


Abstract
Controller (C) proteins regulate the timing of the expression of restriction and modification (R–M) genes through a combination of positive and negative feedback circuits. A single dimer bound to the operator switches on transcription of the C-gene and the endonuclease gene; at higher concentrations, a second dimer bound adjacently switches off these genes. Here we report the first structure of a C protein–DNA operator complex, consisting of two C protein dimers bound to the native 35 bp operator sequence of the R–M system Esp1396I. The structure reveals a role for both direct and indirect DNA sequence recognition. The structure of the DNA in the complex is highly distorted, with severe compression of the minor groove resulting in a 50° bend within each operator site, together with a large expansion of the major groove in the centre of the DNA sequence. Cooperative binding between dimers governs the concentration-dependent activation–repression switch and arises, in part, from the interaction of Glu25 and Arg35 side chains at the dimer–dimer interface. Competition between Arg35 and an equivalent residue of the σ70 subunit of RNA polymerase for the Glu25 site underpins the switch from activation to repression of the endonuclease gene.
INTRODUCTION
Restriction–modification (R–M) systems play a pivotal role in modulating the horizontal transfer of genes in bacterial populations - a major factor in the transmission of antibiotic resistance between bacterial species (1). The emergence of multi-drug-resistant strains through horizontal transfer of antibiotic resistance genes represents an increasing threat to world health. An understanding of R–M systems, and of their regulation, is thus of significant microbiological and biomedical interest.
R–M systems encode a restriction endonuclease and a DNA methyltransferase. The action of the DNA sequence-specific methytransferase (M) protects the host DNA from cleavage by an associated restriction enzyme (R), and the specific methylation pattern of the host R–M system allows the discrimination of ‘self’ from ‘non-self’ DNA (2). This ancient form of innate immunity provides a basis for the selective destruction of foreign DNA. Clearly, expression of the endonuclease prior to protection of the host DNA by the methyltransferase would be lethal. Thus there are a variety of control mechanisms that ensure the correct temporal expression of R–M genes. One common mechanism employs a ‘controller’ (C) protein encoded by a gene downstream of its own promoter, which is co-transcribed with the endonuclease gene from a common promoter (3–7). The C-protein binds to the C/R promoter to regulate transcription of its own gene and the associated endonuclease (R) gene (8).
Biochemical and biophysical analysis in recent years has revealed the general features of this genetic switch (9,10). X-ray crystallographic analysis of the controller protein C.AhdI showed it to be a dimer consisting of two 74 aa subunits, each containing a helix-turn-helix (HTH) motif, and a weak dimer interface consistent with a relatively high Kd (2.5 µM) for dimerization (11). Low-level expression of the C-protein from a weak promoter leads to a delay in transcription until sufficient protein accumulates to form a functional dimer. The C-protein dimer activates transcription of the C/R operon, forming a positive feedback loop, leading to an exponential increase in C-protein expression; at higher concentrations, a second dimer is recruited to the promoter, displacing RNA polymerase and thereby repressing transcription of its own gene (and hence expression of the R gene) in a negative feedback loop (Figure 1).
Figure 1.

The genetic switch regulating the timing and expression of R–M genes. The -35 (green) and -10 (red) transcriptional signals are indicated upstream of the C-gene (blue) and the R-gene (not shown). C-protein dimers are shown in pink and the sigma subunit of RNA polymerase in yellow. Low-level C-protein expression occurs from a weak C-independent promoter (not shown). Occupation of the high affinity OL site by C-protein dimers stimulates transcription of the C-gene via recruitment of RNA polymerase sigma subunit to the -35 site; at higher concentrations of C-protein, occupation of the OR site displaces the subunit and down-regulates the C- and R-genes.
This simple but elegant control circuit has been confirmed by in vitro transcription assays and has been successfully modelled mathematically (12). A similar control circuit is likely to apply to other C-protein-regulated systems, PvuII being the best studied of these (13). The time delay in expression of the C and R genes, relative to the M gene, when establishing a new R–M system has recently been experimentally verified in vivo (14).
Bioinformatic analysis of known and potential C-protein binding sites has identified a repeating symmetrical consensus sequence consisting of four symmetrical ‘C-boxes’ GACTnnnAGTCnnnnGACTnnnAGTC upstream of the C/R genes (6,8). However, the degree of sequence homology between species is moderate and the internal symmetry between ‘C-boxes’ is far from perfect (Figure 2A). The operator sequence includes binding sites (denoted OL and OR) for two C-protein dimers (9). The left-hand operator sequence, OL, is distal to the gene and is believed to activate transcription through favourable interactions with the σ70 subunit of RNA polymerase, bound at the -35 site that overlaps with OR. The right-hand operator sequence, OR, is proximal to the gene and binding of a second dimer to OR blocks σ70 binding to the -35 site, thus switching off the C/R genes (9–11).
Figure 2.

Pseudo-symmetry of the C.Esp1396I DNA binding site. (A) the original C-box symmetry with a pseudo-dyad centred on GT; (B) an alternative symmetry centred on the central T, which is deduced from the structure of the nucleoprotein complex; (C) sequence of the DNA duplex used to form nucleoprotein complexes for crystallography. In (A) and (B), the location of pseudo-dyad axes within operators (blue) and between operators (red) are shown as dotted lines. In (C), nucleotides involved in DNA–protein interactions in the crystal structure of the complex are indicated in blue. When discussed in the text, nucleotides on the lower strand are indicated with a prime (′).
