

Abstract
A therapeutic CMV vaccine incorporating an antigenic repertoire capable of eliciting a cellular immune response has yet to be successfully implemented for patients who already have acquired an infection. To address this problem, we have developed a vaccine candidate derived from modified vaccinia Ankara (MVA) that expresses three immunodominant antigens (pp65, IE1, IE2) from CMV. The novelty of this vaccine is the fusion of two adjacent exons from the immediate-early region of CMV, their successful expression in MVA, and robust immunogenicity in both primary and memory response models. Evaluation of the immunogenicity of the viral vaccine in mouse models shows that it can stimulate primary immunity against all three antigens in both the CD4+ and CD8+ T cell subsets. Evaluation of human PBMC from healthy CMV-positive donors or patients within 6 months of receiving hematopoietic cell transplant shows robust stimulation of existing CMV-specific CD4+ and CD8+ T cells subsets.
Keywords: human cytomegalovirus, viral vaccine, attenuated poxvirus, cellular immunity, transgenic mouse model, memory T cells, regulatory proteins, tegument protein
Introduction
CMV infection is an important complication of transplantation procedures and affects a wide variety of individuals including newborns and HIV patients with advanced disease (Pass et al., 2006;Sinclair et al., 2006;Zaia, 2002). Since the earliest work in the field of CMV infection, it has been a goal to develop a vaccine strategy that would protect at-risk individuals. The goal of developing effective therapeutics to prevent or to control CMV infection have been understood in recent years to likely involve separate classes of host response targets (Khanna & Diamond, 2006). Individuals whom are previously CMV infected or receiving a CMV-infected solid organ or stem cell allograft are candidates for a vaccine strategy that targets the cellular reservoir of the virus (Zaia et al., 2001). In this regard, several antigens have been identified as being associated with protective immunity in transplant recipients. These include the tegument protein pp65 (UL83) and the immediate-early 1 (IE1 or UL123) global gene expression regulator (Boeckh et al., 2006;Cobbold et al., 2005;Cwynarski et al., 2001;Einsele et al., 2002;Gratama et al., 2001). In addition, a recent proteomic study of the whole CMV genome divided into overlapping peptides showed that pp65 stimulated substantial levels of both CD8+ and CD4+ T cells, while IE1 mainly stimulated CD8+ T cells, and the related IE regulator referred to as IE2 (UL122) stimulated a vigorous CD8+ and a smaller CD4+ T cell memory response by a large percentage of asymptomatic CMV-positive adults (Sylwester et al., 2005). While other antigens are also recognized with robust cellular immune responses, the evidence for these three antigens to be highly recognized in a majority of CMV-infected healthy subjects and transplant patients {this manuscript and (Gallez-Hawkins et al., 2005)} is compelling and justifies their inclusion into a vaccine to prevent viremia and control infection.
Evidence from studies of murine CMV (MCMV) point to the important role of IE1 antigens for development of protective immunity in transplantation models (Reddehase et al., 1987). More recently, homologues of the human CMV pp65 antigen assembled into viral or plasmid DNA vectors showed evidence of stimulating protective immunity against MCMV, GPCMV, or RhCMV (Morello et al., 2000;Schleiss et al., 2007;Yue et al., 2007). Our laboratory has developed modified vaccinia Ankara (MVA) into a vaccine for delivering CMV antigens to clinically evaluate which of them exhibit protective qualities in the context of CMV complications resulting from transplant procedures (Song et al., 2007;Wang et al., 2004b;Wang et al., 2004a;Wang et al., 2007). MVA has an extensive history of successful delivery into rodents, Rhesus macaques, and other non-human primates, and more recently as a clinical vaccine in cancer patients (Gilbert et al., 2006;Peters et al., 2007;Rochlitz et al., 2003). Although MVA is able to efficiently replicate DNA in mammalian cells, it is avirulent because of the loss of two important host range genes among >25 mutations and deletions that occurred during its repeated serial passage in chicken cells(Antoine et al., 1998;Meyer et al., 1991). In contrast to NYVAC (attenuated Copenhagen strain) and ALVAC (host-range restricted Avipox), both early and late transcription are unimpaired making MVA a stronger vaccine candidate (Blanchard et al., 1998;Carroll et al., 1997;Carroll & Moss, 1997;Zhang et al., 2007). Studies in rodents and macaques affirm the safety of MVA, including protection against more virulent forms of pox viruses and challenge models and lack of persistence three months beyond initial dosing administration(de Waal et al., 2004;Earl et al., 2007;Hanke et al., 2005). Similarly, a therapeutic vaccination with MVA expressing HIV-nef demonstrated its safety in HIV-1 infected individuals(Cosma et al., 2003). Finally, MVA immunizations of malaria patients coinfected with HIV and/or TB confirm the safety of the vector and its ability to partially protect against a heterologous malaria strain(Gilbert et al., 2006;Moorthy et al., 2003). These properties form the basis for our exploration of MVA as a vaccine vector for CMV antigens in individuals who are severely immunosuppressed and who are experiencing additional complications such as malignancy or organ failure, thereby requiring a transplant for which CMV infection is an important infectious complication.
Our previous work in CMV vaccine development pointed to robust immunity in mouse models using an MVA expressing pp65 and IE1-exon4(Wang et al., 2007). We reasoned that development of an IE fusion protein incorporating the adjacent exon5 would increase the coverage of additional HLA types by the vaccine, because of the different HLA recognition profiles for both IE1 and IE2 genes. Profound sequence differences between the major exons of both IE1 and IE2 genes results in a substantial difference in epitope motifs represented in both genes that is manifested by distinctly different immunologic profile of recognition among individuals who recognize either gene product (see Discussion). Both the IE1 and pp65 antigens have been made into fusion proteins and evaluated as vaccines in primary immunity models as well as stimulators of memory response using human peripheral blood from CMV-seropositive individuals (Rohrlich et al., 2004;Vaz-Santiago et al., 2002). Our approach was to fuse exon4 from IE1 with exon5 from the IE2 gene into a single gene without additional genetic material. The fusion protein comprises a more complete representation of the immediate-early antigens than either protein alone. Our data confirmed the feasibility of fusing both antigens, since we detected robust immunogenicity in mouse models as well as studies in human PBMC. Our analysis of this MVA vaccine is based on measuring CMV-specific IFN-γ+ T cell responses, which previous studies from our laboratory and others have shown to be correlated with cytotoxic function in mouse models and protective immunity in clinical situations (Avetisyan et al., 2007;Sinclair et al., 2006). The extraordinary immunogenicity of the recombinant MVA expressing pp65 and IEfusion antigens provides a strategic approach for developing a CMV vaccine for transplant recipients.
Results
Construction of the IE1/e4-IE2/e5 fusion (IEfusion) gene, cloning into transfer vector pZWIIA, and generation of recombinant MVA
IE1, also known as UL123 is composed of four adjacent exons interspersed with 3 introns while the adjacent UL122 (IE2) gene is composed of the same initial 3 exons of the UL123 gene but as a result of alternate splicing, it also contains a unique adjacent exon 5 (Figure 1A)(White et al., 2007). To approximate the genetic architecture in the CMV genome and to reduce the number of independent transcription units to be inserted into MVA, exon 4 (e4) from IE1 and exon 5 (e5) from IE2 were joined as shown in Figure 1B. Genomic copies of e4 and e5 were amplified from CMV strain AD169 viral DNA, and primers were developed that made use of a newly created restriction site in IE2/e5 that was introduced into the IE1/e4 fragment by PCR methods. The independent exons with overlapping sequence were joined at the newly created Apa I site to create the fusion gene without introduction of new protein sequence. We omitted exons 2-3, because they contain transcriptional activation domains whose activity might cause unexpected gene activation events, and the omission of a relatively short 85 AA segment seems a reasonable compromise with safety(Gyulai et al., 2000;Johnson et al., 1999).
Figure 1. Construction of IEfusion-pZWIIA and pp65-IE1fusion-pZWIIA.



A) The genomic structure of the regulatory immediate early genes IE1 and IE2 of HCMV. IE1 is composed of 4 exons (exon 1, 2, 3 and 4) indicated by solid dark lines and three introns as indicated by intervening thin lines; IE2 is also composed of 4 exons (exon1, 2, 3 and 5) as indicated by solid dark lines and three introns as indicated by intervening thin lines. B) Construction of IEfusion gene. Primers a, b, c, d, e are described in M&M. IE1/e4 was amplified from the IE1 gene using primers a and b, and was further extended using primer a and c to introduce an internal Apa I site, and external Pme I and Asc I sites. IE2/e5 was amplified from the IE2 gene using primers d and e. It was digested at the newly created Apa I and synthetic Asc I site. IE1/e4 and IE2/e5 were joined together by ligation preserving the reading frame. C) Schematic map of IEfusion-pZWIIA and pp65-IEfusion-pZWIIA MVA transfer plasmids. pZWIIA, an ampicillin resistant plasmid (amp shown in light grey) inserts DNA sequence within the boundaries of MVA Deletion II via Flanking regions 1 and 2 shown in red (FL1, FL2). pZWIIA has two vaccinia synthetic E/L promoters (green) of slightly different sequence, arranged head to head to each drive expression of separate genes. IEfusion gene shown in yellow expression is driven by Psyn I promoter(Chakrabarti, Sisler, & Moss, 1997) and pp65 gene shown in yellow expression is driven by Psyn II promoter(Wyatt et al., 2004). gus bacterial marker gene shown in blue used for identifying recombinant MVA is flanked by two direct repeat (DR) sequences shown in dark grey to facilitate gus gene removal by intragenomic recombination from IEfusion-MVA or pp65-IEfusion-MVA. In neither transfer plasmid was pp65 fused to the IEfusion gene. D) Generation of IEfusion-MVA and pp65-IEfusion-MVA. IEfusion-pZWIIA or pp65-IEfusion-pZWIIA was transfected into wtMVA infected CEF cells to generate IEfusion-MVA or pp65-IEfusion-MVA via homologous recombination at Deletion II whose flanking region is contained in the plasmid that is homologous to wtMVA.
The fusion gene was cloned into pZWIIA using unique restriction enzymes that were added by PCR to the ends of each exon (Figure 1C). Versions of the transfer plasmid pZWIIA with UL83 (pp65) were also constructed (Figure 1C). pZWIIA encodes two direct repeats flanking the bacterial marker gene (α-glucoronidase or gus) that facilitates their removal through stochastic recombination as earlier described (Wang et al., 2004a). Both versions of pZWIIA were used in combination with wtMVA to generate rMVA expressing the IEfusion gene alone or co-expressed with pp65 (Figure 1D). Since others have created pp65-IE1 fusion proteins, we did not recapitulate those studies and always kept the pp65 gene separate from the IEfusion gene in the MVA shown in Figure 1D. Each rMVA underwent ≥8 rounds of purification, and was verified to be absent of parental wtMVA using PCR methods (data not shown) (Wang et al., 2007).
