary selection of transformants. In this report we describe new expression vectors that can be used to select directly for P. pastoris transformants using G418 resistance conferred by a modified Tn903kanr gene. Compared to other dominant markers, this system is more economical and offers a higher transformation efficiency, due to the small sizes of the cloning vectors, pKAN B and pKAN α B (GenBank Accession Nos EU285585 and EU285586, respectively). Additionally, multicopy transformants can be generated using these new vectors.
Keywords: heterologous protein expression, methylotrophic yeast, selectable marker, transformation, G418 resistance
Introduction
The methylotrophic yeast, Pichia pastoris, has been used as a host for the successful production of over 600 recombinant proteins (Lin Cereghino and Cregg, 2000; Macauley-Patrick et al., 2005). The major advantages of this yeast expression system include its simplicity, its preference for respiratory growth, the availability of strong promoters available to drive heterologous gene expression and the ability to add eukaryotic post-translational modifications (Cregg et al., 2000; Lin Cereghino and Cregg, 2000).
The generation of multicopy expression strains is a technique routinely used to increase the yield of recombinant proteins in P. pastoris. Generally, two well-established methods for creating multicopy expression strains are used. One method involves the construction of a plasmid with several head-to-tail copies of an expression cassette (Brierley, 1998). The advantage of this approach is that the exact number of expression cassettes is known ahead of time and can be verified by DNA sequencing. However, the cloning of such plasmids can be difficult because of their size.
Another way of generating multicopy strains is simply to screen large numbers of colonies for spontaneously occurring transformants with higher gene dosage or higher product expression (Clare et al., 1991a; Romanos et al., 1991; Wung and Gascoigne, 1996). However, at this time only two dominant markers, the bacterial Sh ble (Zeocinr) or BSD (blasticidinr) genes, can be used to directly select transformants and screen for high-copy strains on increasingly higher concentrations of Zeocin or blasticidin (Miles et al., 1998; Romanos et al., 1991).
Two other selectable markers can be used to generate multicopy strains, the P. pastoris FLD1 gene (which confers resistance to formaldehyde) and the bacterial Tn903kanr (which confers resistance to G418). Using these approaches, strains carrying up to 19 copies of an expression cassette have been isolated (Clare et al., 1991b). However, both of these methods require additional steps and special mutant strains. Direct selection in wild-type P. pastoris is not possible with these markers.
Generation of multicopy strains using resistance to formaldehyde conferred by FLD1 involves an initial selection of transformants on minimal plates containing methylamine chloride as the nitrogen source. Populations of these transformants are then enriched for multicopy strains by selection for increased resistance to formaldehyde (Sunga and Cregg, 2004). However, the formaldehyde resistance gene is limited to use with a fld1 mutant strain.
Expression constructs utilizing wild-type Tn903kanr require cloning of the recombinant gene into a relatively large vector (>9 kb) containing the P. pastoris HIS4 and transformation of the expression vector into a his4 mutant strain. Yeast transformants are initially selected for a HIS+ phenotype and then are replica-plated to plates containing increasing concentrations of G418 to screen for multicopy candidate strains. Plating of transformed colonies directly on G418 medium yields no resistant colonies (Scorer et al., 1994). The major inconvenience of this procedure is that a preliminary selection using another biosynthetic marker, HIS4, must take place before screening for the multicopy strains. We hypothesized that the presence of a bacterial promoter and transcriptional termination region in Tn903kanr hindered its expression in P. pastoris.
In this paper, we describe new G418r vectors designed for the direct selection of P. pastoris expression strains. The modified Tn903kanr gene housed in these plasmids can be used for selection in both bacteria and P. pastoris, allowing cloning vectors to be small and easy to manipulate. Additionally, since G418 is much more affordable than either Zeocin or blasticidin, both time and expense can be saved with this new vector system.