It is notable that the GT in the centre of the proposed consensus sequence is more highly conserved than the proposed tetranucleotide recognition sequences (13), but clearly lacks dyad symmetry. The proposed 3 bp ‘spacers’ within the left and right operator sequences are equally well conserved, the consensus being TAT. Within the symmetrical framework discussed above (Figure 2A), the two TAT ‘spacer’ sequences are not related by dyad symmetry, and nor do they have internal symmetry. If the protein dimer were centred on this sequence, the dyad axis of the dimer would pass through the central A.
However, we note that if the pseudo-dyad axis relating the two operators is shifted by half a base (i.e. centred on T rather than GT), then although the pseudo-dyad between AGTC/GACT sequences is lost, instead there would be perfectly symmetrical TATA sequences at the centre of each operator (Figure 2B). In this case, the dyad axis of each dimer would be located between the central A and T bases of the TATA ‘spacer’. Without structural analysis, it is not possible to predict which of these symmetries is adopted by the nucleoprotein complex.
Since these ‘spacer’ sequences are so strongly conserved, they are likely to play an important structural role. Circular dichroism indicates that a large structural deformation is induced in the DNA when each C.AhdI dimer binds to the operator (15). Circular permutation gel assays show that binding of a C.AhdI dimer results in a substantial (∼50°) bend in the DNA. It was proposed that these structural distortions could arise from the TAT (or TATA) ‘spacers’ since these sequences are frequently found in bent DNA structures (15). However, in the absence of detailed structural data, the precise nature of the conformational changes in the DNA that are induced by C-protein binding remain unknown.
To investigate the structure and symmetry of C-protein/DNA complexes and provide insight into the structural deformation of the DNA and relevant intermolecular contacts, we are carrying out a systematic study of C-protein complexes with a variety of DNA sequences. Here, we investigate the DNA-binding properties of the controller protein of the Esp1396I R–M system, and report the first crystal structure of any C-protein–DNA complex. In addition, we have used site-directed mutagenesis to elucidate the role of key amino acid residues implicated in cooperative binding between adjacent protein dimers on the DNA. Our results reveal the mechanism whereby cooperative binding of dimers to the DNA operator governs the switch from activation to repression of the C and R genes.
MATERIALS AND METHODS
Protein and DNA purification
Large-scale cultures of Escherichia coli BL21 (DE3) cells bearing the plasmid pET-28b/esp1396IC were grown, harvested and disrupted by sonication. Cell lysates were purified by chromatography on a His-Trap HP column, the His-tag removed by thrombin digestion, dialysed and the protein re-applied to the His-Trap column. The final protein sequence following thrombin cleavage includes an N-terminal tripeptide Glycine-Serine-Histidine in addition to the native sequence. Site-directed mutagenesis was performed as previously described (11) and mutant proteins purified as above. DNA duplexes were prepared by annealing equimolar quantities of each oligonucleotide strand, and slowly cooling to room temperature; correct formation of double-stranded DNA was confirmed by native polyacrylamide gel electrophoresis.
Electrophoretic mobility shift assays (EMSAs)
Electrophoretic mobility shift assays were performed using non-denaturing electrophoresis on 8% polyacrylamide gels as previously described (10). C.Esp1396I was incubated at various concentrations with 240 nM γ-33P-labelled DNA duplex at 4°C for a period of 30 min. Gels were run at 100 V for 90 min, dried and analysed on a phosphorimager. The 35 bpWT sequence corresponds to that in Figure 2C. Additional 35 bp oligonucleotide duplexes in which the right or left operator sequences have been mutated to a random sequence (indicated in bold) were also used for EMSAs. The sequences of these were as follows:
OL (OR mutated):
ATGTGACTTATAGTCCGTCTAGCCTAGCCTAGCCT
TACACTGAATATCAGGCAGATCGGATCGGATCGGA
OR (OL mutated):
CTAGCCTAGCCTAGCCGTGTGATTATAGTCAACAT
GATCGGATCGGATCGGCACACTAATATCAGTTGTA
DNA bending assays
DNA bending assays were performed as described elsewhere (15) using the native 35 bp promoter (OL + OR) and an equivalent 35 bp sequence with the intact OL and a randomized OR (as defined above). Relative mobilities (y = Rbound/Rfree) were plotted as a function of the position of the binding site (x) and fitted to the quadratic function y = ax2 − bx + c, where a = −b = 2c(1 cos α), from which the bend angle α could be determined (16).
Crystallization and data collection
Crystallization was carried out at the High Throughput Crystallization Laboratory of the EMBL Grenoble Outstation employing vapour diffusion sitting drops of 100 nl nucleoprotein solution and 100 nl mother liquor. Pure C.Esp1396I protein (0.7 mg/ml) was mixed with purified double-stranded 35-mer DNA at a 4 : 1 molar ratio. Crystals of dimensions 30 × 30 × 10 μm grew within 1 month at 20°C in 50 mM MES, pH 7.5, 25% MPD, 40 mM MgCl2. The crystals were vitrified directly in a cold N2 gas stream at 100 K from an Oxford Cryosystems 700 series Cryostream (Oxford Cryosystems Ltd., Oxford, UK). Data were collected at beamline ID29 at the ESRF, Grenoble, France, employing a custom-designed pinhole (R. Ravelli & F. Felisaz, EMBL Grenoble) producing a 25 μm spherical low divergence beam suitable for small crystals. A total of 100 1° oscillation images were collected at 0.9787 Å wavelength on an ADSC Q315R mosaic CCD detector.
