

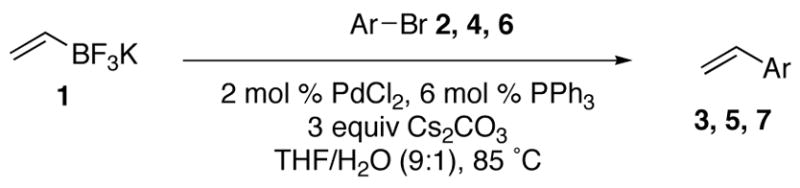
Abstract
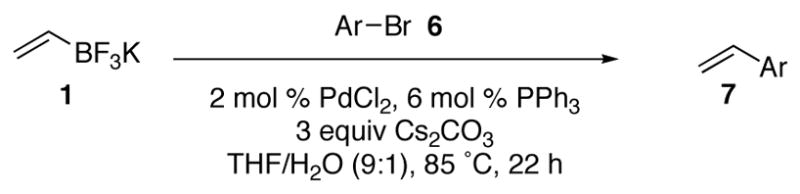
We have previously reported that the palladium catalyzed cross–coupling reaction of potassium vinyltrifluoroborate with aryl electrophiles proceeds with good yields. Herein, we describe recent progress in optimizing the reaction, as well as outlining the scope and limitations of the reaction. The cross–coupling reaction can generally be effected using 2 mol % of PdCl2 and 6 mol % of PPh3 as a catalyst system in THF/H2O with Cs2CO3 as a base. Moderate to good yields are obtained in the presence of a variety of functional groups.
Introduction
Substituted styrene starting materials have seen extensive use in both specialty chemical and polymer synthesis. Not only can styrenes be used in transformations such as olefin metathesis1 or Heck–type reactions,2 the alkene can also be employed as a platform on which to introduce a variety of functionality.3 In regard to polymer synthesis, a multitude of transformations exists for polymerizing styrenes.4 The utility of such transformations has dramatically increased the demand for facile routes to substituted styrenes.
Transition metal–mediated cross–coupling reactions provide efficient routes to functionalized styrenes that complement traditional methods such as dehydration or Hoffman elimination, both of which are incompatible with sensitive functional groups. Although the palladium catalyzed Heck reaction of aryl halides with ethylene provides access to substituted styrenes,5 the high pressures required for the reaction and the tendency for further reaction to give stilbenes make it undesirable.6 This problem has prompted the development of organometallic vinylating agents for use in transition metal–mediated cross–coupling reactions. These reactions generally involve vinylmagnesium,7 –tin,8 –silicon,9 or –boron10 compounds that couple with aryl electrophiles to form the desired styrene product.
The use of vinyltributyltin in the palladium-catalyzed Stille reaction is arguably the most developed of the cross-coupling reactions to access substituted styrenes.8 Making use of fluoride as an activator, conditions have been developed that successfully couple even deactivated aryl chlorides with vinyltributyltin in good yields.8a However, the use of vinyltributyltin also has several drawbacks. Vinyltributyltin suffers from toxicity and can be difficult to remove from the styrene product.10a In addition, vinyltributyltin is quite expensive, and its use contributes to a lack of atom economy.
Vinyltrimethylsilanes9d–e and vinylpolysiloxanes9a–c have also seen use in the palladium-catalyzed vinylation of aryl halides. Using fluoride as an activating agent, vinyltrimethylsilanes can be used in the vinylation of aryl iodides in good yields,9d–e while vinylpolysiloxanes can be used in the vinylation of aryl bromides and aryl iodides in good yields.9a–c
The palladium-catalyzed Suzuki-Miyaura reaction provides another method for the vinylation of aryl halides. Organoboron compounds such as dialkylboranes, boronic acids and boronate esters can generally be utilized for Suzuki–Miyaura cross–coupling reactions. However, vinylboron analogues of such organoboron compounds have notable limitations. For example, vinylboronic acid readily polymerizes and cannot be isolated.11 However, this problem can be solved by converting the vinylboronic acid into its anhydride form, 2,4,6-trivinylcyclotriboroxane-pyridine complex, which can be used to generate vinylboronic acid in situ for use in the coupling reaction.10a Although this method furnishes the desired styrene product in good yields, a 1:1 ratio of halide to 2,4,6-trivinylcyclotriboroxane-pyridine complex is used, equating to three equivalents of vinylating agent. Alternatively, vinyl boronate esters have been shown to participate selectively in Suzuki–type reactions with aryl halides to form styrenes.10f Unfortunately, the diols used to make the boronate esters add considerable expense to the process, and also contribute to a lack of atom economy.