Materials and methods
Strains, media and reagents
Pichia pastoris strains yJC100 (wild-type) and yGS115 (his4) are derivatives of the original wild-type P. pastoris strain NRRL Y11430 (Northern Regional Research Laboratories, US Department of Agriculture, Peoria, IL, USA) and have been described previously (Cregg et al., 1985). Recombinant DNA manipulations were carried out in Escherichia coli strain TOP10 (Invitrogen, Carlsbad, CA, USA). Yeast strains were cultured in YPD medium (1% yeast extract, 2% peptone, 1% glucose) supplemented with 25–10 000 μg/ml G418 or 100–1000 μg/ml Zeocin (Invitrogen), or minimal (YNB) medium (0.17% yeast nitrogen base, 0.5% ammonium sulphate) supplemented with 0.4% glucose (YND) or 0.5% methanol (YNM). Amino acids were added to 50 μg/ml as required. E. coli strains were cultured in Lennox broth (LB) medium supplemented with 100 μg/ml ampicillin, 30 μg/ml kanamycin or 25 μg/ml Zeocin. Restriction enzymes were purchased from MBI Fermentas (Hanover, MD, USA). Oligonucleotides were synthesized by Sigma Genosys (Plano, TX, USA).
Construction of G418-resistant vectors pAC1, pKAN B, pKANα B and pMH1
The vector pAC1 was constructed by using the oligos AJ KAN5 ggcgaattcatgagccatattcaacggg and AJ KAN3 cgggcggccgcttagaaaaactcatcgagc to amplify the coding sequence of the kanamycin resistance gene from pPIC3K (Invitrogen) with flanking EcoRI and NotI restriction sites. This PCR fragment was then cloned into the corresponding sites of the pGAPZ B (Invitrogen) polylinker to form pAC1, a vector that contains genes conferring resistance to both Zeocin and G418.
A PCR product with flanking StuI and BamH I restriction sites containing the GAP promoter, kanamycin resistance coding sequence and AOXI transcriptional termination sequence from pAC1 was generated using the oligos KANFO accaggcctgtgtcctcgtccaatcag and KANBA ccaggatccttgaagctatggtgtgtg. The cloning vectors pKAN B and pKANα B were constructed by inserting the StuI–BamH I digest PCR fragment into the corresponding sites of pPICZ B and pPICZα B (Invitrogen). pKAN B was further modified by removing 39 nucleotides containing non-unique restriction sites from its polylinker and inserting a unique PstI site. This modification was accomplished by using the primer combinations pKanBdeltop caacttgagaagatcaaaacggccgtctcggatcggtacc and pKanBdel-bottom ggtaccgatccgagacggccgttttgatcttctcaagttg as well as pKanBPstIfo gagaagatcaaaaaactgcagtctcggatcggtac and pKanBPstIrev gtaccgatccgagactgcagttttttgatcttctc and the QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA, USA).
The plasmid pMH1 was generated by cloning the ß-lactamase gene into pKAN B. The coding sequence of the ß-lactamase gene was retrieved from pHWGΔ1.0 (Lin-Cereghino et al., 2006) using PCR with the primers CM5BLAC caaggtaccatgtccggtcacccagaaac and CM3BLAC tagccgcggctaccaatgcttaatcagtg. The 800 bp sequence was then ligated into pKAN B after digestion of the product and pKAN B with the restriction enzymes KpnI and SacII, yielding the plasmid, pMH1. Additionally, to create a control for real-time PCR and enzyme assays, the BglII–SacII fragment containing the AOXI promoter fused to the E. coli bla gene from pMH1 was cloned into pBLHIS (Lin Cereghino et al., 2001), yielding the plasmid pBLHIS–bla. This plasmid contains the P. pastoris HIS4 gene for selection in yeast and two copies of the ß-lactamase gene: one for plasmid maintenance in bacteria and one under the control of the methanol-inducible P. pastoris AOX1 promoter.
Pichia pastoris transformation
Transformations of P. pastoris were done by the electrotransformation method described in Cregg and Russell, 1998. Plasmids pAC1, pKAN B and pKANα B were linearized within the AOXI promoter with restriction enzymes XmaJI or SacI, purified with the QIAprep Spin PCR Purification Kit (Qiagen, Valencia, CA, USA) and electroporated into competent yJC100 or yGS115 cells. Transformed cells were allowed to recover for 1 h in 1 ml 50% 1m sorbitol:50% YPD solution at 30 °C and then plated on selective medium. Similarly, pBLHIS-bla was digested, purified and transformed into yGS115; however, no recovery period was necessary before plating. Transformed colonies were then purified by streaking for isolated colonies on selective medium.