Structure determination and refinement
Data were integrated and scaled using XDS and XSCALE (17). Molecular replacement was performed with the program Phaser (18) using a single chain from the C.BclI dimer (PDB entry 2B5A). Four monomers were placed consecutively and, following solvent flattening and density averaging using DM (19), the DNA chains were built manually using COOT (20). Simulated annealing was performed using PHENIX (21) and iterative refinement using REFMAC5 (22) employing non-crystallographic restraints and TLS parameterization. The final model was refined to 2.8 Å and contains four protein chains with residues 2–77 (chains A and D) and residues 2–78 (chains B and C) together with the complete 35-mer DNA duplex. There is a single tetramer–DNA complex in the asymmetric unit with a solvent content of 69%. There are no residues in disallowed regions of the Ramachandran plot. DNA parameters were calculated using CURVES (23,24), contact analysis with NUCPLOT (25) and all structure figures were produced using PyMol (26).
RESULTS
Binding of C.Esp1396I to DNA operator sites
Previous studies on C.AhdI have shown the existence of two operator sequences upstream of the C.AhdI gene (OL and OR). Occupation of the high-affinity distal site (OL) leads to activation of transcription, and binding of a second dimer to the weaker proximal site (OR) represses the gene (9,10). C.AhdI was the first C-protein structure to be determined, but we have been unable to produce suitable crystals of DNA–protein complexes with C.AhdI to date. We therefore turned to a study of the closely related C-protein, C.Esp1396I.
We first investigated by EMSA the interaction of C.Esp1396I with 35 bp DNA sequences corresponding to the operator region upstream of the C/R gene. Titrating in C.Esp1396I to the native operator DNA (Figure 3) shows that tetrameric complexes predominate, as was the case with the related protein C.AhdI (10). Mutation of the OR operator results in the loss of tetrameric complexes so that only dimeric complexes can form; mutation of the OL operator, however, results in complete loss of DNA binding as the protein cannot bind to OR alone. The binding of a second dimer to form tetrameric complexes on the native operator DNA is highly cooperative, since binding to the intrinsically low-affinity OR site cannot occur unless a dimer is already bound to OL.
Figure 3.

Analysis of binding to left and right operators by EMSA. The 35 bp oligonucleotide duplexes (240 nM) containing the native operator (WT), the left-hand operator only (OL) or the right-hand operator only (OR) were incubated with C.Esp1396I at 0, 1, 2, 3, 4, 5, 6, 7 and 8 : 1 protein:DNA ratios. F = free DNA; D = dimer–DNA complex; T = tetramer–DNA complex. The sequences of the mutated operator duplexes are given in Materials and methods section.
DNA bending assays were used to see whether binding of dimers and tetramers induced bending in the DNA. By introducing the 35 bp operator sequence into a plasmid with a series of paired restrictions sites, the position of the DNA binding site across a 1 kb fragment can be varied (15,27). Bending of the DNA results in decreased mobility of complexes when the protein is bound in the centre of the sequence, relative to the mobility when bound at the end of the fragment. From an analysis of the relative mobilities, the bend angle can be estimated (16).
Figure 4 shows the results of the bending assay for dimeric and tetrameric complexes (the former using just the OL sequence and the latter using the intact operator, OL + OR). Clear differential mobilities are observed in both cases, but are more pronounced with binding of the dimer. Fitting the data to the equation derived by Ferrari et al(16) leads to an estimate of a bend angle of 51° for the dimeric complex and 43° for the tetrameric complex. As was the case for C.AhdI, binding of two dimers results in an overall bend angle less than that for binding of a single dimer (15), implying that the two dimers do not bind to the same face of the DNA (as indeed is expected, since the centres of the two binding sites are separated by 15 bp). However, they cannot be on exactly opposite sides of the DNA since the net bend angle should then be close to zero. Indeed, this interpretation is confirmed by the crystal structure (see below).
Figure 4.

DNA bending assay. For complexes with both the native operator (OR + OL) and the distal operator OL, relative mobilities (y = Rb/Rf), were plotted against flexure displacement (x) and fitted to the equation (18): y = ax2 − bx + c, where a = −b = 2c(1 − cosα). For the former a = 0.445, b = −0.444, c = 0.821 and for the latter, a = 0.732, b = −0.660, c = 0.910, giving bend angles of 51° and 43°, respectively.
Crystal structure of the tetramer complex
In order to elucidate the structure of the tetrameric (repression) complex, we crystallized a complex with the native 35 bp operator sequence (Figure 2C) using a 4 : 1 protein:DNA ratio. The best crystals diffracted to a resolution of 2.8 Å. Using molecular replacement, based on the structure of the C.BclI dimer (28), we solved the structure of the protein–DNA complex (Table 1). All the amino acid residues in the protein can be seen in the electron density map, with the exception of 1–2 residues at the N- and C-termini, and all 35 bp of DNA could be located.
Table 1.