The problems associated with the use of traditional organoboron compounds can be solved with the use of potassium organotrifluoroborate salts. These compounds can be readily prepared via the addition of KHF2 to a variety of organoboron intermediates.12 More specifically, potassium vinyl trifluoroborate can be prepared in good yields on large scale through the addition of vinylmagnesium bromide to trimethyl borate followed by treatment with inexpensive KHF2 (eq 1).10c
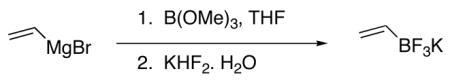 |
(1) |
The use of potassium vinyltrifluoroborate in Suzuki-Miyaura coupling reactions with arenediazonium salts to form styrenes was first reported by Genet in 1998.10d Later, preliminary results of Suzuki–Miyaura cross–coupling reactions between potassium vinyltrifluoroborate and aryl bromides were reported by our research group.10g Herein we provide a full account of our exploration of these reactions, including further optimization of the reaction conditions and expansion of the reaction scope.
Results and Discussion
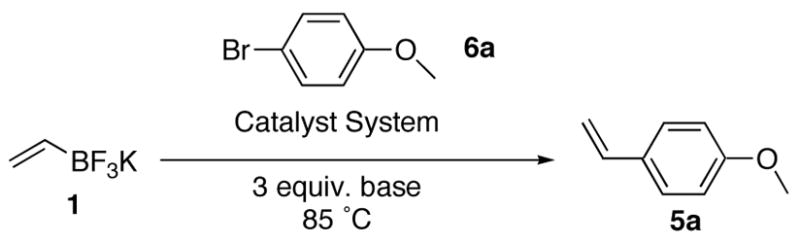
Initially, we focused on optimization of the reaction conditions of the cross coupling reaction of potassium vinyltrifluoroborate and p–bromoanisole. The cross–coupling conditions were optimized using a variety of palladium sources [PdCl2(dppf)·CH2Cl2, PdCl2, Pd(OAc)2, Pd(PPh3)4, PdCl2(CN)2], bases (Cs2CO3, K2CO3, KHCO3, K3PO4, NEt3, pyridine) and solvent systems (i–PrOH/H2O, THF/H2O, toluene/H2O). PPh3 was used as a ligand for the Pd(II) sources (Table 1).
Table 1.