Stability studies
To ensure stability of expression vector DNA in transformants, isolated G418-resistant colonies were grown for 96 h (approximately 50 generations) on non-selective YPD medium, diluted and spread on YPD medium. Colonies were then replica-plated to plates containing YPD + G418 medium and grown for 2 days. The number of viable colonies on each plate was then recorded.
Real-time PCR
Real-time PCR reactions were composed of 10 μl 2× reaction buffer from the DyNAmo SYBR Green qPCR kit distributed by New England Bio-Labs (Beverly, MA, USA), 10 ng genomic DNA and 0.3 μm primers in a 20 μl total reaction volume. PCR reactions were carried out in a MJ MiniOpticon (BioRad Corp, Hercules, CA, USA) with the following parameters: 1 cycle of 95 °C for 5 min, 40 cycles of 94 °C for 1 min, 59 °C for 1 min, 72 °C for 1 min. Primers used to amplify the β-lactamase target gene were blacs5 ctcgccttgatcgttgggaa and blacas3 cagtgctgcaatgataccgc. Primers used to amplify the MET2 (Thor et al., 2005) reference gene were mets100 cgttctcgcaactctttcgaa and metas100 caatggcatcagttatgacggaag. All samples were performed in duplicate, and all samples were tested several times in different experiments. Q-gene software was utilized to analyse real-time PCR data (Muller et al., 2002).
Northern analysis
P. pastoris RNA was prepared by a standard procedure adapted for total yeast RNA isolation, starting with cells harvested at approximately 0.5 OD600 (Cereghino et al., 1995). Samples containing 10 μg total RNA were loaded into the wells of 1.5% agarose/denaturing formaldehyde gels and electrophoresed for separation. Transfer of RNA to MagnaGraph nylon membranes (Osmonics, Westborough, MA, USA), crosslinking, prehybridization, hybridization, high-stringency washing and imaging were all performed following a standard procedure (Sambrook et al., 1989). The nylon membranes were washed and visualized according to the instructions of the Southern-Light Detection Kit (Tropix, Bedford, MA, USA) with an Alpha Innotech ChemiImager 5500 (Alpha Innotech, San Leandro, CA, USA).
Probe generation
A biotinylated DNA probe to the kanamycin resistance gene was synthesized utilizing the Bio-Prime DNA Labeling System (Invitrogen). Template DNAs were generated by PCR with the AJ KAN5 and AJ KAN3 primers described above. Labelled DNA was separated from unincorporated label by using the QIAquick PCR Purification Kit (Qiagen). Approximately 400 ng of purified biotinylated DNA fragment were denatured by heating at 95 °C for 5 min, chilled on ice for 2 min and then added as a hybridization probe.
Miscellaneous methods
Recombinant DNA methods were performed essentially as described in Sambrook et al. (1989). The rapid colony PCR method, used to determine the presence of the kanr gene in cells, has been described previously (Thor et al., 2005). Plasmid DNA was purified from Escherichia coli cultures, using a QIAprep Spin Miniprep Kit (Qiagen). Plasmid DNA digested with restriction enzymes and used for restriction mapping, hybridization probes and cloning of subfragments were separated on TBE agarose gels. DNA fragments were purified from agarose gels by using the Geneclean II kit (Qbiogene, Carlsbad, CA, USA). Chromosomal DNA from P. pastoris transformants was prepared using the OmniPrep™ kit from GenoTechnology Inc. (St. Louis, MO, USA). All mutated sites and ligation junctions in newly synthesized vectors were confirmed by DNA sequencing (Geneway Research, Hayward, CA, USA).