Data collection and refinement statistics
| Tetramer–DNA complex (Native) | |
|---|---|
| Data collection | |
| Space group | P65 |
| Cell dimensions | |
| a, b, c (Å) | 104.48, 104.48, 139.29 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution (Å) | 20.0–2.8 (2.9–2.8) |
| Rmeas | 4.6 (41.8) |
| I / σI | 33.7 (4.8) |
| Completeness (%) | 98.5 (98.5) |
| Redundancy | 6.3 (6.2) |
| Refinement | |
| Resolution (Å) | 19.1–2.80 |
| No. of reflections | 21,157 |
| Rwork/Rfree | 0.207/0.239 |
| No. atoms | |
| Protein | 2496 |
| Ligand/ion | 1431 |
| Water | 4 |
| B-factors | |
| Protein | 94 |
| Ligand/ion | 96 |
| Water | 31 |
| RMSDs | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 1.328 |
The tetramer complex was found to exist in two evenly distributed orientations, related by the central pseudo-dyad (Fig. 2B). The structure therefore represents an average of two orientations. This crystallographic phenomenon is well documented in pseudo-symmetric nucleoprotein complexes (29) and is generally only apparent at high resolution. Given the 2.8 resolution limit of our data, the electron density is of high quality (Supplementary Figure 1) and permits clear interpretation of the general structural features of this complex. In addition, for the 20 bp of the DNA that are related by dyad symmetry, clear details of the protein–DNA interactions can be seen.
The protein structure is similar to that of C.AhdI, consisting of 5 α-helices, but with an extension of helix 5 resulting from the additional 10 residues at the C-terminus (Supplementary Figure 2). The dimer interface includes a network of inter-subunit hydrogen bonds involving the side chain of Asn47, together with a number of main chain interactions (Figure 5A), similar to those seen in the free C.AhdI dimer. However, the more extended helix 5 allows more extensive contacts to be made at the dimer interface (buried surface area, 1900 Å2 c.f. 1400 Å2 for C.AhdI), consistent with the lower Kd (i.e. tighter binding) of C.Esp1396I [Kd ∼ 0.6 µM, c.f. 2.5 µM for C.AhdI (9)].
Figure 5.

Structure of the C.Esp1396I–DNA complex. Protein subunits are shown in yellow (subunit A), green (subunit B), pink (subunit C) and blue (subunit D). Details are shown of (A) hydrogen bonding at the dimer interface; (B) two alternative R35 contacts from the central subunits B and C, involving both protein–protein and protein–DNA interactions; (C) protein–DNA interactions stabilizing the compressed minor groove around TATA; (D) R35 contacts to the conserved guanine, G3 (outer subunits, A and D).
The overall structure of the complex (Figure 5) comprises two dimers bound to the DNA, each centred on the pseudo-dyad at the TATA sequence that is found at the centre of each operator site. The two dimers are bound to approximately opposite faces of the DNA, as anticipated from the DNA bending assays. The two dimers are related by a 150° rotation and a 53 Å translation along the DNA helix, the latter being close to the expected value for a separation of 15 bp between centres (Figure 6). However, the 150° rotation, rather than 180°, implies a 30° unwinding of the DNA between the two dimer binding sites. The overall organization of the DNA–protein complex is quite different to that of the tetrameric λ CII complex (30), where the protein dimers are on the same face of the DNA helix, resulting in a large interaction interface between dimers in the tetrameric complex.
Figure 6.
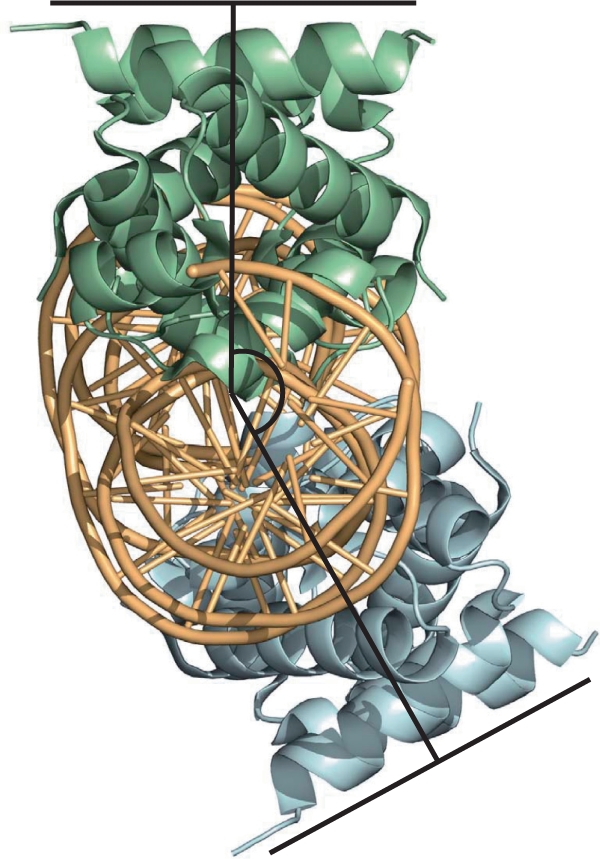
Topology of the tetramer–DNA complex. The relative position of the two dimers (shown in green and blue) is illustrated with respect to the approximate DNA axis. The overall bend angle of ∼43° results from the two individual bends imposed by each dimer, which are related by a 150° rotation about the helix axis (rather than 180° that would cancel the overall bend).
Each subunit interacts with the DNA by inserting helix 3 of the classical HTH motif into the major groove of DNA, either side of the central TATA within each operator. The two protein dimers are related by a dyad axis that coincides with the pseudo-dyad axis lying within the central T:A base pair of the 35 bp duplex.