Optimization of Conditions for Cross-Coupling Reaction
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | catalyst system | catalyst loading | solvent | molarity | base | reaction time | isolated yield | ratio 5a:6aa |
| 1 | PdCl2(dppf)·CH2Cl2 | 2 mol % | THF/H2O (9:1) | 0.1 | Cs2CO3 | 22 h | 63 | 100:0 |
| 2 | PdCl2(dppf)·CH2Cl2 | 2 mol % | i-PrOH/H2O (9:1) | 0.1 | NEt3 | 22 h | 23 | 100:0 |
| 3 | PdCl2(dppf)·CH2Cl2 | 2 mol % | Toluene/H2O (9:1) | 0.1 | Cs2CO3 | 22 h | -- | 20:80 |
| 4 | PdCl2(dppf)·CH2Cl2 | 5 mol % | THF/H2O (9:1) | 0.1 | Cs2CO3 | 18 h | 30 | 100:0 |
| 5 | Pd(PPh3)4 | 2 mol % | THF/H2O (9:1) | 0.1 | Cs2CO3 | 40 h | 62 | 100:0 |
| 6 | Pd(OAc)2 + 2 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.1 | Cs2CO3 | 45 h | 78 | 92:8 |
| 7 | Pd(OAc)2 + 3 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.1 | Cs2CO3 | 45 h | 72 | 97:3 |
| 8 | PdCl2(CN)2 + 2 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.1 | Cs2CO3 | 45 h | 69 | 95:5 |
| 9 | PdCl2(CN)2 + 3 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.1 | Cs2CO3 | 45 h | 70 | 96:4 |
| 10 | PdCl2 + 2 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.1 | Cs2CO3 | 45 h | 72 | 95:5 |
| 11 | PdCl2 + 3 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.5 | Cs2CO3 | 22 h | 72 | 100:0 |
| 12 | PdCl2 + 2 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.5 | Cs2CO3 | 22 h | 68 | 100:0 |
| 13 | PdCl2 + 3 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.5 | K3PO4 | 22 h | 67 | 92:8 |
| 14 | PdCl2 + 3 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.5 | KHCO3 | 22 h | 62 | 82:18 |
| 15 | PdCl2 + 3 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.5 | K2CO3 | 22 h | 67 | 97:3 |
| 16 | PdCl2 + 3 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.5 | pyridine | 22 h | -- | 20:80 |
| 17 | PdCl2 + 3 equiv PPh3 | 2 mol % | THF/H2O (9:1) | 0.5 | NEt3 | 22 h | -- | 55:45 |
Ratio of 5a:6a in isolated material by GC assay.
The reaction proceeded most effectively using 2 mol % of PdCl2 and 6 mol % of PPh3 as a catalyst system in THF/H2O (9:1) in the presence of 3 equivalents of Cs2CO3 as a base to furnish 4–methoxystyrene in 72% yield (entry 11).
Through the optimization process, it was observed that the reaction was rather specific to the THF/H2O solvent system, as it was the only system tested that offered satisfactory results. In contrast, a number of catalyst systems tested provided satisfactory results. Using 2 mol % of PdCl2(dppf)·CH2Cl2 as a catalyst system with the optimized solvent and base conditions furnished the desired 4–methoxystyrene product in 63% yield, while using 2 mol % of Pd(OAc)2 and 6 mol % of PPh3 as a catalyst system with the optimized solvent and base conditions furnished the desired 4–methoxystyrene in a 72% yield. The PdCl2 system was chosen over the Pd(OAc)2 system because of its lower price. With regard to the base, only Cs2CO3 and K2CO3 provided acceptable results.
With the optimized conditions in hand, the scope of the cross–coupling reaction in the presence of a variety of functional groups was explored. As shown in Tables 2 and 3, the cross coupling reaction proceeds with good to excellent yields in the presence of a large variety of functional groups including nitriles, ketones, esters, aldehydes, alcohols, and nitro groups. Furthermore, both electron–deficient and electron–rich bromides react with the vinyltrifluoroborate to yield the desired products in good yields; however, the rate of reaction of the electron deficient-bromides is higher than that of the electron–rich bromides. Although the reaction time for each bromide tested was not optimized, and the general protocol was to run the reactions for 22 hours, minimized reaction times were compiled for a number of subtrates (Table 4). The increased rate of the electron deficient-bromides in comparison to the electron rich-bromides is presumably due to the fact that electron–deficient bromides undergo oxidative addition more rapidly than electron–rich bromides.
Table 2.
Cross–Coupling of Electron Deficient Bromides with Potassium Vinyltrifluoroborate 1
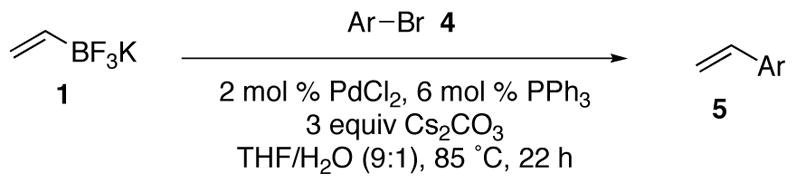 | ||||
|---|---|---|---|---|
| entry | ArBr | coupling product | % isolated yield | |
| 1 |
|
4a | 5a | 83 |
| 2 |
|
2c | 3a | 85 (79)a |
| 3 |
|
4b | 5b | 64 |
| 4 |
|
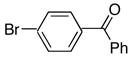
4c | 5c | 85 |
| 5 |
|
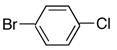
4d | 5d | 77 |
| 6 |
|
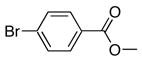
4e | 5e | 87 |
| 7 |
|
4f | 5f | 83 |
| 8 |
|
4g | 5g | 84 |
8 mmol scale reaction.
