

Abstract
The NMR and EPR spectra of two bis-imidazole and three bis-pyridine complexes of tetraphenylchlorinatoiron(III), [(TPC)Fe(L)2]+ (L = Im-d4, 2-MeHIm, 4-Me2NPy, Py and 4-CNPy) have been investigated. The full resonance assignments of the [(TPC)Fe(L)2]+ complexes of this study have been made from COSY and NOESY experiments and ADF calculations. Unlike the [(OEC)Fe(L)2]+ complexes reported previously (Cai, S.; Lichtenberger, D. L.; Walker, F. A. Inorg. Chem. 2005, 44, 1890-1903), the NMR data for the [(TPC)Fe(L)2]+ complexes of this study indicate that the ground state is S = ½ for each bis-ligand complex, whereas a higher spin state was present at NMR temperatures for the Py and 4-CNPy complexes of (OEC)Fe(III). The pyrrole-8,17 and pyrroline-H of all [TPCFe(L)2]+ show large magnitude chemical shifts (hence indicating large spin density on the adjacent carbons that are part of the π system), while pyrrole-12,13-CH2 and -7,18-CH2 protons show much smaller chemical shifts, as predicted by the spin densities obtained from ADF calculations. The magnitude of the chemical shifts decreases with decreasing donor ability of the substituted pyridine ligands, with the non-hindered imidazole ligand having slightly larger magnitude chemical shifts than the most basic pyridine, even though its basicity is significantly lower (4-Me2NPyH+ pKa = 9.7, H2Im+ pKa = 6.65 (adjusted for the statistical factor of 2 protons)). The temperature dependence of the chemical shifts of all but the 4-Me2NPy bis-ligand complexes studied over the temperature range of the NMR investigations shows that most of them have mixed (dxy)2(dxz,dyz)3/(dxzdyz)4(dxy)1 electron configurations that cannot be resolved by temperature-dependent fitting of the proton chemical shifts, with a S = 3/2 excited state in each case that in most cases lies at more than kT at room temperature above the ground state. The observed pattern of chemical shifts of the 4-CNPy complex and analysis of the temperature dependence indicates that it has a pure (dxzdyz)4(dxy)1 ground state and that it is ruffled, because ruffling mixes the a2u(π)-like orbital of the chlorin into the SOMO. This mixing accounts for the negative chemical shift of the pyrroline-H (−6.5 ppm at −40 °C), and thus the negative spin density at the pyrroline-α-carbons, but the mixing is not to the same extent as observed for [(TPC)Fe(t-BuNC)2]+, whose pyrroline-H chemical shift is −36 ppm at 25 °C (Simon-neaux, G.; Kobeissi, M. Dalton Trans. 2001, 1587-1592). Peak assignments for high-spin (TPC)FeCl have been made by saturation transfer techniques that depend upon chemical exchange between this complex and its bis-4-Me2NPy adduct.
Introduction
There are a number of “green” hemes that catalyze biological reactions, including the heme d of the cytochrome bd terminal oxidase of E. coli,1-4 the heme d of the catalase HPII from E. coli,5-7 and the sulfhemes,8,9 as well as heme d1 of the bacterial cytochrome cd1 nitrite reductase/cytochrome oxidase from Pseudomonas aeruginosa, Paracoccus denitrificans and Thiobacillus denitrificans (a dioxoisobacteriochlorin10) and siroheme of E. coli sulfite reductase heme protein (an isobacteriochlorin11,12). All of these pigments are iron chlorin or isobacteriochlorin or dioxoisobacteriochlorin complexes which have uniquely different structures. There has previously been some confusion regarding the classification of reduced hemes and the expectations as to their electronic properties, and a number of careful structural investigations were required to elucidate the geometric and stereoisomeric structures of these macrocycles.1-12 There have been a number of previous studies of synthetic and natural iron chlorin and/or isobacteriochlorin complexes by magnetic resonance techniques, including the seminal paper of Stolzenberg, Strauss and Holm in 198113 and additional papers from these authors,14-18 as well as other researchers.19-31 Understanding the electronic properties of the iron(III) complexes of each of the individual “green” hemes is an important step in understanding their mechanisms of action.