As discussed earlier, previous predictions had placed the pseudo-dyad at the centre of the conserved GT dinucleotide step (G17-T18), since this conforms to the symmetry of the idealized GACT—AGTC—GACT—AGTC repeating C-box sequence (6). However, this is clearly not the case in the crystal structure of the C.Esp1396I–DNA complex. Instead, the central pseudo-dyad at the T18/A18’ base pair relates the two TATA sequences by a two-fold rotation (as in Figure 2B). It had previously been noted (13) that the TAT element of this sequence was at least as highly conserved between C-proteins as the GACT/AGTC repeat (6,8). The structural basis for this conservation is now clear. Moreover, the TG sequence (T2–G3) on the 5′ side of the operator sequence and the CA on the 3′ side (C33–A34) are also highly conserved between C-proteins, although on the original scheme, they were not symmetrically related (Figure 2A). With T18 as the centre of symmetry, as observed in the crystal structure of the complex, these four base pairs are now related by the pseudo-dyad axis (Figure 2B).
Structural distortion of DNA occurs at TATA sites
The DNA in the complex displays a major kink at the centre of each of the operator sequences (Figure 5), in accordance with the results of the circular permutation assays. From the crystal structure, the overall bend at each operator is estimated as ∼50°, which is close to that observed in the gel assay (51°). Figure 7 plots the major and minor groove width across the DNA sequence. The minor groove width is remarkably narrow (∼2 Å) at each TATA site, compared to typical values of around 7 Å elsewhere. In addition, there is a smaller local narrowing of the minor groove in the centre of the 35 bp sequence (GTG), where the groove width changes locally from ∼8.5 Å to ∼6 Å. In contrast, the major groove width is only slightly increased (to ∼13 Å) around the TATA sequences, but increases quite drastically to 16 Å at the centre of the DNA binding site.
Figure 7.

DNA distortion in the complex. Variation in major (red) and minor (blue) groove widths in the crystal structure of the C.Esp1396I DNA–protein complex are plotted across the 35 bp DNA sequence. Groove parameters were obtained using the program CURVES.
The severe compression of the minor groove at the TATA sites is stabilized by interactions of the phosphodiester backbone (at the 3′ end of each strand in both TATA sequences) with the side chains of Ser52 and Tyr37 from each of the four subunits (Figure 5C). There may be additional interactions with the DNA backbone in this region involving amino acid residues at the dimer interface (Asn47 and Thr49) but the details are indistinct at this resolution. In addition, the bent conformation of the DNA is stabilized by interactions from Gln24 and Arg43 (and possibly Ser39) of each of the four subunits with the phosphodiester backbone at the extremities of each operator site (nucleotides 1, 2 and 17, 18 on each strand).
The role of Glu25 and Arg35 in stabilizing the tetrameric complex
The relative orientation of the two protein dimers in the complex is such that Arg35 and Glu25 of neighbouring subunits (Subunits B and C) can make contacts between positively and negatively charged side chains to stabilize the tetrameric complex (Figure 5B). To test whether these interactions were indeed stabilizing the tetrameric complex and contributing to the cooperativity of binding, we separately mutated Arg35 and Glu25 and checked the DNA binding activity of the mutant proteins by EMSA (Figure 8). In order to confirm their structural integrity prior to binding studies, C.Esp1396I wild-type, E25A and R35A proteins were purified and crystallized individually in the absence of DNA. Preliminary diffraction studies indicate that all three crystallize with identical unit cell dimensions in the same space group.
Figure 8.

Analysis of E25 and R35 mutants. Wild-type C.Esp1396I, and mutant proteins E25A and R35A were analysed by EMSA. Proteins were incubated with a 35 bp dsDNA fragment (240 nM) corresponding to the native operator sequence at protein:DNA ratios of 0, 1, 2, 3, 4, 5, 6, 7 and 8:1. Free DNA (F), dimer–DNA complex (D) and tetramer–DNA (T) complex are indicated. The E25 mutation causes the formation of dimers rather than tetramers. The R35 mutation abolishes all specific nucleoprotein complexes.
As anticipated, the E25A mutant shows greatly reduced cooperativity, and thus destabilizes tetramer formation on the intact operator DNA. More surprisingly, for R35A, DNA binding was completely abolished. The reason for the inability of the R35A mutant to bind to the operator DNA becomes apparent from the structure of the complex; the R35 side chains of the outer subunits (A and D) are in contact with the conserved G3 base on each strand, through paired hydrogen-bond interactions between the guanidinium group of Arg35 and the O6 and N7 H-bond acceptors of guanine G3 in the major groove (Figure 5D). The loss of two strong H-bonds at each site will severely weaken the interaction with the operator.
The E25 mutation, however, does not completely abolish cooperativity (Figure 8), and a further contribution almost certainly arises from distortion of the DNA. The major groove is significantly widened where the HTH motif inserts into the major groove of the DNA and this is most noticeable at the centre of the sequence (Figure 7), where two adjacent HTH motifs are located in the tetrameric complex (Figure 5). This is consistent with the unwinding of the DNA helix between the two operator sites, as deduced from the DNA bending assays. Binding of subunit B of the first dimer would assist the second dimer to bind DNA by opening up the major groove to more easily accommodate the HTH motif from subunit C. This distortion of the major groove between the two operators therefore represents an additional contribution to cooperativity.
DISCUSSION
Our understanding of the recognition of DNA sequences by proteins is far from complete and it is becoming clear that there is no simple ‘read-out’ code involving passive DNA and protein structures (31,32). Frequently both ‘direct’ and ‘indirect’ read-out mechanisms are employed to achieve specificity e.g. as seen in the bacterial transcriptional activator catabolite activator protein (AP) (33). In eukaryotic transcription factors, a combination of read-out mechanisms can also be found; for example, Hox proteins detect DNA shape in the minor groove, in addition to base pair recognition in the major groove (31,34). We propose that a similar dual read-out mechanism applies to C-protein recognition of the operator sequences preceding the C/R genes, but with some unusual features.