Evaluation of expression of recombinant antigens using Western blot approach
We tested whether the artificial joint between IE1/e4 and IE2/e5 would allow continuous translation of the predicted full length protein product, by infecting CEF with wtMVA and simultaneous transfection with pZWIIA containing the IEfusion gene. The results show a 125 Kda protein band composed solely of the IE1/e4 and IE2/e5 exons, detected using the IE1/e4 specific mAB that also detected the expected 60Kd band after infection of CEF with IE1/e4-MVA(Wang et al., 2007) (data not shown). Virus plaques expressing the IEfusion gene with and without separately co-expressed pp65 were amplified, and titered viruses were used to make lysates that were separated using SDS-PAGE, followed by WB analysis using antibodies to detect pp65 (Figure 2A) and IEfusion proteins (Figure 2B). The results confirm that the IEfusion protein can be highly expressed alone or in combination with pp65 (Figure 2).
Figure 2. Western blot (WB) detection of pp65 and IEfusion protein antigens.
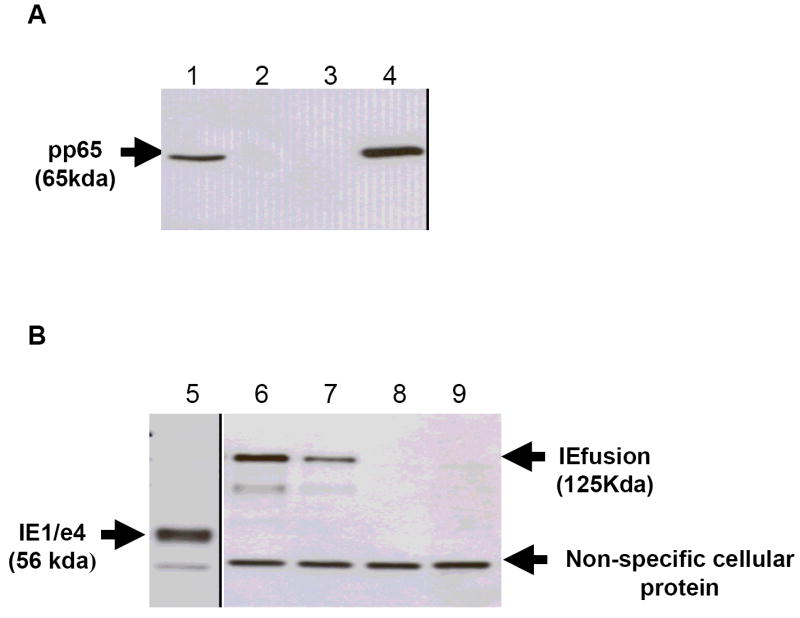
A) Lane 1: CEF cell lysate infected with pp65-rMVA as (+) control; Lanes 2 and 3: cell lysate from wtMVA infected and uninfected CEF as (-) controls; Lane 4: cell lysate of pp65-IEfusion-MVA infected CEF cells. The WB in Panel A was incubated with mAB 28-103 against pp65. B) Lane 5: cell lysate of CEF infected with rMVA expressing IE1/e4 as (+) control; Lanes 6 and 7: cell lysate from wtMVA infected and uninfected CEF as (-) controls; Lane 8: cell lysate of pp65-IEfusion-MVA infected CEF cells and Lane 9: cell lysate of IEfusion-MVA infected CEF cells. The WB in Panel B was incubated with mAB p63-27 against IE1. See M&M for details of MVA construction that clarifies that pp65 and IEfusion are separate proteins of 65 and 125 Kda, respectively.
Immunogenicity in transgenic HLA A2 mice
To establish whether rMVA would elicit primary immunity in a CMV-naïve host, experiments were performed in transgenic (Tg) mice naïve to all antigens expressed from the rMVA. HHDII mice which are Tg for the HLA A2 gene and focus presentation on the human MHC were immunized with the IEfusion-MVA or pp65-IEfusion-MVA for three weeks (Pascolo et al., 1997). Spleens were processed and IVS was set up for a period of 7 days followed by ICC to detect IFN-γ expression (see Material and Methods for details). To evaluate the HLA A2-restricted CD8+ T cell response we used the immunodominant HLA A2-restricted pp65 and IE1 CTL epitopes, and the IE2 peptide library, since no HLA A2-restricted IE2 epitopes have been defined (Wang et al., 2004b;Wang et al., 2007). To measure MHC Class II CD4+ T cell responses, peptide libraries specific for the pp65, IE1, and IE2 antigens were used both during the IVS and ICC stimulations. The results presented in Figure 3 demonstrate robust immunogenicity of the rMVA after infection in the HHDII mouse. Levels of specific IFN-γ produced by CD8+ T cells were significantly higher than for CD4+ T cells for all 3 CMV antigens. In contrast, both a robust CD4+ and CD8+ T cell response was found for pp65 (Figure 3). Likewise, there was substantial recognition of the IE1/e4 portion of the IEfusion protein demonstrated by a potent CD8+ T cell response using the IE1 peptide library (Figure 3). Finally there was a good CD8+ T cell response to the IE2 library, and a lesser response by CD4+ T cells (Figure 3). We verified that the immunogenicity of the IEfusion protein was not dependent on the presence of the pp65 antigen by immunizing HHDII mice with an MVA that included the IEfusion protein without co-expression of pp65 (data not shown). The HHDII mice responded similarly to the IE2 library, and also appropriately responded to the HLA-A2 restricted epitope of IE1 in a robust manner. These experiments confirm the strong immunogenicity of the IEfusion protein, and also verify that the immunogenicity of the IE1 portion of the molecule is not disrupted when the IE2 portion is fused to it. The immunogenicity of the IE1 portion compares favorably to constructs in which IE1/e4 is expressed as a single exon without fusion(Wang et al., 2004b;Wang et al., 2007).
Figure 3. Immunogenicity of pp65-IEfusion-MVA in HHDII mice.

A) Percentage of IFN-γ producing splenocytes specific for pp65, IE1 and IE2 (x axis) in three mice immunized with 50 million pfu of pp65-IEfusion-MVA. Grey bars represent pp65-, IE1- and IE2-specific IFN-γ production by CD8+ T-cells using either peptide epitopes or libraries (identified below the x axis) during IVS and ICC stimulations. Unfilled bars represent simultaneous pp65-, IE1- and IE2-specific IFN-γ production by CD4+ T-cells, following IVS and ICC stimulation with the corresponding CMV libraries indicated below each set of bars. IVS and ICC stimulation conditions are described in M&M. In all graphs, error bars represent standard error of the mean among the immunized mice (N=3). In all experiments, IFN-γ production to mock stimulated cells was subtracted. P values indicate statistically significant differences measured by T-test.
Immunization of Tg HLA B7 Kbko/Dbko (B7 mice) with pp65-IEfusion-MVA
The success of the immunogenicity trial in HHDII mice prompted us to investigate a Tg model expressing a different HLA allele to generalize the scope of immunogenicity of the rMVA in different HLA backgrounds. B7 mice are deficient in both Kb and Db murine genes, and mainly process Class I antigens using the Tg MHC molecule, HLA B*0702 (Rohrlich et al., 2003). Immunization conditions were similar as we described for HHDII mice, and after three weeks, mice splenocytes were stimulated during both IVS and ICC procedures with HLA B*0702 pp65265-275 epitope to evaluate the Tg CD8+ T cell response. Since in B7 mice the recognition of HLA B*0702 IE1 epitopes is minimal (unpublished results) we used the IE1 peptide library to measure the Tg CD8+ T cell response (Figure 4). Peptide libraries specific for the pp65, IE1, and IE2 antigens were also used to evaluate the MHC Class II responses. Similar to the findings in HHDII mice, higher levels of CD8+ and lower levels of CD4+ T cell responses were elicited against all 3 CMV antigens (Figure 4). This evaluation demonstrates that both the pp65 and IEfusion genes are functional and immunologically recognized in the Tg HLA B7 mouse model.
Figure 4. T-cell responses in B7 mice.

Percentage of IFN-γ producing splenocytes assessed by flow cytometry specific for pp65 (CTL epitope or library), IE1 and IE2 peptide libraries (x axis) in three mice immunized with 50 million pfu of pp65-IEfusion-MVA, using methods as described in the legend to Figure 3. In all graphs, error bars represent standard error of the mean among the immunized mice (N=3). In all experiments, IFN-γ production to mock stimulated cells was subtracted. P values indicate statistically significant differences measured by T-test.
Ex-vivo response to CMV-pp65, IE1, and IE2 peptide libraries in healthy volunteers and HCT recipients
To best gauge the strength of the rMVA to stimulate CMV-specific T cells from PBMC of CMV-positives, we first examined the ex vivo recognition of the three peptide libraries (pp65, IE1, IE2) corresponding to the cognate expressed proteins in rMVA. We conducted measurements on 22 CMV-positive (Figure 5) and 8 CMV-negative healthy adults (data not shown). We classified individuals as responders if they had antigen-specific T cell frequencies of greater magnitude than levels found in CMV-negatives, which averaged 0.05% of CD8+ and 0.08% of CD4+ T cells for each of the 3 peptide libraries (data not shown). The number of individuals classified as responders was highest for pp65 in both the CD8 (16/22) and CD4 (10/22) subsets, and lower numbers of responders (9/22) for both the IE1 and IE2 library in CD8, but far fewer (3/22) in the CD4 subset. The number of individuals responding to the 3 peptide libraries is qualitatively similar to the only other comparable dataset (Sylwester et al., 2005). We then calculated the CMV-specific CD8+ and CD4+ T cell frequencies for each of the 22 CMV-positives, and found roughly equivalent response levels to all 3 libraries in the CD8+ T cell subset (Figure 5A). In contrast, there was a dichotomy of response in the CD4+ T cell subset such that pp65 responses were on average >3-fold higher than found in IE responders, which agrees with findings from previous investigators (Sylwester et al., 2005). In summary, the T cell responses of this randomly chosen group of CMV-positives is qualitatively in accord with the previous study, confirming the reliability of our approach and the legitimacy of using results from our healthy volunteer group as a benchmark for comparisons with less well characterized HCT patients.
Figure 5. Ex vivo response to pp65, IE1, and IE2 peptide libraries.