Table 3.
Cross–Coupling of Electron Rich Bromides with Potassium Vinyltrifluoroborate 1
 | ||||
|---|---|---|---|---|
| entry | ArBr | coupling product | % isolated yield | |
| 1 |
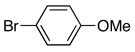
|
6a | 7a | 72 |
| 2 |
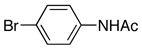
|
6b | 7b | 78 |
| 3 |
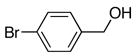
|
6c | 7c | 82 |
| 4 |
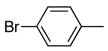
|
6d | 7d | 76 |
| 5 |
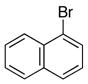
|
6e | 7e | 82 |
| 6 |
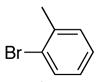
|
6f | 7f | 82a |
| 7b |
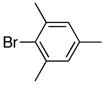
|
6g | 7g | 81 |
| 8b |
|
6h | 7h | 93 |
7% o-bromotoluene was present.
RuPhos was used as a ligand.
Table 4.
Minimized Reaction Times for Cross-Coupling of Representative Aryl Bromides with Potassium Vinyltrifluoroborate 1
 | |||||
|---|---|---|---|---|---|
| entry | ArBr | coupling product | reaction time (h) | % isolated yield | |
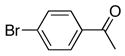
| 1 |
|
2c | 3a | 3 | 90 |
| 2 |
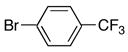
|
4b | 5b | 5 | 62 |
| 3 |
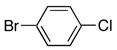
|
4d | 5d | 4 | 81 |
| 4 |
|
6a | 7a | 22 | 72 |
| 5 |
|
6d | 7d | 7 | 79 |
| 6 |
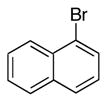
|
6e | 7e | 8 | 81 |
Through the investigation of functional group tolerance, we found the optimized conditions to be quite general, affording good yields of the desired products in almost all cases. A few cases, however, required further ligand optimization. Following the general procedure and using PPh3 as the ligand, entries 7 and 8 of Table 3 only afforded 35% and 40% conversion, respectively, to the desired product by gas chromatography assay. This poor conversion was most likely due to the extremely electron rich nature of both bromides and the steric hinderance of the mesityl bromide.13 Using the highly hindered mesityl bromide, Buchwald’s SPhos, XPhos, and RuPhos ligands (Figure 1) were screened (Table 5).14
Figure 1.
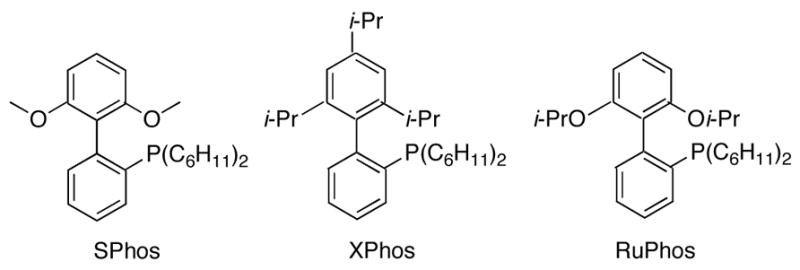
Buchwald Ligands.
Table 5.
Ligand Screening for 2–Bromomesitylene
 | ||
|---|---|---|
| entry | Ligand | Ratio 6g:7g:7ja |
| 1 | PPh3 | 3.5:6.5:0 |
| 2 | SPhos | 0:3:1 |
| 3 | XPhos | 0:1:1 |
| 4 | RuPhos | 1:20:1 |
By GC assay.