Indirect read-out
Indirect readout by C.Esp1396I is based on a number of structural features in the DNA backbone that must be recognized (or induced) by the protein: severe compression of the minor groove at TATA sequences where there is a major kink at the centre of each operator, and expansion of the major groove around the central GTG between operator sites. In total, we can identify 36 interactions to the DNA backbone (8–10 from each subunit), including a number from positively charged arginine side chains.
These interactions are concentrated into two areas: (i) at the extremities of each operator and (ii) either side of the TATA sequences at the centre of each operator. The compression around TATA is accomplished by phosphodiester backbone interactions with Ser52 and Tyr37 (Figure 5C). The side chains of Arg17, Gln24, Ser39 and Arg43 of subunits B and C around the central GTG stabilize the conformation of the phosphodiester backbone defining the wide major groove. Some, but not all, of the equivalent side chains from subunits A and D also make symmetry-equivalent interactions to the phosphodiester backbone near the 5′ end of each DNA strand. All of the above amino acid residues are very highly conserved between C-proteins, with the exception of Ser39 and Ser52, which are frequently replaced by Gly or Asn, respectively (11).
There is evidence from circular permutation gel assays that the DNA is not intrinsically bent, as differential mobility is not seen in the free DNA; moreover, the pronounced structural deformation of the DNA observed by circular dichroism is clearly induced by C-protein binding (15). Thus the interactions of the C-protein with the DNA backbone are responsible for deforming its structure; the sequence of the DNA is also crucial, as it must be capable of assuming this bent conformation without too big an energy penalty. TATA sequences are known to be easily deformable and are frequently found in bent DNA structures in DNA–protein complexes (35). It is notable that the compression of the minor groove that we see at the TATA sites is in stark contrast to the expansion of the minor groove in TATA box recognition by TATA-box binding protein (TBP) (36). In the latter case, the expansion is caused by insertion of aromatic side chains of the protein into the DNA minor groove; for C.Esp1396I, the minor groove of the DNA contracts due to the interactions of amino acid residues of the protein with the phosphodiester backbone of the DNA, which pulls the two strands together across the minor groove.
Direct read-out
Direct readout is provided by the insertion of helix 3 of C.Esp1396I into the major groove of DNA. However, there is likely to be some plasticity in these interactions to accommodate the lack of true symmetry between adjacent operators. Indeed, across species there is also substantial variation in C-box sequences, even when the recognition helices of the C-proteins are identical. The clearest interactions to the DNA that are visible at this resolution are from the side chains of Arg35, and interactions from Arg46 and Thr36 are also likely. Again, it is notable that all three residues are highly conserved in this family of C-proteins, although occasionally conservative changes (Lys and Ser, respectively) are found at the latter two sites (11).
The Arg35 guanidinium group (subunits A and D) interacts with G3 of each DNA strand, each forming two H bonds to the base (Figure 5D). These guanines are strongly conserved across a wide variety of C-protein binding sites (6,8), as is Arg35 in the recognition helix of the C-protein (11). The adjacent base T2 is also highly conserved, perhaps because of the very limited intra-stand base stacking (but significant inter-strand stacking of the purines) that is typical of TpG (= CpA) steps (37). This allows the planar guanidinium group of Arg35 to stack with the exposed face of the thymine base whilst forming hydrogen bonds to the edge of G3 (Figure 9).
Figure 9.

Interaction of Arg35 of subunits A and D with conserved TG (=CA) sequences. Inter-strand stacking of purines A34 and G3, allows T2 to stack with the planar guanidinium group of Arg35, which also hydrogen-bonds to G3 in the major groove.
The symmetrically equivalent interactions from the Arg35 side chains of subunits B and C, however, cannot occur with this DNA sequence (or indeed any other) as a guanine would be required at position 18 on both strands (i.e. a GG base pair at the centre of the 35 bp sequence). In fact, a T:A base pair is located at this site; moreover, this T, together with the G on its 5′ side, is very highly conserved, suggesting that the GT might be essential for the function of the genetic switch (13). We note that the side chain of arginine is long and sufficiently flexible to make alternative contacts in the major groove of the DNA, possibly to the O6 and N7 of G17 (Figure 5B). Indeed, a small displacement and a rotation of the guanidinium group of Arg35 of subunit B would allow it to hydrogen bond simultaneously with both the O4 of T18 and the O6 of G17. Although the electron density map in this region is not of sufficient clarity to be unambiguous, such an interaction could provide a plausible explanation for the high conservation of the central GT. The equivalent interaction from subunit C, however, cannot occur since the major groove on the opposite strand is populated by amino groups, rather than carbonyl groups. Instead, we propose that the Arg35 side chain from subunit C contacts Glu25 of subunit B (Figure 5B), to bridge the two dimers and stabilize the tetramer, rather than interacting with DNA (as discussed below).
Stabilization of the repression complex
Repression of transcription requires the occupation of OR by a C-protein dimer. Since the intrinsic binding of C.Esp1396I to this site is weak, the ‘off switch’ is dependent (at least in part) upon stabilizing the tetrameric complex through energetically favourable interactions at the dimer–dimer interface, resulting in binding of the second dimer. However, there is very little contact between the two dimers when bound to DNA, and there is no buried surface area at the ‘interface’. In the crystal structure of the complex, the only contact between dimers is between Glu25 (subunit B) and Arg35 (subunit C). This electrostatic interaction between positive and negative amino acid side chains of the ion pair contributes significantly to the strength of the dimer–dimer interaction and, together with conformational changes in the DNA induced by binding of the first dimer to OL, is a major contribution to the observed cooperativity.