A) Responses in healthy volunteers. PBMC were obtained from N=22 healthy volunteers for which we had complete HLA typing. Five million PBMC were divided into four aliquots and were individually co-incubated with peptide libraries at 1μg/ml/peptide in single use aliquots as described in Materials and Methods. PBMC from each individual were treated in separate cultures with each peptide library at the same time, but not all individuals were evaluated on the same day. Standard gating procedures were employed for each individual flow acquisition, such that conditions were standardized for all evaluations. Separate aliquots from the ICC assay were incubated with CD4+, CD8+ or isotype control antibodies as described in Materials and Methods. The plots show the percentage of IFN-γ producing T Cells for each of the antigen-specific peptide libraries. Error bars represent the standard error of the mean calculated using Microsoft Excel statistical package. B) Ex vivo response of PBMC from HCT recipients. Three examples from each of three separate risk categories of HCT shown in 3 separate plots (L-R; D+/R+, D-/R+, D-R+) based on CMV status were evaluated for response against peptide libraries using the same technical approach as described in A). Data from all 3 individuals was averaged in each category, and the error bars represent the standard error of the mean.
We followed up these studies in healthy adults by investigating the immune response of stem cell transplant (HCT) recipients to all three peptide libraries in three combinations of donor (D) and recipient (R) pairs with increasing risk for complications of CMV infection (D+/R+, D+/R- and D-/R+) at 180 days post-transplant (Figure 5B). All 9 recipients that we chose were part of a study of natural development of immunity to CMV and were known responders to CMV antigens (Gallez-Hawkins et al., 2005;Lacey et al., 2006). All 9 patients responded to the 3 peptide libraries by producing a CD8+ T cell response of similar magnitude to healthy adults with chronic CMV infection (Figure 5A). Similar to the results for healthy volunteers, the pp65 library stimulated a strong response in both the CD4+ and CD8+ T cell subset, while the IE1 and IE2 libraries were most effective for stimulating a CD8+ T cell response (Figure 5B). The low level of CD4+ T cell response to both the IE1 and IE2 libraries is also consistent with previous reports and our results in healthy volunteers. These observations indicate that both the magnitude and quality of the T cell response to the pp65, IE1, and IE2 antigens is similar in recovering HCT recipients as it is in healthy CMV-positive volunteers.
IEfusion-MVA stimulates CMV-specific T cells in human PBMC
We examined the immunogenicity of the IEfusion protein as a single immunogen or co-expressed with pp65 in rMVA. Autologous APC are matured to be optimally receptive to MVA infection and antigen presentation by the use of a CpG DNA cocktail(La Rosa et al., 2006). Following three days of maturation, APC are infected with rMVA containing the IEfusion gene or rMVA containing both the IEfusion and pp65 genes, followed by irradiation to inactivate the APC for proliferation. We examined IEfusion-MVA in PBMC from three CMV-positive healthy donors and one CMV-negative donor. ex vivo recognition of either the IE1 or IE2 peptide libraries was first conducted as a comparison to the MVA IVS study (Figure 6A). The average increase was quite substantial after IVS with IEfusion-MVA, nearing 5-fold in each of the three CMV-positive individuals evaluated in either the CD4+ or CD8+ subset as detected with the IE1 or IE2 peptide libraries (Figure 6A). In contrast, there was no evidence for ex vivo recognition of peptide libraries in the CMV-negative individual, nor was there any significant stimulation of either IE-specific T cell population (data not shown). As expected, no evidence of pp65-specific stimulation beyond ex vivo levels was found in both CMV-positives and negatives, because the rMVA did not express pp65.
Figure 6. rMVA stimulates CMV-specific T cells in human PBMC.


A) Using the IEfusion MVA, as described in Materials and Methods, APC were infected for 5-6 hours, irradiated, and then co-incubated with unmanipulated PBMC from the autologous individual. The time course and conditions of the IVS are described in Materials and Methods. Four separate evaluations were conducted with each IVS culture as shown in Panel A. After treatment with the peptide library, and ICC was performed, and aliquots of PBMC were either stained with CD4 or CD8 antibodies as described in Figure 5A. Results shown are averages of measurements from three CMV-positive individuals selected randomly from our cohort of blood donors. Not shown is a comparison with a CMV-negative donor who showed no specific recognition of any of the three peptide libraries after IVS with IEfusion- and pp65-IEfusion-MVA. B) Similar to A), but using the pp65-IEfusion-MVA, IVS was carried out using conditions identical as in A) and fully described in Materials and Methods. PBMC from 8 healthy CMV-positive blood donors were evaluated both ex vivo without manipulation and post-IVS following infection with rMVA as described in A). Statistical differences between ex vivo levels of CMV-specific T Cells versus post-IVS were calculated as described in Materials and Methods, and when a P-value is ≤0.05, it is shown above the error bar for each evaluation of individual peptide libraries. All methods for IVS, ICC, and flow cytometry are described in Materials and Methods.
The immunogenicity of pp65-IEfusion-MVA was assessed by comparison to ex vivo measurements of the autologous PBMC populations using all three peptide libraries (Figure 6B). In all individuals examined, there was brisk stimulation of antigen-specific T cell populations that often exceeded levels found with IEfusion-MVA (Figure 6A). In the case of the CD8+ T cell subset, IVS caused substantial increase in all three antigen-specific T cell populations. As expected, the ex vivo level of the CD4+ subset recognizing pp65 was far greater than for the IE antigens, which was also reflected in the amplified frequencies after IVS with rMVA. Using the same CMV-negative healthy donor that we investigated with IEfusion-MVA, we found no evidence of pp65 or IEfusion-specific immunity after in vitro immunization with pp65-IEfusion-MVA (data not shown). Results from both vaccine viruses establish that rMVA stimulation does not substantially alter the relationship of the T cell subset proportion measured ex vivo for all three antigens; it amplifies ex vivo levels to a higher level after IVS (Figures 6A and B). As a further control for specificity of CMV antigen recognition, we investigated in vitro immunization of PBMC from 3 healthy donors shown in Figure 6B with an MVA only expressing the Gus gene (Gus-MVA) that was constructed using different transfer vectors and described in a previous report(Wang et al., 2004a). As expected, there was no incremental increase in CMV-specific recognition of all 3 peptide libraries greater than what was measured ex vivo (data not shown).
rMVA stimulates CMV-specific effectors in PBMC from transplant recipients
The success of the studies in healthy adults prompted us to evaluate the capability of the three-antigen rMVA to stimulate memory responses in PBMC from HCT recipients. In our evaluation, two examples were chosen from three different risk categories of patients that were also examined ex vivo: D+/R+, D-/R+ and D+/R- (Figure 5B). Results of the IVS with MVA are shown juxtaposed to the ex vivo response to demonstrate the magnitude of the stimulation of CMV-specific T cell responses in all patient risk groups (Figure 7). The CD8+ was more substantial than the CD4+ T cell stimulation which reflected the ex vivo profile, which shows substantial over-representation of CD8 versus CD4 responses (Figure 7). The levels of rMVA amplification of CMV-specific T cells in many cases exceed those found in healthy volunteers (Figures 6 and 7). This is evident in both the CD4 and CD8 T cell populations, and is observed in all three patient groups with different combinations of CMV serostatus. While not all antigens were equally stimulated in all patients, the majority of measurements demonstrate a substantial amplification from ex vivo levels in both the CD4+ and CD8+ T cell population. The specificity of the immune responses to CMV antigens was confirmed by including an additional in vitro immunization culture using Gus-MVA, from two of the six HCT patients that we were fortunate to have sufficient PMBC to conduct this additional control (data not shown). There was no evidence of CMV-specific immune stimulation, beyond what was measured ex vivo from both individuals (Figure 7 and data not shown).
Figure 7. rMVA stimulates CMV-specific T cells in PBMC from HCT recipients.

Six examples of patients that were evaluated for response to peptide libraries shown in Figure 5B were also evaluated after IVS with pp65-IEfusion-MVA. Methods including conditions for IVS, post-IVS analysis of cell population, ICC, and flow cytometry are identical as described in Figure 6. A comparison was made between the ex vivo level versus post-IVS for each stimulation, and each category of donor and recipient serostatus is shown in 3 separate plots as discussed in the Legend to Figure 5B.
Discussion
The goal of deriving pp65-IEfusion-MVA is to expand the diversity of cellular immune responses against CMV to counter viremia in the immuno-suppressed patient. The rationale of this vaccine virus is to include antigens from CMV that are expressed early to disrupt the viral life cycle. In contrast to previous efforts, we sought to maximize the representation of IE specific immunity by including both the IE1 and IE2 antigens. pp65, IE1, followed by IE2, rank among the best recognized antigens in the CD8 subset, and with the largest proportion of the T cell memory response to CMV (Sylwester et al., 2005). Interestingly, there is no region of homology greater than 5 amino acids between the major exons of both proteins. Individually, both antigens are recognized broadly by almost 70% of the general population (Sylwester et al., 2005). While few epitopes have been mapped to unique sequence positions of either gene, the divergent sequence of both IE1/e4 and IE2/e5 used in our construction predicts an entirely different subset of HLA binding peptides using publicly available Class I and II motif algorithms (Peters & Sette, 2007). Human subjects that we empirically evaluated for recognition of both IE1 and IE2 antigens were found in many instances to recognize one or the other but not both. Among the research subjects we analyzed, 24% recognized IE2 with or without pp65 to the exclusion of IE1 (data not shown). This result strongly suggests that the recognition elements for both antigens are unique, and by including both of them in the vaccine, we extend the breadth of individuals with disparate HLA types that will recognize and develop an immune response to the vaccine. We potentially double the level of recognition of IE genes based on our own results and what was previously reported (Sylwester et al., 2005). The fusion of major exons from both antigens achieves the twin goal of reducing the number of separate inserts and eliminating the need for a third insert promoter. The advantages of this approach include placement of all vaccine antigens in one vector, and diminishing the dose of virus needed to attain sufficient immunity simultaneously against all of the included antigens.