Results and discussion
Generation of kanamycin/G418 resistance vectors with control regions from P. pastoris
G418 resistance has been used previously as a secondary selection in P. pastoris. Transformants of pPIC3K or pPIC9K (Invitrogen) cannot be directly selected on YPD-G418 plates. We hypothesized that because the Tn903kanr gene in these vectors has a bacterial promoter and transcriptional termination sequences, its expression in yeast was poor. As described in the Materials and Methods, the vector pAC1 (derived from the plasmid pGAPZ B) was constructed to test the functionality of the Tn903kanr gene under the control of a 5′ P. pastoris GAP promoter and fused to the AOXI transcriptional terminator at the 3′ end. Both Tn903kanr and Zeocin resistance genes are present on pAC1. P. pastoris strains, such as yJC100 or yGS115, transformed with pAC1, yielded transformants when directly selected on YPD supplemented with 0.5 mg/ml G418 or 100 μg/ml Zeocin. pAC1 transformants selected on G418 grew more quickly. Furthermore, transformation efficiency was approximately 50% greater with G418 selection than Zeocin selection. Recovery time, the period between electroporation and plating, was varied from 0 to 16 h, and it was found that cells needed at least 1 h to demonstrate sufficient G418 resistance, as is the case for transformations with Zeocin. Although kanamycin and neomycin sulphate are structurally similar to G418, these compounds could not be used for selection in P. pastoris because of their lack of toxicity to the yeast. Nevertheless, kanamycin can be used for selection of plasmids in E. coli when initially constructing these vectors.
Expression of Tn903kanr RNA
Total RNA from yJC100 strains carrying pPIC3K and pAC1 were isolated to compare the amount of specific Tn903kanr mRNA generated in P. pastoris. Figure 1 demonstrates that the amount of transcript from the pAC1 (Figure 1A, lane 3) was approximately 20-fold more than that from pPIC3K (Figure 1A, lane 2). This result suggests that by replacing the original transcriptional control elements of the Tn903kanr gene with those from yeast, expression of the Tn903kanr gene in P. pastoris was improved significantly.
Figure 1.

Northern blot analysis of modified Tn903kanr in P. pastoris. (A) Total RNA was probed with an 800 bp fragment derived from Tn903kanr in: non-transformed yJC100 (lane 1); yJC100 carrying pPIC3K (lane 2); and yJC100 with pAC1 (lane 3). (B) Ethidium-stained RNA corresponding to lanes 1–3 above to show equal loading
Construction of expression vectors for the production of intracellular and extracellular proteins
In order to test the modified Tn903kanr gene more thoroughly as a dominant marker, the expression vectors pKAN B and pKANα B were constructed. pKAN B is designed for the expression of intra-cellular proteins and is shown in Figure 2. Foreign coding sequences inserted into this plasmid must provide a start codon. pKANα B is structurally identical to pKAN B, except that the 266 bp α-factor signal sequence precedes the multiple cloning site. Therefore, foreign coding sequences must be cloned in frame with the S. cerevisiae α-factor prepro peptide (α-MF) in order for the resultant protein to be exported into the extracellular medium.
Figure 2.

Map of pKAN B. A small multiple cloning site after the AOXI promoter allows for cloning of foreign genes. The modified kanamycin resistance gene allows for selection of transformants with kanamycin in E. coli and G418 in P. pastoris
Characteristics of P. pastoris transformants selected with G418
Resulting primary transformants of pKAN B and pKANα B displayed two distinctive colony types, as shown in Figure 3, after 48 h of growth at 30 °C. Sizes of transformants were in the range 0.5–6 mm. As the concentration of G418 in the selection plates was raised from 0.50 to 10.0 mg/ml, the number of these colonies (<2 mm) declined, while the number of larger colonies remained stable. In order to investigate the genetic nature of these various types of transformants, they were subjected to colony PCR. It was found that <20% of small colonies <2 mm in diameter actually contained integrated copies of the expression cassette. Most of the smaller colonies are believed to be transiently transformed with non-integrated extrachromosomal copies of the expression vector. Similar results have been observed with kanMX6, a modified dominant resistance marker used in S. cerevisiae (Wach, 1996). None of these tiny colonies could tolerate G418 concentrations above 2 mg/ml. In contrast, over 95% of the colonies >2 mm contained one or several integrated genomic copies of the expression cassette and could grow on G418 concentrations up to 10 mg/ml. To prove the stability of the large G418r transformants, they were grown for more than 50 generations on YPD, spread on YPD plates, then replica-plated onto YPD plates containing G418. Because all colonies grew under selective conditions, we concluded that the resistance marker was stably integrated into the cells, since growth on medium lacking the antibiotic did not trigger the loss of the marker gene. Since both size differences and possible multicopy transformants were evident at 500 μg/ml, this concentration of G418 was used as the standard level in selection plates.