In all three cases, nearly all of the mesityl bromide was consumed. While the reaction employing RuPhos gave almost entirely the desired product, the reactions employing SPhos and XPhos gave significant amounts of both the desired product and the Heck product resulting from the reaction of the Suzuki–Miyaura coupling product with the mesityl bromide. No homocoupled product was observed in any case, most likely due to the steric hinderance. Clearly, the RuPhos ligand was the most desirable for the reaction, and subsequently supplied the desired product in good yields (entry 7, Table 3). The successful coupling of the mesityl bromide prompted the use of RuPhos in the coupling of the bromoaniline, which also yielded the desired product in excellent yields (entry 8, Table 3).
At this point, the viability of the cross–coupling reaction was tested on a larger scale. While the general protocol was to run the reaction on a 1 mmol scale, the cross–coupling reaction of 4–bromoacetophenone and potassium vinyltrifluoroborate was found to be comparable in utility on a gram scale, as an 8 mmol reaction afforded the desired product in 79% yield.
Having demonstrated the utility of the cross–coupling reaction conditions on a number of functional groups, mostly at the para position, we chose to test the generality of the functional group tolerance at the ortho and meta positions. As shown in Table 6, the acetyl– and nitrile–substituted bromides were chosen as representative electron–deficient bromides and the methoxy–substituted bromide was chosen as a representative electron–rich bromide.
Table 6.
Cross–Coupling of ortho–, meta–, and para–Functionalized Bromides with Potassium Vinyltrifluoroborate 1
 | ||||
|---|---|---|---|---|
| entry | ArBr | coupling product | % isolated yield | |
| 1 |
|
8a | 9a | 82 |
| 2 |
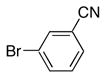
|
8b | 9b | 78 |
| 3 |
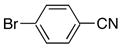
|
4a | 5a | 83 |
| 4 |
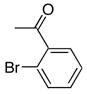
|
8c | 9c | 92a |
| 5 |
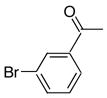
|
8d | 9d | 79 |
| 6 |
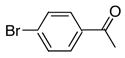
|
2c | 3a | 85 |
| 7 |
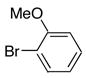
|
8e | 9e | 71 |
| 8 |
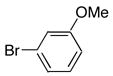
|
8f | 9f | 74 |
| 9 |
|
6a | 7a | 72 |
8% 2′-bromoacetophenone was present
In all cases, the tolerance of the functional group was general as the desired product was obtained in comparable yields at all three positions for each functional group tested.
We continued our investigation by exploring the reaction of potassium vinyltrifluoroborate with heteroaryl bromides. A variety of diverse heteroaryl bromides were reacted with potassium vinyltrifluoroborate (Table 7).
Table 7.
Cross–Coupling of Heteroaryl Bromides with Potassium Vinyltrifluoroborate 1
 | ||||
|---|---|---|---|---|
| entry | ArBr | coupling product | % isolated yield | |
| 1 |
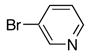
|
10a | 11a | 72 |
| 2 |
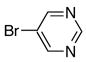
|
10b | 11b | 79b |
| 3 |
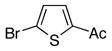
|
10c | 11c | 66 |
| 4 |
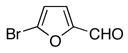
|
10d | 11d | 70 |
| 5 |
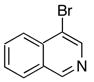
|
10e | 11e | 88b |
| 6 |
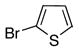
|
10f | 11f | 64 |
| 7 |
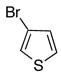
|
10g | 11g | 70a |
| 8 |
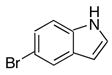
|
10h | 11h | 38 |
| 9 |
|
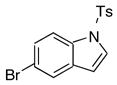
10i | 11i | 88 |
| 10 |
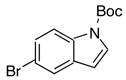
|
10j | 11j | 77 |
| 11 |
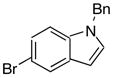
|
10k | 11k | 71 |
2% THF and 8% 3-bromothiophene were present.
Contaminated by unknown impurity.