The IEfusion gene has not been previously evaluated as an immunogen. We conducted extensive analysis to establish parameters of expression and immunogenicity to qualify it for potential clinical use. Two forms of rMVA were designed to test the IEfusion protein, either as a single antigen or combined with pp65. This dual evaluation allowed conclusions about the capacity of various immunologic tests to accurately score the antigen as being recognized as a single entity or in combination with another strongly immunogenic antigen. We demonstrated strong expression of the IEfusion protein as a single antigen MVA or in combination with pp65. In either virus, the antigen was strongly expressed behind the synthetic E/L promoter (Psyn I). This test demonstrates that the presence of pp65 did not interfere with IEfusion protein expression, since there were earlier reports of interference (Gilbert et al., 1993;Gilbert et al., 1996). This result establishes that the IEfusion protein is immunologically recognized, either alone or in combination with the pp65 antigen. Equally important to the success of the MVA vaccine is the reduced levels of inflammation it stimulates allowing for repeated boosting, and the role of pre-existing immunity to the vector, perhaps caused by an earlier smallpox vaccination using the Dryvax™ vaccine. Our work and the work of others suggest that limited repeated exposures to MVA in a defined vaccine period of months can be tolerated (Ramirez et al., 2000;Wang et al., 2004a;Yang et al., 2003). Hammerlund et al commented on the longevity of poxvirus-specific T cell response decades after Dryvax™ immunization, and point out that cellular immunity decays more rapidly than humoral immunity (Hammarlund et al., 2003). The ability to re-stimulate pox-specific memory responses is dictated by the length of time since the last exposure to the virus.
Humanized Tg mice which do not express murine Class I alleles (Lemonnier, 2002) are available in a variety of forms that express human HLA A2, B7, A11, providing the most direct way to assess HLA recognition of vaccines in a mouse model(Firat et al., 2002). We first examined the processing of both rMVAs utilizing HHD II mice, which are known to be effective in processing and recognition of poxviruses specific for a wide variety of infectious pathogens, including CMV(Daftarian et al., 2003;Gomez et al., 2007;Wang et al., 2004b). Our results substantiate that the IEfusion antigen in MVA is processed and immunologically recognized throughout both exons, and the fusion join does not impede this process. A limitation of this study was the exclusive use of IFN-γ to assess T cell recognition of CMV antigens expressed from the vaccine, though we and others have shown a strong correlation between IFN-γ expression and cytotoxic function in mouse models (Daftarian et al., 2003;Song et al., 2007;Wang et al., 2004a). In addition, measurement of IFN-γ production has been relied upon to assess CMV immunity in CMV infected healthy individuals (Ghanekar et al., 2001;Sinclair et al., 2004;Sylwester et al., 2005).
To further generalize the capacity of the rMVA vaccine to be recognized in a variety of HLA context, B7 mice with a similar C57BL/6 background as the HHDII mice were also immunized with the pp65-IEfusion-MVA and investigated for immunogenicity using the same approach as with the HHDII mice. As expected, we found highly effective recognition of the pp65 antigen as well as a CD8 response to the IE2 antigen using the peptide library. This series of experiments illustrates that rMVA is processed efficiently by multiple HLA alleles, and provides further support for its utility as a clinical vaccine strategy. While HLA Class I Tg mice serve a fundamental and irreplaceable role to demonstrate the immunogenicity of the MVA constructs; however, they cannot be directly compared with the human in vitro clinical results, since the latter were obtained exclusively using CMV-specific peptide libraries. Humans express a diversity of HLA alleles, and the purpose for developing a multi-antigen vaccine is to encompass as many as possible to broaden the applicability of the vaccine to outbred human populations. While the Tg mice are a valuable tool to evaluate HLA Class I restricted CD8 T cell responses, they have an intact full complement of murine MHC Class II genes and cannot be directly compared to humans who possess a different repertoire of Class II MHC. While not an optimal comparison, it is noteworthy that of the three antigens, pp65 elicits the strongest CD4 response in both mice and humans. In contrast both IE1 and IE2 do not elicit strong CD4-based immunity in both mice and humans (compare Figures 3 and 5).
Prior to conducting experiments with rMVA in clinical samples, we assessed the capacity for stimulation of both CD4+ and CD8+ T cells using the commercially available pp65 and IE1 library and our newly designed IE2 peptide library. We found relationships among the T cell populations to be similar to what has been published, as pp65 promotes a substantial CD4 and CD8 response in over 70% of participants, while IE1 and IE2 are recognized less frequently and mainly in the CD8+ T cell compartment (Khan et al., 2002;Khan et al., 2007;Sylwester et al., 2005). The conclusion from the library studies suggest that our IE2 formulation is a reagent of equal potency to the commercially available pp65 and IE1 peptide libraries to assess memory T cell responses in the volunteer pool, and should be an effective detection reagent of memory immune responses to rMVA. We also evaluated recognition of all three libraries in transplant recipients in all three risk groups including those with CMV-positive or –negative donors or themselves being CMV-negative with a CMV-positive donor. The uniqueness of this study is that no previous evaluation of peptide libraries has been carried out with HCT recipients using all three antigens simultaneously (Lacey et al., 2006). We examined patient samples at day 180 post-transplant to minimize the effects of myeloablation and incomplete immune reconstitution on the recognition of the peptide libraries. The immune recognition of all three libraries was successful in all patients, and interestingly, the relative proportion of T cells that responded to each library also mirrored what was found in the healthy volunteers.
We evaluated both rMVA expressing the IEfusion antigen in PBMC from healthy volunteers to establish their recognition properties using a fully human system. The results showed that the memory T cell expansion stimulated by the rMVA for both the IEfusion and pp65 antigens, followed the proportions found ex vivo for the same volunteers using the peptide library approach. While there was substantial amplification of the relevant T cell populations, the stimulation did not skew the population towards a particular subset or antigen specificity. The data also confirms that the IEfusion protein is processed and presented appropriately to stimulate existing T cell populations in a manner that maintains the phenotypic distribution as expected in the ex vivo analysis. This outcome is most favorable towards using the rMVA as a vaccine in both CMV positives and negatives, since it is preferable to maintain the proportion of T cells that are associated with an asymptomatic phenotype and hopefully induce that same proportion in CMV-negative subjects. In this report, we succeeded in stimulating primary immunity in CMV-naïve mice using in vivo immunization, but failed to elicit primary responses from clinical samples in the CMV-naïve subject we investigated. The conditions of in vitro immunization are insufficient in most cases to drive primary immunity, because the architecture of the lymph node, thymus and dendritic cell systems is missing, so the T cells precursors must pre-exist or form in culture. Developing primary immunity to CMV post-transplant is often delayed in the CMV-naïve recipient or donor in the case of stem cell transplant, and is thought to be a risk factor for CMV disease (Limaye et al., 2006;Ljungman et al., 2006). Precedent for poxvirus-based CMV vaccines to stimulate primary immunity was established with a single-antigen pp65-ALVAC used in a clinical trial conducted with CMV-negative healthy volunteers (Berencsi et al., 2001).
The inability to rely on in vitro immunization for eliciting primary immunity is a key motivation in pursuing a vaccine, since the pioneering approaches of adoptive transfer would be infeasible when considering a CMV-negative donor for HCT, or a CMV-negative recipient of a CMV-infected donor organ (Walter et al., 1995). Recently, our group has shown that MVA vaccines composed of CMV subunit antigens (e.g. pp65, IE1, and gB) can elicit primary immunity in CMV-naïve rhesus macaques, even offering partial protection against a challenge dose of rhesus CMV (Yue et al., 2008). Consequently, since rMVA or ALVAC expressing CMV antigens predictably expand T cell populations in both CMV-naïve (mice and macaques) and experienced (human) hosts, one application of a CMV vaccine is in the high-risk CMV-negative transplant recipient for protection against the effects of a CMV-infected organ. One scenario would be to introduce the pp65-IEfusion-MVA as a vaccine into the CMV-negative recipient as a precaution several months before transplant (Khanna & Diamond, 2006;La Rosa et al., 2007). Another application is to use pp65-IEfusion-MVA as a vehicle to expand T cell populations from CMV-positive donors of HCT, and infuse the amplified T cells into a transplant recipient with active viremia. The proof of principle for that application has been demonstrated in HCT using different approaches, though the level of amplification and short time frame provided by rMVA makes it an attractive candidate for this application (Cobbold et al., 2005;Einsele et al., 2002;Walter et al., 1995).
In contrast, CMV is problematic to serve as a stimulator for characterizing memory responses. The concurrent activating and immunosuppressive properties of CMV can confound interpretation of immunologic methods using it for in vitro stimulation (Manley et al., 2004). In fact, one needs to artificially remove the immune-evasion genes from CMV in order to elicit a diverse immune response that includes the IE antigens, a fact that has been stressed in the literature (Khan et al., 2005;Manley et al., 2004). Laboratory strains of CMV that are the only practical approach for growing the virus to assess recall immunity are plagued with an artificial excessive accumulation of the pp65 protein that interferes with the recognition of IE proteins which also has been discussed in the literature and has been the source of controversy in the field (Gilbert et al., 1996;Kern et al., 1999;Wills et al., 1996). Therefore, it is to be expected that the profile of immune responses that are stimulated by MVA will be different than what could be elicited using CMV as a viral stimulator in culture methods.
The most rigorous evaluation of the processing of the rMVA for T cell response is using PBMC from transplant patients. Evaluating PBMC from HCT recipients in all three risk categories, we found an equivalently strong recognition of both rMVAs, in some cases more vigorous than in the PBMC of healthy adults. We found no interference with the recognition of the IE antigen by the co-expressed pp65 antigen from the same rMVA, which further confirms that the recognition of both antigens can take place at the same time and derived from the same vector. Prime-boost strategies utilizing heterologous vaccines, including DNA and viral vectors, suggests improved immunogenicity in several pathogen models, including malaria and HIV(Barouch et al., 2003;Gilbert et al., 2006;Goonetilleke et al., 2006;Peters et al., 2007). The ongoing evaluation of a DNA vaccine against CMV suggests a worthwhile strategy of combining MVA with a plasmid DNA vaccine. The excellent track record of MVA used as a vaccine in the immunosuppressed population makes it an ideal candidate as a therapeutic in HCT recipients(Cosma et al., 2003;Mayr & Danner, 1978;Stittelaar et al., 2001).
Materials and Methods
Human patient specimens
The study protocols were approved by the institutional review board at City of Hope Medical Center (COH), and specimens and data were obtained prospectively after informed consent was obtained from subjects. PBMC from healthy donors and 9 HCT recipients at d180 post-transplant were collected at COH and cryopreserved by standard methods (Maecker et al., 2005). HLA typing was performed at the COH HLA Laboratory by standard SSOP methods. Haplotypes for both Class I and II alleles were determined (data not shown). No intentional bias occurred in selection of HLA types to be included in this study, as all patients or volunteers were randomly chosen. The CMV serostatus of these subjects was determined as previously described by using latex agglutination (CMV-SCAN; Becton Dickinson) (La Rosa et al., 2007).
Mouse strains and conditions of immunization
HHD II (Pascolo et al., 1997) and HLA-B*0702 (Rohrlich et al., 2003) Tg mice were bred and maintained under pathogen-free conditions in the AALAC-approved animal care facility at COH. Eight to ten-week old groups of Tg mice (3-4) were immunized i.p. with different rMVA constructs (as described in Figure legends).