Figure 3.
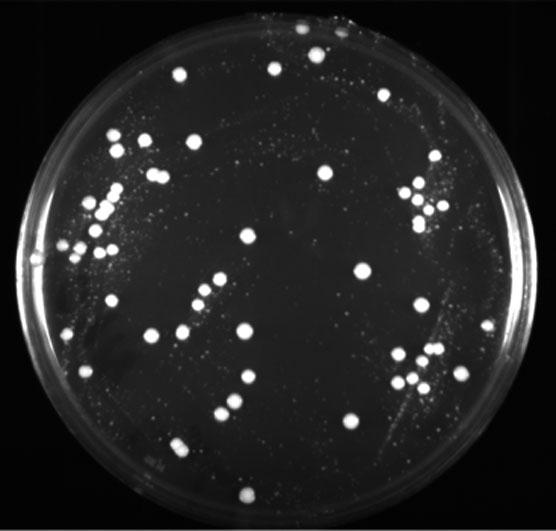
P. pastoris transformed with pKAN B. Transformants were selected on 0.5 mg/ml G418. The plate was grown for 48 h at 30 °C
Multicopy expression strains resistant to G418
The plasmid pMH1 was constructed to test the ability of pKAN B to act as a heterologous expression vector of E. coli ß-lactamase. E. coli cells containing pMH1 were selected on 30 μg/ml kanamycin, and the isolated pMH1 was linearized and electroporated into P. pastoris. G418-resistant transformants of pMH1 were directly selected on YPD plates supplemented with 0.5 mg/ml G418. Genomic DNA from several transformants was isolated to assess their multicopy nature. Using a strain containing two copies of the β-lactamase gene (generated from pBLHIS-bla) as a normalization standard, real-time PCR was used to analyse the number of copies in over 20 different transformants. In these real-time experiments, relative quantification was performed by comparing the CT values of the β-lactamase target gene and the single-copy reference gene, MET2. Sample copy numbers from representative transformants are shown in Figure 4. Transformants bearing 1–4 copies of the β-lactamase target gene were routinely present in transformant populations selected with 0.5 mg/ml G418. The same range in copy number was observed in colonies selected on higher concentrations of G418 (2–5 mg/ml). Very rarely, transformants containing up to eight copies of the target gene were found. These results were also confirmed by Southern analysis of the same genomic DNA samples (data not shown).
Figure 4.

Estimated copy number. Genomic DNA from various transformants was subjected to real-time PCR analysis. Relative quantification of copy number was performed by comparing the CT values of the β-lactamase target gene and the single-copy reference gene, P. pastoris MET2. Values were normalized to the pBLHIS-bla sample, which contains two copies of the β-lactamase gene
In contrast, selection of high-copy strains using Zeocin resistance requires plating transformants on high concentrations (>1000 μg/ml) of Zeocin ($200/g). Because the pKAN B and pKANα B vectors are smaller and easier to manipulate, they allow for more rapid and economical isolation of P. pastoris expression strains.
Acknowledgements
This work was supported by NIH–AREA Grant No. GM65882-02 to J.L.-C. and G.P.L.-C.