As shown in Table 7, the cross–coupling reaction of potassium vinyltrifluoborate proceeds with a wide variety of heteroaryl bromides to give the desired product in good yields. Although the unprotected indole (entry 8) does not provide the desired product in acceptable yields, simple Boc, tosylate, or benzyl protection15 allows the cross–coupling reaction to proceed, furnishing good yields of the desired product. The desirable reactivity of all three protecting group indicates that the poor results seen in the case of the unprotected indole were a result of the N-H functionality, and not the electron rich nature of the system (entries 9–11, Table 7).
With the generality of the coupling demonstrated in the presence of diverse functionality, we focused our attention on the scope of the leaving group. As outlined in Table 8, iodide, triflate, bromide and chloride were all tested as leaving groups. While the triflate, iodide, and bromide all afforded comparable yields of the desired product, the chloride failed to produce any of the desired product.
Table 8.
Electrophile Compatibility
 | ||||
|---|---|---|---|---|
| entry | ArX | coupling product | % isolated yield | |
| 1 |
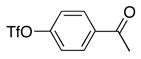
|
2a | 3a | 82 |
| 2 |
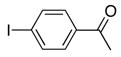
|
2b | 3a | 89 |
| 3 |
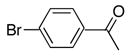
|
2c | 3a | 85 |
| 4 |
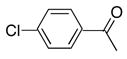
|
2d | 3a | 0 |
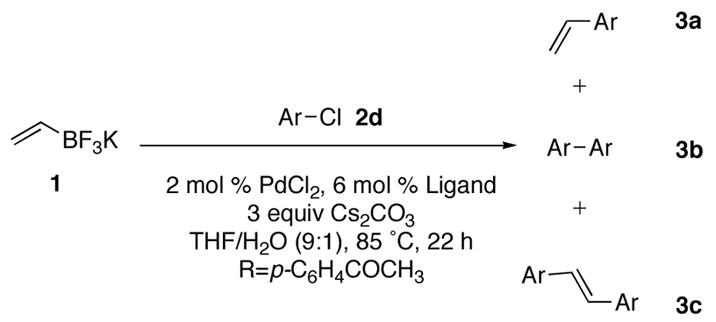
The poor conversion observed using the chloride (entry 4), prompted the screening of alternative ligands for the coupling reaction. In this case again, Buchwald’s SPhos, XPhos, and RuPhos were examined (Table 9).
Table 9.
Ligand Screening for Chloroacetophenone
 | ||
|---|---|---|
| entry | Ligand | Ratio 2d:3a:3b:3ca |
| 1 | PPh3 | 1:0:0:0 |
| 2 | SPhos | 0:20:4:3 |
| 3 | XPhos | 0:27:8:3 |
| 4 | RuPhos | 0:17:4:1.5 |
By GC assay
Although all of the Buchwald ligands tested were far more effective than PPh3, all three of these ligands yielded not only the desired product, but also two undesired byproducts. Byproduct 3b is a result of homocoupling between two chloroacetophenone molecules, while byproduct 3c is a result of a Heck–type reaction between the desired product and chloroacetophenone. The screening process showed RuPhos as the most effective ligand for the coupling reaction, and upon isolation, the desired product was obtained in 65% yield (eq 2). The yield is significantly reduced due to the subsequent homocoupling and Heck chemistry.
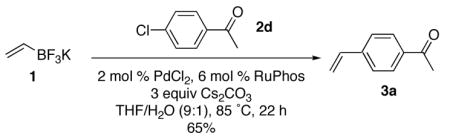 |
(2) |
Although the cross–coupling proceeded readily with the activated, electron–deficient chloroacetophenone, the conditions utilizing RuPhos as the ligand were not general for deactivated, electron–rich chlorides. Neither 4–chloroanisole or 4–chlorotoluene reached completion after 22 hours at 85 °C.
Conclusions
The Suzuki–Miyaura crosss–coupling reaction of aryl- and heteroaryl electrophiles with potassium vinyltrifluoroborate has proven to be an efficient route for accessing functionalized styrenes. The reaction proceeds readily in the presence of a wide variety of functional groups and can be used with iodides, bromides, chlorides, and triflates as electrophiles.