Synthetic peptides and monoclonal antibodies
HLA-A*0201 pp65495-503 (Diamond et al., 1997), HLA-B*0702 pp65265-275 (Longmate et al., 2001) and HLA-A*02 IE-1316-324 (Khan et al., 2002) restricted CMV peptides were used as previously described (La Rosa et al., 2001). Overlapping 15-AA peptides (PepMix™) spanning full length CMV pp65 and IE1 proteins were purchased from JPT Peptide Technologies GmbH (Berlin, Germany). Splenic murine cell suspensions were evaluated in flow cytometry using the following antibodies: CD8-FITC (Clone Ly-2), CD4-PE (Clone L3T4), and IFN-γ-APC (Clone XMG1.2). Antibodies used for flow cytometry of clinical specimens include anti-human CD8-PE (Clone RPA-T8), CD4-FITC (Clone RPA-T4) and IFN-γ-APC (Clone B27), purchased from BD-Pharmingen.
Construction of synthetic IE2 peptide library
The 580 amino acid primary sequence of the IE2 protein [Swiss Prot #P19893] was divided into 15mer stretches that overlap successive peptides by 11 amino acids using an online program which excludes impermissible amino acids at the amino (Q) and carboxyl (GPEDQNTSC) terminus of each 15mer peptide based on synthetic considerations. 133 peptides were predicted using the algorithm with an average length of 15-AA, but tolerating up to 5-AA length variance to eliminate forbidden terminal amino acids. A total of 123 peptides were synthesized in our laboratory with a Symphony™ peptide synthesizer (Protein Technologies, Inc., Tucson, AZ) using standard 9-fluorenylmethoxy carbonyl protocols and purified by high performance liquid chromatography (Agilent Technologies 1200 series) (Daftarian et al., 2005). The mass of each peptide was confirmed by matrix assisted laser desorption ionization time of flight analysis using a Kompact Probe™ mass spectrometer (Shimadzu Corp., Chestnut Ridge, NY). The library was sub-divided into ~20 peptides/pool, and subsequently combined into one super-pool containing all the component peptides. Several peptides were impractical to synthesize, and they did not enter into the pool including a 32-AA stretch between 251-282 of IE2 sequence. All peptides were individually solubilized in 30% acetonitrile/water, except 6 with pK’s >8, which were dissolved in 0.1% ammonium bicarbonate in 30% acetonitrile/water. The solubilized peptides, sub-divided into six pools, were further combined into a single super-pool, with each peptide at a concentration of 200 μg/ml, dissolved in 50% DMSO/water, and kept at -80° C in single use aliquots. The single use aliquots were diluted into cell culture medium at a final concentration of 1μg/ml/peptide for all cellular immunology assays based on previous titration studies (data not shown).
Construction of recombination plasmids for derivation of rMVA
The MVA transfer plasmid named pZWIIA with dual vaccinia synthetic promoters (Psyn I and II) was constructed to facilitate the derivation of bacterial marker gene-free rMVA(Wang et al., 2007). To construct the IEfusion gene, the following primers were designed with synthetic restriction enzyme sites shown as underlined: Primer a: 5’ AGCTTTGTTTAAACGCCACCACCATGGTCAAACAGATTAAGGTTCG 3’; Primer b: 5’GGCATGATTGACAGCCTGGGCGAGGATGTCACCCTGGTCAGCCTTGCTTCTAGTCACCAT 3’; Primer c: 5’TGTTAGCGTGGGCCCGGTGCTACTGGAATCGATACCGGCATGATTGACAGCCTGGGCGAGG ATGTCACC 3’; Primer d: 5’ TAGCACCGGGCCCACGC TAACAACCCAC 3’; Primer e: 5’ TTGGCGCGCCTTTATTTTACTGAGACTTGTTCCTCAGGT 3’. Primers a and b were used to amplify IE1/e4, and after gel purification, the IE1/e4 PCR product was amplified again using Primers a and c and digested with Pme I and Apa I. Primer b overlaps the junction between IE/e4 and IE2/e5 without adding any non-CMV genomic sequence. Primers c (G to C) and d (C to G) contains a single nucleotide change (double underline) that creates an Apa I site, but does not alter the amino acid sequence. The resulting fragment was cloned into pNEB193 to yield IE1/e4-pNEB193. IE2/e5 was amplified using Primers d and e and PCR products were digested with Apa I and Asc I and cloned into IE1/e4-pNEB193 to yield IE1-IE2-pNEB193. IE1-IE2 fusion gene (IEfusion) was excised from IE1-IE2-pNEB193 with Pme I and Asc I restriction enzymes and cloned into pZWIIA behind vaccinia Psyn I promoter (Chakrabarti et al., 1997) to yield IEfusion-pZWIIA (Figure 1C). To construct pp65-IEfusion-pZWIIA, the 1.7 Kb CMV pp65 gene was PCR amplified from an existing plasmid and cloned into Nhe I and Asc I site of pZWIIA behind vaccinia Psyn II promoter (Wyatt et al., 2004) to yield pp65-IEfusion-pZWIIA (Figure 1C). In neither MVA was pp65 fused to the IEfusion gene. MVA transfer plasmids were verified by restriction enzyme digestion and DNA sequencing.
Generation of IEfusion-MVA and pp65-IEfusion-MVA
pp65-IEfusion-MVA was generated in CEF cells via homologous recombination by transfection/infection method as described previously (Wang et al., 2004a;Wang et al., 2007). pp65-IEfusion-MVA was isolated based on blue color selection by nine consecutive rounds of plaque purification on CEF cells in the presence of β-glucoronidase substrate (X-glcA). IEfusion-MVA was generated similarly to pp65-IEfusion-MVA. The gus selection marker gene in rMVA virus was screened out using a limiting dilution method as described previously (Wang et al., 2007). Wt MVA-free and color marker gene-free recombinant IEfusion-MVA and pp65-IEfusion-MVA virus were expanded and purified by 36% sucrose density ultracentrifugation, resuspended in PBS containing 7.5% lactose, re-tested for protein expression by western blot (WB), aliquoted, and frozen at -80°C.
Western blot detection of protein expression
The pp65 and IEfusion protein expression levels, measured as separate proteins with distinct molecular weights from IEfusion-MVA and pp65-IEfusion-MVA-infected cells was determined by WB using an enhanced chemiluminescence-based ECL Plus™ detection kit (Amersham Pharmacia Biotech, Buckinghamshire, United Kingdom). After electro-transfer of proteins from the gel onto PVDF membranes (Bio-Rad, Hercules, CA), the membranes were incubated first with purified mAb 28-103 (Britt & Auger, 1985) to detect pp65 as a separate protein, or mAb p63-27 (Plachter et al., 1993) to detect the IE1 or IEfusion protein, followed with HRP-labeled goat anti–mouse Ab according to the manufacturer’s instructions.
Ex vivo and in vitro stimulation conditions for human PBMC
Cryopreserved PBMC were rapidly thawed and immediately cultured in 15-ml Falcon tubes at a density of 1 million/ml in RPMI 1640 medium (Invitrogen) supplemented with 10% FCS (Omega Scientific Inc, Tarzana, CA) and containing either pp65 PepMix™, IE-1 PepMix™ or IE-2 peptide library (at a final concentration of 1μg/ml of the individual peptides) at 37°C in a 5% CO2-gassed incubator. After 1h in culture, brefeldin A (GolgiPlug; Becton Dickinson Biosciences) was added, and incubation continued for an additional 11 h under the same conditions. IVS using rMVA was modified from a published method (La Rosa et al., 2006). Briefly, cryopreserved PBMCs were rapidly thawed and immediately dispensed in a 12-well plate at a concentration of 2 × 106 cells/mL in RPMI 1640 medium (2.5 mL/well) supplemented with 10% human AB serum (COH Blood Bank) and were incubated with 5 μg/mL of both CpG-A ODN 2216 and CpG-B ODN 2006 (TriLink BioTechnologies, San Diego, CA, USA). After 3 days, ODN-treated PBMCs were infected with rMVA expressing pp65 and IEfusion antigens (pp65-IEfusion-MVA), or rMVA expressing pp65 and IE1 exon4 (pp65-IE1-MVA), or rMVA expressing only IEfusion antigen (IEfusion-MVA) at a multiplicity of infection of 5, for 6 hours in RPMI 1640 medium with reduced (2%) human AB serum. Infected PBMCs were γ-irradiated (2500 rads) and used as APC. APC were plated in a 24-well plate (1.5 × 106/well), co-incubated with autologus PBMC (3 × 106/well), in a final volume consisting of 2ml/well RPMI 1640 medium with 10% human AB serum and human rIL-2 (NIH AIDS Research and Reference Reagent Program, 10 units/ml). Every 2 days, 50% of the culture medium was removed, and replaced with fresh medium containing rIL2. Cells were incubated for 8 days and split into additional wells when necessary. At day 8, cells were collected and washed with medium without rIL-2 and transferred into 15ml Falcon tubes. The same stimulation conditions for intracellular cytokine (ICC) assays performed on ex vivo PBMC were used for PBMC after IVS.
Intracellular cytokine staining of human PBMC
After 12 hours of incubation, PBMC were harvested, washed, labeled with PE–conjugated anti-CD8 and FITC-conjugated anti-CD4 antibodies, fixed, and permeabilized (Cytofix-Cytoperm; Becton Dickinson Biosciences) before they were labeled with APC–conjugated antibody to IFN-γ. The stained cells were analyzed on a FACSCanto™ (BD Immunocytometry Systems, San Jose, CA), and data were analyzed using FCS Express (version 3.0; DeNovo Software). 0.5×106 events were acquired for each sample. Lymphocytes were initially gated using forward vs side scatter, then CD4+ and CD8+ lymphocytes cells were gated separately. The number of IFN-γ expressing cells is shown as a percentage of the CD8+ or CD4+ lymphocyte population.
In vitro stimulation of mouse splenocytes and detection of cellular responses
Three weeks after immunization, spleens were aseptically removed and splenocytes from individual or pooled mice were stimulated in vitro (IVS) for 1 week with syngeneic LPS blasts as APC, loaded either with the relevant CMV-CTL epitope or CMV-peptide library (La Rosa et al., 2001;La Rosa et al., 2006). The immunological activity of the stimulated murine cultures was determined after assessing the levels of IFN-γ+/CD4+ or IFN-γ+/CD8+ T cells by ICC staining (La Rosa et al., 2001;La Rosa et al., 2006). For CD4, CD8, and IFN-γ labeling, APC-conjugated antibody to IFN-γ, PE-conjugated CD4, and FITC-conjugated CD8 were used (BD, San Jose, CA). Flow cytometric acquisition was performed on a FACSCanto™ (BD Immunocytometry Systems). Between 0.80 to 1.0×106 events were acquired for each sample. FACS analysis was performed using FCS Express version 2 software (De Novo, Ontario, Canada). The number of double-positive cells is expressed as a percentage of the CD8+ T-cell population.