We have shown that Glu25 is important for cooperativity in binding two consecutive C.Esp1396I dimers. The equivalent acidic amino acid, Glu34, in λ repressor is essential for transcription activation through its interaction with σ70 (Arg588) and the equivalent residue has been suggested to play this role in activation of the C/R promoter (11). We envisage that competition between the Arg35 side chain of subunit C of C.Esp1396I and Arg588 (or its equivalent) in the σ70 subunit of RNA polymerase for the negatively charged side chain of Glu25 leads to competition for the -35 promoter site. It is this competition that is primarily responsible for switching off transcription of the C/R genes at high concentrations of C-protein.
Given the high level of sequence conservation between the majority of C-proteins so far studied (8,11), we anticipate that our findings will be generally applicable to this family of proteins (although there may be some subtle variations in sequence recognition). The ‘spacer’ sequences originally identified within C-protein binding sites (6,8) are almost invariably Py-Pu-Py (usually TAT or CAT). Moreover, these trinucleotide sequences are generally followed by another purine, and all such tetranucleotide sites (Py-Pu-Py-Pu) would be predicted to be bending sites (15), even if they lacked the perfect symmetry of the TATA sequences in the Esp1396I operators. Likewise, the GT between operator sites is highly conserved, and is likely to play a similar role in cooperative binding to that we propose for C.Esp1396I, in which Arg35 plays a crucial role.
Further understanding of the detailed molecular interactions responsible for the specificity (and promiscuity) of sequence recognition by C-proteins will require higher resolution crystallographic studies of a variety of DNA sequences, including smaller dimeric complexes, and these are currently in progress. The current structure highlights for the first time the principal structural features that underpin the genetic switch that regulates gene expression in R–M systems, and provides a paradigm for a new class of protein–DNA complexes.
Accession code
Atomic coordinates and structure factor files have been deposited in the Protein Data Bank with the accession code 3CLC.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The research was funded by a BBSRC project grant to G.G.K. (Grant reference BB/E000878/1). We thank RCUK for provision of an Academic Fellowship (J.E.M.), the University of Portsmouth for a PhD bursary (to N.B.) and NWO-CW for a vidi grant (RBGR). We thank ESRF, Grenoble, for the allocation of synchrotron beam time. We are grateful to K. Severinov and E. Bogdanova for providing the expression plasmid pET-28b/esp1396IC and to D. Wigley for advice on refinement of the structure. Funding to pay the Open Access publication charges for this article was provided by BBSRC.
Conflict of interest statement. None declared.
REFERENCES
- 1.Akiba T, Koyama K, Ishiki Y, Kimura S, Fukushima T. On the mechanism of the development of multiple-drug-resistant clones of Shigella. Jpn. J. Microbiol. 1960;4:219–227. doi: 10.1111/j.1348-0421.1960.tb00170.x. [DOI] [PubMed] [Google Scholar]
- 2.Wilson GG, Murray NE. Restriction and modification systems. Ann. Rev. Genet. 1991;25:585–627. doi: 10.1146/annurev.ge.25.120191.003101. [DOI] [PubMed] [Google Scholar]
- 3.Tao T, Bourne JC, Blumenthal RM. A family of regulatory genes associated with type II restriction-modification systems. J. Bacteriol. 1991;173:1367–1375. doi: 10.1128/jb.173.4.1367-1375.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ives CL, Nathan PD, Brooks JE. Regulation of the BamHI restriction-modification system by a small intergenic open reading frame, bamHIC, in both Escherichia coli and Bacillus subtilis. J. Bacteriol. 1992;174:7194–7201. doi: 10.1128/jb.174.22.7194-7201.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rimseliene R, Vaisvila R, Janulaitis A. The eco72IC gene specifies a trans-acting factor which influences expression of both DNA methyltransferase and endonuclease from the Eco72I restriction-modification system. Gene. 1995;157:217–219. doi: 10.1016/0378-1119(94)00794-s. [DOI] [PubMed] [Google Scholar]
- 6.Vijesurier RM, Carlock L, Blumenthal RM, Dunbar JC. Role and mechanism of action of C·PvuII, a regulatory protein conserved among restriction-modification systems. J. Bacteriol. 2000;182:477–487. doi: 10.1128/jb.182.2.477-487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cesnaviciene E, Mitkaite G, Stankevicius K, Janulaitis A, Lubys A. Esp1396I restriction-modification system: structural organization and mode of regulation. Nucleic Acids Res. 2003;31:743–749. doi: 10.1093/nar/gkg135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knowle D, Lintner RE, Touma YM, Blumenthal RM. Nature of the promoter activated by C.PvuII, an unusual regulatory protein conserved among restriction-modification systems. J. Bacteriol. 2005;187:488–97. doi: 10.1128/JB.187.2.488-497.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Streeter SD, Papapanagiotou I, McGeehan JE, Kneale GG. DNA footprinting and biophysical characterisation of the controller protein C.AhdI suggests the basis of a genetic switch. Nucleic Acids Res. 2004;32:6445–6453. doi: 10.1093/nar/gkh975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGeehan JE, Papapanagiotou I, Streeter SD, Kneale GG. Cooperative binding of the C.AhdI controller protein to the C/R promoter and its role in endonuclease gene expression. J. Mol. Biol. 2006;358:523–531. doi: 10.1016/j.jmb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 11.McGeehan JE, Streeter S, Papapanagiotou I, Fox GC, Kneale GG. High-resolution crystal structure of the restriction-modification controller protein C.AhdI from Aeromonas hydrophila. J. Mol. Biol. 2005;346:689–701. doi: 10.1016/j.jmb.