Experimental Section
General Procedure for Suzuki–Miyaura Cross–Coupling Reactions. Preparation of 1-(4-vinyl-phenyl)-ethanone (3a)
A solution of potassium vinyltrifluoroborate (134 mg, 1.00 mmol), PdCl2 (3.5 mg, 0.02 mmol), PPh3 (16 mg, 0.06 mmol), Cs2CO3 (978 mg, 3.00 mmol), and 4′–bromoacetophenone (199 mg, 1.00 mmol) in THF/H2O (9:1) (2 mL) was heated at 85 °C under a N2 atmosphere in a sealed tube. The reaction mixture was stirred at 85 °C for 22 h, then cooled to rt and diluted with H2O (3 mL) followed by extraction with CH2Cl2 (10 mL × 3). The solvent was removed in vacuo, and the crude product was purified by silica gel chromatography (eluting with 20:1 n-pentane:ether) to yield 1-(4-vinyl-phenyl)-ethanone as a pale yellow solid (123 mg, 0.848 mmol, 85%) mp 33–34 °C. The spectral data were in accordance with those described in the literature.9a
Supplementary Material
Full experimental details and copies of all NMR spectra (1H and 13C). This material is available free of charge via the internet at http://pubs.acs.org.
Acknowledgments
The authors thank the NIH (GM35249), Amgen, Merck Research Laboratories, and the Vagelos Program in the Molecular Life Sciences for support. We thank Johnson Matthey for donation of catalysts, and Prof. Stephen L. Buchwald (MIT) for a donation of ligands.
References
- 1.Grubbs RH. Tetrahedron. 2004;60:7117–7140. [Google Scholar]
- 2.(a) Heck RF. J Am Chem Soc. 1968;90:5518–5526. [Google Scholar]; (b) Braese S, de Meijere A. In: Metal-Catalyzed Cross-Coupling Reactions. de Meijere A, Diederich F, editors. Wiley-VCH; Weinheim: 2004. pp. 217–315. [Google Scholar]; (c) Beller M, Zapf A, Riermeier TH. In: Transition Metals for Organic Synthesis. Beller M, Bolm C, editors. Wiley-VCH; Weinheim: 2004. pp. 271–305. [Google Scholar]
- 3.(a) Rodriguez AL, Bunlaksananusorn T, Knochel P. Org Lett. 2000;2:3285–3287. doi: 10.1021/ol000174l. [DOI] [PubMed] [Google Scholar]; (b) Ikeda Y, Nakamaru T, Yorimitsu H, Oshima K. J Am Chem Soc. 2002;124:6514–6515. doi: 10.1021/ja026296l. [DOI] [PubMed] [Google Scholar]; (c) Terao J, Saito K, Nii S, Kambe N, Sonada N. J Am Chem Soc. 1998;120:11822–11823. [Google Scholar]; (d) Crudden CM, Hleba YB, Chen AC. J Am Chem Soc. 2004;126:9200–9201. doi: 10.1021/ja049761i. [DOI] [PubMed] [Google Scholar]; (e) RajanBabu TV, Nomura N, Jin J, Nandi M, Park H, Sun X. J Org Chem. 2003;68:8431–8446. doi: 10.1021/jo035171b. [DOI] [PubMed] [Google Scholar]; (f) Agbossou F, Carpentier J-F, Mortreux A. Chem Rev. 1995;95:2485–2506. [Google Scholar]
- 4.(a) Lou Y, Baldamus J, Huo Z. J Am Chem Soc. 2004;126:13910–13911. doi: 10.1021/ja046063p. [DOI] [PubMed] [Google Scholar]; (b) Guo N, Li L, Marks TJ. J Am Chem Soc. 2004;126:6542–6543. doi: 10.1021/ja048761f. [DOI] [PubMed] [Google Scholar]
- 5.Plevyak JE, Heck RF. J Org Chem. 1978;43:2454–2456. [Google Scholar]