Synthesis of 5,10,15,20-tetraphenylchlorinatoiron (III) chloride ((TPC)FeCl, Scheme I) and trans-2,3,7,8,12,13,17,18-octaethylchlorinatoiron (III) chloride ((OEC)FeCl) was first reported and the complexes studied by 1H NMR spectroscopy some 25 years ago.13 However, because of the broad lines and correspondingly short proton relaxation times, until now full peak assignments of either the high-spin chloride complex or low-spin complexes of (TPC)Fe(III) with axial ligands have been reported, even though the 1D 1H NMR spectra of tetraphenylchlorinatoiron(III) (TPC), octaethylchlorinatoiron(III) (OEC),30 mono-oxooctaethylchlorinatoiron(III) (oxo-OEC),31 and of substituted tetraphenylporphyrinatoiron(III) (TPP,32 TMP33-35 and the octaalkyltetraphenylporphyrins36-39) have been reported. In this work we report detailed NMR studies of high-spin (TPC)FeCl and its low-spin complexes with axial ligands (imidazole, 2-methylimidazole, 4-dimethylaminopyridine, pyridine, 4-cyanopyridine). We have made full assignments, with the help of ADF calculations, of the resonances for both low-spin and high-spin complexes, studied the effect of axial ligands on the electronic structure of the iron center, and compared our findings to those reported recently for (OEC)Fe(III)30 and oxo-(OEC)Fe(III)31 complexes. The EPR spectra of these complexes are also reported. By comparing and contrasting the electronic properties of these molecules with the electronic properties of the others, this work provides additional information on the factors that control the electronic properties of all of these systems.
Scheme I.

(TPC)FeCl with the numbering convention of the atoms of the chlorin ligand used for labeling the proton resonances.
Experimental
The 5,10,15,20-tetraphenylchlorin free base (H2TPC) was purchased from Porphyrin Products Inc. The tetraphenylchlorinatoiron(III) chloride ((TPC)FeCl) was synthesized using a modification of a method developed in this laboratory:40 20 mg of H2TPC was dissolved in a minimum amount of chloroform (about 5 ml). An approximate 50-fold excess of ferrous chloride was dissolved in 30 ml of mixed solvent of 3:1 chloroform:methanol (v/v). The ferrous chloride solution was mixed with the chlorin free-base solution in a round-bottom flask. The flask with the reactant was purged with nitrogen for 5 minutes and several crystals of sodium dithionite were added to remove any traces of oxidizing agents in the solution. The mixture was refluxed under nitrogen for 2 hours, cooled to room temperature and washed with water 3 times. The product was bubbled with HCl gas to convert any possible dimer to (TPC)FeCl. Finally, the solvent was evaporated to dryness and the solid product was pumped under vacuum overnight and kept in a dry box for future use.
All samples for NMR studies were prepared in 5 mm NMR tubes by addition of methylene chloride-d2 (Cambridge Isotopes) in the glove box. Complexes with axial ligands were made by directly adding an excess of desired axial ligand (iron chlorin:axial ligand 1:4 or larger). Axial ligands used in this work include deuterated imidazole (Im-d4, Cambridge Isotopes), 2-methylimidazole (2-MeHIm, Aldrich), 4-dimethylaminopyridine (4-Me2NPy, Aldrich), pyridine (Py, Aldrich), pyridine-d5 (Py-d5, Cambridge Isotopes) and 4-cyanopyridine (4-CNPy, Aldrich). For complexes with pyridine and 4-cyanopyridine as axial ligands, an excess of silver trifluoromethanesulfonate (AgOSO2CF3, 99%, Alfa ÆSAR) was also added to improve the binding ability of the ligands by removing chloride ion. All NMR tubes were sealed tightly before removal from the glove box.