Statistical analysis methods
Ex vivo IFN-γ production versus post-IVS with rMVA by PBMC against pp65, IE-1 and IE-2 peptide libraries were compared using Friedman’s test with 2 degrees of freedom, followed by Wilcoxon’s rank-sum test for pairwise comparisons. Comparison of paired data before and after IVS with rMVA was performed using the student t-test.
Acknowledgments
Support for this work was from Public Health Service grants CA030206 (DJD, SJF), CA077544 (DJD), and CA114889 (DJD) from the National Cancer Institute and grants AI062496 (DJD), AI058148 (JAZ), and AI049357 (WJB) from the National Institute of allergy and Infectious Disease. The GCRC is supported by M01-RR00043-39 and the City of Hope Comprehensive Cancer Center is supported by CA033572.
The authors thank Linda Wyatt and Bernard Moss for their gifts of plasmid vectors and advice on the construction of rMVA. IL-2 was the gift of the NIH AIDS Research and Reference Reagent Program.
The authors acknowledge the technical assistance of Joybelle Martinez, Aparna Krishnan, Heang Ly, and Zhongqui Li. We gratefully acknowledge assistance in human sample collection and preparation from Ghislaine-Gallez Hawkins, Lia Thao, Brenda Williams, and the support staff of the GCRC. The assistance of Donna Isbell and the support staff of the City of Hope Animal Research Center are gratefully acknowledged. The secretarial and administrative assistance of Donna Packer and Denise Marsano is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antoine G, Scheiflinger F, Dorner F, Falkner FG. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology. 1998;244:365–396. doi: 10.1006/viro.1998.9123. [DOI] [PubMed] [Google Scholar]
- Avetisyan G, Aschan J, Hagglund H, Ringden O, Ljungman P. Evaluation of intervention strategy based on CMV-specific immune responses after allogeneic SCT. Bone Marrow Transplant. 2007;40:865–869. doi: 10.1038/sj.bmt.1705825. [DOI] [PubMed] [Google Scholar]
- Barouch DH, McKay PF, Sumida SM, Santra S, Jackson SS, Gorgone DA, Lifton MA, Chakrabarti BK, Xu L, Nabel GJ, Letvin NL. Plasmid chemokines and colony-stimulating factors enhance the immunogenicity of DNA priming-viral vector boosting human immunodeficiency virus type 1 vaccines. The Journal of Virology. 2003;77:8729–8735. doi: 10.1128/JVI.77.16.8729-8735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berencsi K, Gyulai Z, Gonczol E, Pincus S, Cox WI, Michelson S, Kari L, Meric C, Cadoz M, Zahradnik J, Starr S, Plotkin S. A canarypox vector-expressing cytomegalovirus (cmv) phosphoprotein 65 induces long-lasting cytotoxic t cell responses in human cmv- seronegative subjects. J Infect Dis. 2001;183:1171–1179. doi: 10.1086/319680. [DOI] [PubMed] [Google Scholar]
- Blanchard TJ, Alcami A, Andrea P, Smith GL. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vaccine. J Gen Virol. 1998;79(Pt 5):1159–1167. doi: 10.1099/0022-1317-79-5-1159. [DOI] [PubMed] [Google Scholar]
- Boeckh M, Nakamura R, Cornelissen JJ, Zaia JA, Forman SJ, Gaal K, Brooimans RA, Gratama JW, Gasior GH, Sullivan LA. Immune monitoring with iTAg(TM) MHC tetramers for prediction of recurrent or persistent cytomegalovirus (CMV) infection in allogeneic stem cell transplant (SCT) recipients: A prospective multicenter clinical trial. Biology of Blood and Marrow Transplantation. 2006;12:79. doi: 10.1182/blood-2010-03-273508. [DOI] [PubMed] [Google Scholar]
- Britt WJ, Auger D. Identification of a 65 000 dalton virion envelope protein of human cytomegalovirus. Virus Res. 1985;4:31–36. doi: 10.1016/0168-1702(85)90018-8. [DOI] [PubMed] [Google Scholar]
- Carroll MW, Moss B. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology. 1997;238:198–211. doi: 10.1006/viro.1997.8845. [DOI] [PubMed] [Google Scholar]
- Carroll MW, Overwijk WW, Chamberlain RS, Rosenberg SA, Moss B, Restifo NP. Highly attenuated modified vaccinia virus Ankara (MVA) as an effective recombinant vector: a murine tumor model. Vaccine. 1997;15:387–394. doi: 10.1016/s0264-410x(96)00195-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Sisler JR, Moss B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. Biotechniques. 1997;23:1094–1097. doi: 10.2144/97236st07. [DOI] [PubMed] [Google Scholar]
- Cobbold M, Khan N, Pourgheysari B, Tauro S, McDonald D, Osman H, Assenmacher M, Billingham L, Steward C, Crawley C, Olavarria E, Goldman J, Chakraverty R, Mahendra P, Craddock C, Moss PA. Adoptive transfer of cytomegalovirus-specific CTL to stem cell transplant patients after selection by HLA-peptide tetramers. The Journal of Experimental Medicine. 2005;202:379–386. doi: 10.1084/jem.20040613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma A, Nagaraj R, Buhler S, Hinkula J, Busch DH, Sutter G, Goebel FD, Erfle V. Therapeutic vaccination with MVA-HIV-1 nef elicits Nef-specific T-helper cell responses in chronically HIV-1 infected individuals. Vaccine. 2003;22:21–29. doi: 10.1016/s0264-410x(03)00538-3. [DOI] [PubMed] [Google Scholar]
- Cwynarski K, Ainsworth J, Cobbold M, Wagner S, Mahendra P, Apperley J, Goldman J, Craddock C, Moss PA. Direct visualization of cytomegalovirus-specific T-cell reconstitution after allogeneic stem cell transplantation. Blood. 2001;97:1232–1240. doi: 10.1182/blood.v97.5.1232. [DOI] [PubMed] [Google Scholar]
- Daftarian P, Ali S, Sharan R, Lacey SF, La Rosa C, Longmate J, Buck C, Siliciano RF, Diamond DJ. Immunization with Th-CTL Fusion Peptide and Cytosine-Phosphate-Guanine DNA in Transgenic HLA-A2 Mice Induces Recognition of HIV-Infected T Cells and Clears Vaccinia Virus Challenge. J Immunol. 2003;171:4028–4039. doi: 10.4049/jimmunol.171.8.4028. [DOI] [PubMed] [Google Scholar]
- Daftarian P, Sharan R, Haq W, Ali S, Longmate J, Termini J, Diamond DJ. Novel conjugates of epitope fusion peptides with CpG-ODN display enhanced immunogenicity and HIV recognition. Vaccine. 2005;23:3453–3468. doi: 10.1016/j.vaccine.2005.01.093. [DOI] [PubMed] [Google Scholar]
- de Waal L, Wyatt LS, Yuksel S, van Amerongen G, Moss B, Niesters HG, Osterhaus AD, de Swart RL. Vaccination of infant macaques with a recxombinant MVA expressing the RSV F and G genes does not predispose for immunopathology. Vaccine. 2004;22:923–926. doi: 10.1016/j.vaccine.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Diamond DJ, York J, Sun J, Wright CL, Forman SJ. Development of a candidate HLA A*0201 restricted peptide-based vaccine against human cytomegalovirus infection. Blood. 1997;90:1751–1767. [PubMed] [Google Scholar]
- Earl PL, Americo JL, Wyatt LS, Eller LA, Montefiori DC, Byrum R, Piatak M, Lifson JD, Amara RR, Robinson HL, Huggins JW, Moss B. Recombinant modified vaccinia virus Ankara provides durable protection against disease caused by an immunodeficiency virus as well as long-term immunity to an orthopoxvirus in a non-human primate. Virology. 2007;366:84–97. doi: 10.1016/j.virol.2007.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einsele H, Roosnek E, Rufer N, Sinzger C, Riegler S, Loffler J, Grigoleit U, Moris A, Rammensee HG, Kanz L, Kleihauer A, Frank F, Jahn G, Hebart H. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood. 2002;99:3916–3922. doi: 10.1182/blood.v99.11.3916. [DOI] [PubMed] [Google Scholar]
- Firat H, Cochet M, Rohrlich PS, Garcia-Pons F, Darche S, Danos O, Lemonnier FA, Langlade-Demoyen P. Comparative analysis of the CD8(+) T cell repertoires of H-2 class I wild-type/HLA-A2.1 and H-2 class I knockout/HLA-A2.1 transgenic mice. Int Immunol. 2002;14:925–934. doi: 10.1093/intimm/dxf056. [DOI] [PubMed] [Google Scholar]
- Gallez-Hawkins G, Thao L, Lacey SF, Martinez J, Li X, Franck AE, Lomeli NA, Longmate J, Diamond DJ, Spielberger R, Forman SJ, Zaia JA. Cytomegalovirus immune reconstitution occurs in recipients of allogeneic hematopoietic cell transplants irrespective of detectable cytomegalovirus infection. Biol Blood Marrow Transplant. 2005;11:890–902. doi: 10.1016/j.bbmt.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Ghanekar SA, Nomura LE, Suni MA, Picker LJ, Maecker HT, Maino VC. Gamma interferon expression in CD8(+) T cells is a marker for circulating cytotoxic T lymphocytes that recognize an HLA A2-restricted epitope of human cytomegalovirus phosphoprotein pp65. Clin Diagn Lab Immunol. 2001;8:628–631. doi: 10.1128/CDLI.8.3.628-631.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert MJ, Riddell SR, Li CR, Greenberg PD. Selective interference with class I major histocompatibility complex presentation of the major immediate-early protein following infection with human cytomegalovirus. The Journal of Virology. 1993;67:3461–3469. doi: 10.1128/jvi.67.6.3461-3469.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert MJ, Riddell SR, Plachter B, Greenberg PD. Cytomegalovirus selectively blocks antigen processing and presentation of its immediate-early gene product. Nature. 1996;383:720–722. doi: 10.1038/383720a0. [DOI] [PubMed] [Google Scholar]