References
- Brierley RA. Secretion of recombinant human insulin-like growth factor I IGF-1. Methods Mol Biol. 1998;103:149–177. doi: 10.1385/0-89603-421-6:149. [DOI] [PubMed] [Google Scholar]
- Cereghino GP, Atencio DP, Saghbini M, et al. Glucose-dependent turnover of the mRNAs encoding succinate dehydrogenase peptides in Saccharomyces cerevisiae: sequence elements in the 5′ untranslated region of the Ip mRNA play a dominant role. Mol Biol Cell. 1995;6:1125–1143. doi: 10.1091/mbc.6.9.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clare JJ, Rayment FB, Ballantine SP, et al. High-level expression of tetanus toxin fragment C in Pichia pastoris strains containing multiple tandem intergrations of the gene. Bio/Technology. 1991a;9:455–460. doi: 10.1038/nbt0591-455. [DOI] [PubMed] [Google Scholar]
- Clare JJ, Romanos MA, Rayment FB, et al. Production of mouse epidermal growth factor in yeast: high-level secretion using Pichia pastoris strains containing multiple gene copies. Gene. 1991b;105:205–212. doi: 10.1016/0378-1119(91)90152-2. [DOI] [PubMed] [Google Scholar]
- Cregg JM, Barringer KJ, Hessler AY, Madden KR. Pichia pastoris as a host system for transformations. Mol Cell Biol. 1985;5:3376–3385. doi: 10.1128/mcb.5.12.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregg JM, Cereghino JL, Shi J, Higgins DR. Recombinant protein expression in Pichia pastoris. Mol Biotechnol. 2000;16:23–52. doi: 10.1385/MB:16:1:23. [DOI] [PubMed] [Google Scholar]
- Cregg JM, Russell KA. Transformation. In: Higgins DR, Cregg JM, editors. Pichia Protocols. Humana; Totowa, New Jersey: 1998. pp. 27–39. [Google Scholar]
- Lin-Cereghino GP, Godfrey L, de la Cruz BJ, et al. Mxr1p, a key regulator of the methanol utilization pathway and peroxisomal genes in Pichia pastoris. Mol Cell Biol. 2006;26:883–897. doi: 10.1128/MCB.26.3.883-897.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Cereghino GP, Lin Cereghino J, Sunga AJ, et al. New selectable marker/auxotrophic host strain combinations for molecular genetic manipulation of Pichia pastoris. Gene. 2001;263:159–169. doi: 10.1016/s0378-1119(00)00576-x. [DOI] [PubMed] [Google Scholar]
- Lin Cereghino J, Cregg JM. Heterologous protein expression in the methylotrophic yeast Pichia pastoris. FEMS Microbiol Rev. 2000;24:45–66. doi: 10.1111/j.1574-6976.2000.tb00532.x. [DOI] [PubMed] [Google Scholar]
- Macauley-Patrick S, Fazenda ML, McNeil B, Harvey LM. Heterologous protein production using the Pichia pastoris expression system. Yeast. 2005;22:249–270. doi: 10.1002/yea.1208. [DOI] [PubMed] [Google Scholar]
- Miles D, Busser K, Stadler C, Higgins D. Isolation of nucleic acids. In: Higgins D, Cregg J, editors. Pichia Protocols. Humana; Totowa, New Jersey: 1998. pp. 73–80. [DOI] [PubMed] [Google Scholar]
- Muller PT, Janovjak H, Miserez AR, Dobbie Z. Processing of gene expression data generated by quantitative real-time RT–PCR. BioTechniques. 2002;32:1372–1379. [PubMed] [Google Scholar]
- Romanos MA, Clare JJ, Beesley KM, et al. Recombinant Bordetella pertussis pertactin P69 from the yeast Pichia pastoris: high-level production and immunological properties. Vaccine. 1991;9:901–906. doi: 10.1016/0264-410x(91)90011-t. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Handbook. 2nd edn. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Scorer CA, Clare JJ, McCombie WR, et al. Rapid selection using G418 of high copy number transformants of Pichia pastoris for high-level foreign gene expression. Bio/Technology. 1994;12:181–184. doi: 10.1038/nbt0294-181. [DOI] [PubMed] [Google Scholar]
- Sunga AJ, Cregg JM. The Pichia pastoris formaldehyde dehydrogenase gene FLD1 as a marker for selection of multicopy expression strains of P. pastoris. Gene. 2004;330:39–47. doi: 10.1016/j.gene.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Thor D, Xiong S, Orazem C, et al. Cloning and characterization of the Pichia pastoris MET2 gene as a selectable marker. FEMS Yeast Res. 2005;5:935–942. doi: 10.1016/j.femsyr.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast. 1996;12:259–265. doi: 10.1002/(SICI)1097-0061(19960315)12:3%3C259::AID-YEA901%3E3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Wung JL, Gascoigne NR. Antibody screening for secreted proteins expressed in Pichia pastoris. BioTechniques. 1996;21(5):808–812. doi: 10.2144/96215bm11. [DOI] [PubMed] [Google Scholar]