- 6.Spencer A. J Organomet Chem. 1983;258:101–108. [Google Scholar]
- 7.Bumagin NA, Luzikova EV. J Organomet Chem. 1997;532:271–273. [Google Scholar]
- 8.(a) Littke AF, Schwartz L, Fu GC. J Am Chem Soc. 2002;124:6343–6348. doi: 10.1021/ja020012f. [DOI] [PubMed] [Google Scholar]; (b) Grasa GA, Nolan SP. Org Lett. 2001;3:119–122. doi: 10.1021/ol006827f. [DOI] [PubMed] [Google Scholar]; (c) McKean DR, Parrinello G, Renaldo AF, Stille JK. J Org Chem. 1987;52:422–424. [Google Scholar]; (d) Shirakawa E, Yamasaki K, Tamerjiro H. Synthesis. 1998:1544–1549. [Google Scholar]; (e) Krolski ME, Renaldo AF, Rudisill DE, Stille JK. J Org Chem. 1988;53:1170–1176. [Google Scholar]
- 9.(a) Denmark SE, Butler CR. Org Lett. 2006;8:63–66. doi: 10.1021/ol052517r. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Wang Z. Synthesis. 2000:999–1003. [Google Scholar]; (c) Denmark SE, Wang Z. J Organomet Chem. 2001;624:372–375. [Google Scholar]; (d) Hatanaka Y, Hiyama T. J Org Chem. 1988;53:918–920. [Google Scholar]; (e) Jeffrey T. Tetrahedron Lett. 1999;40:1673–1676. [Google Scholar]
- 10.(a) Kerins FO, O’Shea DF. J Org Chem. 2002;67:4968–4971. doi: 10.1021/jo020074o. [DOI] [PubMed] [Google Scholar]; (b) Peyroux E, Berthiol F, Doucet H, Santelli M. Eur J Org Chem. 2004:1075–1082. [Google Scholar]; (c) Darses S, Michaud G, Genet J-P. Eur J Org Chem. 1999:1875–1883. [Google Scholar]; (d) Darses S, Michaud G, Genet J-P. Tetrahedron Lett. 1998;39:5045–5048. [Google Scholar]; (e) Stewart SK, Whiting A. J Organomet Chem. 1994;482:293–300. [Google Scholar]; (f) Lightfoot AP, Twiddle SJR, Whiting A. Synlett. 2005:529–531. [Google Scholar]; (g) Molander GA, Rivero MR. Org Lett. 2002;4:107–109. doi: 10.1021/ol0169729. [DOI] [PubMed] [Google Scholar]; (g) Grisorio R, Mastrorillo CFN, Romanazzi G, Suranna GP. Tetrahedron Lett. 2005;46:2555–2558. [Google Scholar]
- 11.Matteson DS. J Am Chem Soc. 1960;82:4228–4233. [Google Scholar]
- 12.(a) Vedejs E, Chapman RW, Fields SC, Lin S, Schrimpf MR. J Org Chem. 1995;60:3020–3027. [Google Scholar]; (b) Vedejs E, Fields SC, Hayashi R, Hitchcock SR, Powell DR, Schrimpf MR. J Am Chem Soc. 1999;121:2460–2470. [Google Scholar]
- 13.Carter RR, Wyatt JK. Tetrahedron Lett. 2006;47:6091–6094. [Google Scholar]
- 14.(a) Milne JE, Buchwald SL. J Am Chem Soc. 2004;126:13028–13032. doi: 10.1021/ja0474493. [DOI] [PubMed] [Google Scholar]; (b) Barder TE, Walker SD, Martinelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 15.(a) Prieto M, Zurita E, Rosa E, Munoz L, Loyd-Williams P, Giralt E. J Org Chem. 2004;69:6812–6820. doi: 10.1021/jo0491612. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Fandrick KR, Song H-J. J Am Chem Soc. 2005;127:8942–8943. doi: 10.1021/ja052433d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental details and copies of all NMR spectra (1H and 13C). This material is available free of charge via the internet at http://pubs.acs.org.