- Gilbert SC, Moorthy VS, Andrews L, Pathan AA, McConkey SJ, Vuola JM, Keating SM, Berthoud T, Webster D, McShane H, Hill AV. Synergistic DNA-MVA prime-boost vaccination regimes for malaria and tuberculosis. Vaccine. 2006;24:4554–4561. doi: 10.1016/j.vaccine.2005.08.048. [DOI] [PubMed] [Google Scholar]
- Gomez CE, Najera JL, Jimenez EP, Jimenez V, Wagner R, Graf M, Frachette MJ, Liljestrom P, Pantaleo G, Esteban M. Head-to-head comparison on the immunogenicity of two HIV/AIDS vaccine candidates based on the attenuated poxvirus strains MVA and NYVAC co-expressing in a single locus the HIV-1BX08 gp120 and HIV-1(IIIB) Gag-Pol-Nef proteins of clade B. Vaccine. 2007;25:2863–2885. doi: 10.1016/j.vaccine.2006.09.090. [DOI] [PubMed] [Google Scholar]
- Goonetilleke N, Moore S, Dally L, Winstone N, Cebere I, Mahmoud A, Pinheiro S, Gillespie G, Brown D, Loach V, Roberts J, Guimaraes-Walker A, Hayes P, Loughran K, Smith C, De Bont J, Verlinde C, Vooijs D, Schmidt C, Boaz M, Gilmour J, Fast P, Dorrell L, Hanke T, McMichael AJ. Induction of Multifunctional Human Immunodeficiency Virus Type 1 (HIV-1)-Specific T Cells Capable of Proliferation in Healthy Subjects by Using a Prime-Boost Regimen of DNA- and Modified Vaccinia Virus Ankara-Vectored Vaccines Expressing HIV-1 Gag Coupled to CD8+ T-Cell Epitopes. The Journal of Virology. 2006;80:4717–4728. doi: 10.1128/JVI.80.10.4717-4728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratama JW, van Esser JW, Lamers CH, Tournay C, Lowenberg B, Bolhuis RL, Cornelissen JJ. Tetramer-based quantification of cytomegalovirus (CMV)-specific CD8+ T lymphocytes in T-cell-depleted stem cell grafts and after transplantation may identify patients at risk for progressive CMV infection. Blood. 2001;98:1358–1364. doi: 10.1182/blood.v98.5.1358. [DOI] [PubMed] [Google Scholar]
- Gyulai Z, Endresz V, Burian K, Pincus S, Toldy J, Cox WI, Meri C, Plotkin S, Berencsi K. Cytotoxic T Lymphocyte (CTL) Responses to Human Cytomegalovirus pp65, IE1-Exon4, gB, pp150, and pp28 in Healthy Individuals: Reevaluation of Prevalence of IE1-Specific CTLs. J Infect Dis. 2000;181:1537–1546. doi: 10.1086/315445. [DOI] [PubMed] [Google Scholar]
- Hammarlund E, Lewis MW, Hansen SG, Strelow LI, Nelson JA, Sexton GJ, Hanifin JM, Slifka MK. Duration of antiviral immunity after smallpox vaccination. Nat Med. 2003;9:1131–1137. doi: 10.1038/nm917. [DOI] [PubMed] [Google Scholar]
- Hanke T, McMichael AJ, Dennis MJ, Sharpe SA, Powell LA, Mcloughlin L, Crome SJ. Biodistribution and persistence of an MVA-vectored candidate HIV vaccine in SIV-infected rhesus macaques and SCID mice. Vaccine. 2005;23:1507–1514. doi: 10.1016/j.vaccine.2004.08.050. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Yurochko AD, Poma EE, Zhu L, Huang ES. Domain mapping of the human cytomegalovirus IE1-72 and cellular p107 protein-protein interaction and the possible functional consequences. J Gen Virol. 1999;80(Pt 5):1293–1303. doi: 10.1099/0022-1317-80-5-1293. [DOI] [PubMed] [Google Scholar]
- Kern F, Surel IP, Faulhaber N, Frommel C, Schneider-Mergener J, Schonemann C, Reinke P, Volk HD. Target structures of the CD8(+)-T-cell response to human cytomegalovirus: the 72-kilodalton major immediate-early protein revisited. The Journal of Virology. 1999;73:8179–8184. doi: 10.1128/jvi.73.10.8179-8184.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Best D, Bruton R, Nayak L, Rickinson AB, Moss PA. T cell recognition patterns of immunodominant cytomegalovirus antigens in primary and persistent infection. The Journal of Immunology. 2007;178:4455–4465. doi: 10.4049/jimmunol.178.7.4455. [DOI] [PubMed] [Google Scholar]
- Khan N, Bruton R, Taylor GS, Cobbold M, Jones TR, Rickinson AB, Moss PA. Identification of Cytomegalovirus-Specific Cytotoxic T Lymphocytes In Vitro Is Greatly Enhanced by the Use of Recombinant Virus Lacking the US2 to US11 Region or Modified Vaccinia Virus Ankara Expressing Individual Viral Genes. The Journal of Virology. 2005;79:2869–2879. doi: 10.1128/JVI.79.5.2869-2879.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Cobbold M, Keenan R, Moss PA. Comparative analysis of CD8+ T cell responses against human cytomegalovirus proteins pp65 and immediate early 1 shows similarities in precursor frequency, oligoclonality, and phenotype. J Infect Dis. 2002;185:1025–1034. doi: 10.1086/339963. [DOI] [PubMed] [Google Scholar]
- Khanna R, Diamond DJ. Human cytomegalovirus vaccine: time to look for alternative options. Trends Mol Med. 2006;12:26–33. doi: 10.1016/j.molmed.2005.11.006. [DOI] [PubMed] [Google Scholar]
- La Rosa C, Krishnan R, Markel S, Schneck JP, Houghten R, Pinilla C, Diamond DJ. Enhanced immune activity of cytotoxic T-lymphocyte epitope analogs derived from positional scanning synthetic combinatorial libraries. Blood. 2001;97:1776–1786. doi: 10.1182/blood.v97.6.1776. [DOI] [PubMed] [Google Scholar]
- La Rosa C, Limaye AP, Krishnan A, Longmate J, Diamond DJ. Longitudinal Assessment of Cytomegalovirus (CMV)-Specific Immune Responses in Liver Transplant Recipients at High Risk for Late CMV Disease. J Infect Dis. 2007;195:633–644. doi: 10.1086/511307. [DOI] [PubMed] [Google Scholar]
- La Rosa C, Wang Z, Lacey SF, Lalimarmo MM, Krishnan A, Longmate J, Diamond DJ. In vitro expansion of polyclonal T-cell subsets for adoptive immunotherapy by recombinant modified vaccinia Ankara. Exp Hematol. 2006;34:497–507. doi: 10.1016/j.exphem.2005.12.018. [DOI] [PubMed] [Google Scholar]
- Lacey SF, La Rosa C, Zhou W, Sharma MC, Martinez J, Krishnan A, Gallez-Hawkins G, Thao L, Longmate J, Spielberger R, Forman SJ, Limaye A, Zaia JA, Diamond DJ. Functional Comparison of T Cells Recognizing Cytomegalovirus pp65 and Intermediate-Early Antigen Polypeptides in Hematopoietic Stem-Cell Transplant and Solid Organ Transplant Recipients. J Infect Dis. 2006;194:1410–1421. doi: 10.1086/508495. [DOI] [PubMed] [Google Scholar]
- Lemonnier FA. The utility of H-2 class I knockout mice. Virus Res. 2002;82:87–90. doi: 10.1016/s0168-1702(01)00392-6. [DOI] [PubMed] [Google Scholar]
- Limaye AP, Bakthavatsalam R, Kim HW, Randolph SE, Halldorson JB, Healey PJ, Kuhr CS, Levy AE, Perkins JD, Reyes JD, Boeckh M. Impact of cytomegalovirus in organ transplant recipients in the era of antiviral prophylaxis. Transplantation. 2006;81(12):1645–52. doi: 10.1097/01.tp.0000226071.12562.1a. [DOI] [PubMed] [Google Scholar]
- Ljungman P, Perez-Bercoff L, Jonsson J, Avetisyan G, Sparrelid E, Aschan J, Barkholt L, Larsson K, Winiarski J, Yun Z, Ringden O. Risk factors for the development of cytomegalovirus disease after allogeneic stem cell transplantation. Haematologica. 2006;91:78–83. [PubMed] [Google Scholar]
- Longmate J, York J, La Rosa C, Krishnan R, Zhang M, Senitzer D, Diamond DJ. Population coverage by HLA class-I restricted cytotoxic T-lymphocyte epitopes. Immunogenetics. 2001;52:165–173. doi: 10.1007/s002510000271. [DOI] [PubMed] [Google Scholar]
- Maecker HT, Moon J, Bhatia S, Ghanekar SA, Maino VC, Payne JK, Kuus-Reichel K, Chang JC, Summers A, Clay TM, Morse MA, Lyerly HK, DeLaRosa C, Ankerst DP, Disis ML. Impact of cryopreservation on tetramer, cytokine flow cytometry, and ELISPOT. BMC Immunol. 2005;6:17. doi: 10.1186/1471-2172-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley TJ, Luy L, Jones T, Boeckh M, Mutimer H, Riddell SR. Immune evasion proteins of human cytomegalovirus do not prevent a diverse CD8+ cytotoxic T-cell response in natural infection. Blood. 2004;104:1075–1082. doi: 10.1182/blood-2003-06-1937. [DOI] [PubMed] [Google Scholar]
- Mayr A, Danner K. Vaccination against pox diseases under immunosuppressive conditions. Dev Biol Stand. 1978;41:225–234. [PubMed] [Google Scholar]
- Meyer H, Sutter G, Mayr A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J Gen Virol. 1991;72(Pt 5):1031–1038. doi: 10.1099/0022-1317-72-5-1031. [DOI] [PubMed] [Google Scholar]
- Moorthy VS, Pinder M, Reece WH, Watkins K, Atabani S, Hannan C, Bojang K, McAdam KP, Schneider J, Gilbert S, Hill AV. Safety and immunogenicity of DNA/modified vaccinia virus ankara malaria vaccination in African adults. J Infect Dis. 2003;188:1239–1244. doi: 10.1086/378515. [DOI] [PubMed] [Google Scholar]
- Morello CS, Cranmer LD, Spector DH. Suppression of murine cytomegalovirus (MCMV) replication with a DNA vaccine encoding MCMV M84 (a homolog of human cytomegalovirus pp65) The Journal of Virology. 2000;74:3696–3708. doi: 10.1128/jvi.74.8.3696-3708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascolo S, Bervas N, Ure JM, Smith AG, Lemonnier FA, Perarnau B. HLA-A2.1-restricted education and cytolytic activity of CD8(+) T lymphocytes from beta2 microglobulin (beta2m) HLA-A2.1 monochain transgenic H-2Db beta2m double knockout mice. The Journal of Experimental Medicine. 1997;185:2043–2051. doi: 10.1084/jem.185.12.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pass RF, Fowler KB, Boppana SB, Britt WJ, Stagno S. Congenital cytomegalovirus infection following first trimester maternal infection: symptoms at birth and outcome. J Clin Virol. 2006;35:216–220. doi: 10.1016/j.jcv.2005.09.015. [DOI] [PubMed] [Google Scholar]