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 12.Bogdanova E, Djordjevic M, Papapanagiotou I, Heyduk T, Kneale G, Severinov K. Transcription regulation of the type II restriction-modification system AhdI. Nucleic Acids Res. 2008;36:1429–1442. doi: 10.1093/nar/gkm1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mruk I, Rajesh P, Blumenthal RM. Regulatory circuit based on autogenous activation-repression: roles of C-boxes and spacer sequences in control of the PvuII restriction-modification system. Nucleic Acids Res. 2007;35:6935–6952. doi: 10.1093/nar/gkm837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mruk I, Blumenthal RM. Real-time kinetics of restriction modification gene expression after entry into a new host cell. Nucleic Acids Res. 2008;36:2581–2593. doi: 10.1093/nar/gkn097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papapanagiotou I, Streeter SD, Cary PD, Kneale GG. DNA structural deformations in the interaction of the controller protein C.AhdI with its operator sequence. Nucleic Acids Res. 2007;35:2643–2650. doi: 10.1093/nar/gkm129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferrari S, Harley VR, Pontiggia A, Goodfellow PN, Lovell-Badge R, Bianchi ME. SRY, like HMG1, recognizes sharp angles in DNA. EMBO J. 1992;11:4497–4506. doi: 10.1002/j.1460-2075.1992.tb05551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993;26:795–800. [Google Scholar]
- 18.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallog. D Biol. Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 19.CCP4. The CCP4 suite: programs for crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 20.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallog. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 21.Adams PD, Gopal K, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Pai RK, Read RJ, Romo TD, et al. Recent developments in the PHENIX software for automated crystallographic structure determination. J. Synchrotron Radiat. 2004;11:53–55. doi: 10.1107/s0909049503024130. [DOI] [PubMed] [Google Scholar]
- 22.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 23.Lavery R, Sklenar HJ. The definition of generalized helicoidal parameters and of axis curvature for irregular nucleic acids. J. Biomol. Struct. Dyn. 1988;6:63–91. doi: 10.1080/07391102.1988.10506483. [DOI] [PubMed] [Google Scholar]
- 24.Stofer E, Lavery R. Measuring the geometry of DNA grooves. Biopolymers. 1994;34:337–346. doi: 10.1002/bip.360340305. [DOI] [PubMed] [Google Scholar]
- 25.Luscombe NM, Laskowski RA, Thornton JM. NUCPLOT: a program to generate schematic diagams of protein-DNA interactions. Nucleic Acids Res. 1997;25:4940–4945. doi: 10.1093/nar/25.24.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 27.Zwieb C, Adhya S. Improved plasmid vectors for the analysis of protein-induced DNA bending. Methods Mol. Biol. 1994;30:281–294. doi: 10.1385/0-89603-256-6:281. [DOI] [PubMed] [Google Scholar]
- 28.Sawaya M, Zhu Z, Mersha F, Chan S, Dabur R, Xu S, Balendiran G. Crystal structure of the restriction-modification system control element C.BclI and mapping of its binding site. Structure. 2005;13:1837–1847. doi: 10.1016/j.str.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 29.Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3β homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- 30.Jain D, Kim Y, Maxwell KL, Beasley S, Zhang R, Gussin GN, Edwards AM, Darst SA. Crystal structure of bacteriophage λcII and its DNA complex. Mol. Cell. 2005;19:259–269. doi: 10.1016/j.molcel.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Harrison SC. Three-dimensional intricacies in protein-DNA recognition and transcriptional control. Nat. Struct. Mol. Biol. 2007;14:1118–1119. doi: 10.1038/nsmb1207-1118. [DOI] [PubMed] [Google Scholar]
- 32.Albright RA, Matthews BW. How Cro and lambda repressor distinguish between operators: the structural basis underlying a genetic switch. Proc. Natl Acad. Sci. 1998;95:3431–3436. doi: 10.1073/pnas.95.7.3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawson CL, Swigo D, Murakami KS, Darst SA, Berman HM, Ebright RH. Catabolite activator protein: DNA binding and transcription activation. Curr. Opin. Struct. Biol. 2004;14:10–20. doi: 10.1016/j.sbi.2004.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joshi R, Passner JM, Rohs R, Jain R, Sosinsky A, Crickmore MA, Jacob V, Aggarwal AK, Honig B, Mann RS. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell. 2003;131:530–543. doi: 10.1016/j.cell.2007.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dickerson RE. DNA bending: the prevalence of kinkiness and the virtues of normality. Nucleic Acids Res. 1998;26:1906–1926. doi: 10.1093/nar/26.8.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Juo ZS, Chiu TK, Leiberman PM, Baikalov I, Berk AJ, Dickerson RE. How proteins recognize the TATA box. J. Mol. Biol. 1996;261:239–254. doi: 10.1006/jmbi.1996.0456. [DOI] [PubMed] [Google Scholar]
- 37.Calladine CR, Drew HR. Understanding DNA: The Molecule & How It Works, Ch. 3. Academic Press, London; 1992. p. 52. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.