- Peters B, Sette A. Integrating epitope data into the emerging web of biomedical knowledge resources. Nat Rev Immunol. 2007;7:485–490. doi: 10.1038/nri2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters BS, Jaoko W, Vardas E, Panayotakopoulos G, Fast P, Schmidt C, Gilmour J, Bogoshi M, Omosa-Manyonyi G, Dally L, Klavinskis L, Farah B, Tarragona T, Bart PA, Robinson A, Pieterse C, Stevens W, Thomas R, Barin B, McMichael AJ, McIntyre JA, Pantaleo G, Hanke T, Bwayo J. Studies of a prophylactic HIV-1 vaccine candidate based on modified vaccinia virus Ankara (MVA) with and without DNA priming: Effects of dosage and route on safety and immunogenicity. Vaccine. 2007;25:2120–2127. doi: 10.1016/j.vaccine.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Plachter B, Britt W, Vornhagen R, Stamminger T, Jahn G. Analysis of proteins encoded by IE regions 1 and 2 of human cytomegalovirus using monoclonal antibodies generated against recombinant antigens. Virology. 1993;193:642–652. doi: 10.1006/viro.1993.1172. [DOI] [PubMed] [Google Scholar]
- Ramirez JC, Gherardi MM, Rodriguez D, Esteban M. Attenuated modified vaccinia virus Ankara can be used as an immunizing agent under conditions of preexisting immunity to the vector. The Journal of Virology. 2000;74:7651–7655. doi: 10.1128/jvi.74.16.7651-7655.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddehase MJ, Mutter W, Munch K, Buhring HJ, Koszinowski UH. CD8-positive T lymphocytes specific for murine cytomegalovirus immediate-early antigens mediate protective immunity. The Journal of Virology. 1987;61:3102–3108. doi: 10.1128/jvi.61.10.3102-3108.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochlitz C, Figlin R, Squiban P, Salzberg M, Pless M, Herrmann R, Tartour E, Zhao Y, Bizouarne N, Baudin M, Acres B. Phase I immunotherapy with a modified vaccinia virus (MVA) expressing human MUC1 as antigen-specific immunotherapy in patients with MUC1-positive advanced cancer. J Gene Med. 2003;5:690–699. doi: 10.1002/jgm.397. [DOI] [PubMed] [Google Scholar]
- Rohrlich PS, Cardinaud S, Firat H, Lamari M, Briand P, Escriou N, Lemonnier FA. HLA-B*0702 transgenic, H-2KbDb double-knockout mice: phenotypical and functional characterization in response to influenza virus. Int Immunol. 2003;15:765–772. doi: 10.1093/intimm/dxg073. [DOI] [PubMed] [Google Scholar]
- Rohrlich PS, Cardinaud S, Lule J, Montero-Julian FA, Prodhomme V, Firat H, Davignon JL, Perret E, Monseaux S, Necker A, Michelson S, Lemonnier FA, Charneau P, Davrinche C. Use of a lentiviral vector encoding a HCMV-chimeric IE1-pp65 protein for epitope identification in HLA-Transgenic mice and for ex vivo stimulation and expansion of CD8(+) cytotoxic T cells from human peripheral blood cells. Hum Immunol. 2004;65:514–522. doi: 10.1016/j.humimm.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Schleiss MR, Lacayo JC, Belkaid Y, McGregor A, Stroup G, Rayner J, Alterson K, Chulay JD, Smith JF. Preconceptual Administration of an Alphavirus Replicon UL83 (pp65 Homolog) Vaccine Induces Humoral and Cellular Immunity and Improves Pregnancy Outcome in the Guinea Pig Model of Congenital Cytomegalovirus Infection. J Infect Dis. 2007;195:789–798. doi: 10.1086/511982. [DOI] [PubMed] [Google Scholar]
- Sinclair E, Black D, Epling CL, Carvidi A, Josefowicz SZ, Bredt BM, Jacobson MA. CMV antigen-specific CD4+ and CD8+ T cell IFNgamma expression and proliferation responses in healthy CMV-seropositive individuals. Viral Immunol. 2004;17:445–454. doi: 10.1089/0882824041857049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair E, Tan QX, Sharp M, Girling V, Poon C, Natta MV, Jabs DA, Inokuma M, Maecker HT, Bredt B, Jacobson MA. Protective Immunity to Cytomegalovirus (CMV) Retinitis in AIDS Is Associated with CMV-Specific T Cells That Express Interferon-gamma and Interleukin-2 and Have a CD8+ Cell Early Maturational Phenotype. J Infect Dis. 2006;194:1537–1546. doi: 10.1086/508997. [DOI] [PubMed] [Google Scholar]
- Song GY, Gibson G, Haq W, Huang EC, Srivasta T, Hollstein M, Daftarian P, Wang Z, Diamond D, Ellenhorn JD. An MVA vaccine overcomes tolerance to human p53 in mice and humans. Cancer Immunol Immunother. 2007;56:1193–1205. doi: 10.1007/s00262-006-0270-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stittelaar KJ, Kuiken T, de Swart RL, van Amerongen G, Vos HW, Niesters HG, van Schalkwijk P, van der KT, Wyatt LS, Moss B, Osterhaus AD. Safety of modified vaccinia virus Ankara (MVA) in immune-suppressed macaques. Vaccine. 2001;19:3700–3709. doi: 10.1016/s0264-410x(01)00075-5. [DOI] [PubMed] [Google Scholar]
- Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. The Journal of Experimental Medicine. 2005;202:673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz-Santiago J, Lule J, Rohrlich P, Kravtzoff R, Le Roy E, Davignon JL, Betbeder D, Davrinche C. IE1-pp65 recombinant protein from human CMV combined with a nanoparticulate carrier, SMBV, as a potential source for the development of anti-human CMV adoptive immunotherapy. Cytotherapy. 2002;4:11–19. doi: 10.1080/146532402317251482. [DOI] [PubMed] [Google Scholar]
- Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl JMed. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- Wang Z, La Rosa C, Li Z, Ly H, Krishnan A, Martinez J, Britt WJ, Diamond DJ. Vaccine properties of a novel marker gene-free recombinant modified vaccinia Ankara expressing immunodominant CMV antigens pp65 and IE1. Vaccine. 2007;25:1132–1141. doi: 10.1016/j.vaccine.2006.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, La Rosa C, Maas R, Ly H, Brewer J, Mekhoubad S, Daftarian P, Longmate J, Britt WJ, Diamond DJ. Recombinant modified vaccinia virus Ankara expressing a soluble form of glycoprotein B causes durable immunity and neutralizing antibodies against multiple strains of human cytomegalovirus. The Journal of Virology. 2004a;78:3965–3976. doi: 10.1128/JVI.78.8.3965-3976.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, La Rosa C, Mekhoubad S, Lacey SF, Villacres MC, Markel S, Longmate J, Ellenhorn JD, Siliciano RF, Buck C, Britt WJ, Diamond DJ. Attenuated Poxviruses Generate Clinically Relevant Frequencies of CMV-Specific T cells. Blood. 2004b;104:847–856. doi: 10.1182/blood-2003-10-3469. [DOI] [PubMed] [Google Scholar]
- White EA, Del Rosario CJ, Sanders RL, Spector DH. The IE2 60-kilodalton and 40-kilodalton proteins are dispensable for human cytomegalovirus replication but are required for efficient delayed early and late gene expression and production of infectious virus. The Journal of Virology. 2007;81:2573–2583. doi: 10.1128/JVI.02454-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills MR, Carmichael AJ, Mynard K, Jin X, Weekes MP, Plachter B, Sissons JG. The human CTL response to cytomegalovirus is dominanted by structural protein pp65: Frequency, specificity, and T Cell Receptor usage of pp65-Specific CTL. The Journal of Virology. 1996;70:7569–7579. doi: 10.1128/jvi.70.11.7569-7579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt LS, Earl PL, Liu JY, Smith JM, Montefiori DC, Robinson HL, Moss B. Multiprotein HIV type 1 clade B DNA and MVA vaccines: construction, expression, and immunogenicity in rodents of the MVA component. AIDS Res Hum Retroviruses. 2004;20:645–653. doi: 10.1089/0889222041217428. [DOI] [PubMed] [Google Scholar]
- Yang ZY, Wyatt LS, Kong WP, Moodie Z, Moss B, Nabel GJ. Overcoming immunity to a viral vaccine by DNA priming before vector boosting. The Journal of Virology. 2003;77:799–803. doi: 10.1128/JVI.77.1.799-803.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Kaur A, Eberhardt MK, Kassis N, Zhou SS, Tarantal AF, Barry PA. Immunogenicity and protective efficacy of DNA vaccines expressing rhesus cytomegalovirus glycoprotein B, phosphoprotein 65-2, and viral interleukin-10 in rhesus macaques. The Journal of Virology. 2007;81:1095–1109. doi: 10.1128/JVI.01708-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Wang Z, Abel K, Li J, Strelow L, Mandarino A, Eberhardt MK, Schmidt KA, Diamond DJ, Barry PA. Evaluation of recombinant modified vaccinia Ankara virus-based rhesus cytomegalovirus vaccines in rhesus macaques. Med Microbiol Immunol. 2008;197:117–123. doi: 10.1007/s00430-008-0074-5. [DOI] [PubMed] [Google Scholar]
- Zaia JA. Prevention and management of CMV-related problems after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2002;29:633–638. doi: 10.1038/sj.bmt.1703407. [DOI] [PubMed] [Google Scholar]
- Zaia JA, Sissons JG, Riddell SR, Diamond DJ. Status of cytomegalovirus prevention and treatment in 2000. Hematology 2000. 2001:339–355. doi: 10.1182/asheducation-2000.1.339. [DOI] [PubMed] [Google Scholar]
- Zhang X, Cassis-Ghavami F, Eller M, Currier J, Slike BM, Chen X, Tartaglia J, Marovich M, Spearman P. Direct comparison of antigen production and induction of apoptosis by canarypox virus- and modified vaccinia virus ankara-human immunodeficiency virus vaccine vectors. The Journal of Virology. 2007;81:7022–7033. doi: 10.1128/JVI.02654-06. [DOI] [PMC free article] [PubMed] [Google Scholar]