1H NMR spectra of the free-base H2TPC were recorded on a Varian Unity-300 spectrometer operating at 299.955 MHz with the variable temperature unit set at 10 °C, referenced to the resonance of the residual solvent protons of CD2Cl2 (5.32 ppm relative to TMS). NOESY and COSY spectra were acquired with the normal Varian pulse sequences with spectral width 3904 Hz in each dimension, acquisition time 254 ms, relaxation delay 550 ms, mixing time of 900 ms (NOESY), and 120 t1 increments. HMQC spectra were recorded on a Bruker DRX-500 NMR spectrometer at ambient temperatures using the standard Bruker pulse sequence.
For 1H NMR measurements of the iron chlorin complexes at low temperature, the spectra were recorded on the Varian Unity-300 spectrometer over a temperature range from −90 °C to +20 °C, and again referenced to the resonance from residual solvent protons. The temperature was calibrated using the standard Wilmad methanol and ethylene glycol samples. For other experiments, the spectra were acquired on the Varian Unity-300 or Bruker DRX-500 NMR spectrometers. 1D saturation transfer experiments were carried out on both the Unity-300 and DRX-500 NMR spectrometers using the normal NOE difference pulse sequence. On both instruments, a selective intermediate level pulse was irradiated on the specific peaks of the fast relaxing (e.g. high-spin) species with an irradiation time of 50 ms. On the Unity-300, the decoupler power was set to 35–40 dB (63 dB is the maximum power level). On the DRX-500, the pulse level was 45–50 dB (−6 dB is the maximum power level).
For EPR spectroscopy the low-spin bis-ligand complexes of (TPC)Fe(III) were again prepared in dry CD2Cl2 immediately before the experiments using the same method as for the NMR studies, and frozen in liquid nitrogen. The EPR spectra were obtained on a CW Bruker ESP-300E EPR spectrometer operating at X-band using 0.2 mW microwave power and 100 kHz modulation amplitude of 2 G. A Systron-Donner microwave counter was used for frequency calibration. The EPR measurements were performed at 4.2 K using an Oxford continuous flow helium cryostat, ESR 900.
Results
NMR spectra and peak assignments of H2TPC
The 1D 300 MHz 1H NMR spectrum and the NOESY spectrum of H2TPC in CD2Cl2, recorded at 10 °C over the 3.6–9.2 ppm region are shown in Figure 1. The only resonance of the complex not shown in this spectrum is the single broad N-H resonance, which occurs at −1.31 ppm. As can be seen, the three pyrrole-H resonances are found between 8.19 and 8.57 ppm; the two doublets at those extreme chemical shifts show NOEs with each other. They also show cross peaks in the DQF-COSY spectrum shown in Supporting Information Figure S1, which clearly identifies them as arising from the 7,18- and 8,17-pyrrole-H, although which is which cannot be determined from this minimal information. The 3JH-H coupling constant between these protons on adjacent β-pyrrole carbons is 4.7 Hz. The singlet at 8.57 ppm thus is due to the 12,13-pyrrole-H. The singlet at 4.16 ppm with relative intensity 4 as compared to 2 for each of the pyrrole-H resonances is clearly the pyrroline-H of the chlorin ring. Between these pyrroline-H and pyrrole-H regions are the phenyl-H of H2TPC, and the multiplets have chemical shifts and relative intensities (in parentheses) ∼7.70 (12), 7.89 (4), and 8.10 (4) ppm. In comparison, H2TPP has overlapping meta- and para-H resonances at 7.79 (8) and 7.77 (4) ppm, respectively, and the ortho-H doublet of doublets at 8.23 (8) ppm (not shown). From the doublet of doublets coupling patterns of the multiplets at 7.89 and 8.09 ppm of H2TPC shown in Figure 1 and the DQF-COSY cross peak pattern shown in Supporting Information Figure S1 it is clear that the cross peaks observed are between each of the two types of ortho-H with the strongly overlapping meta- and para-H of the two types of phenyl rings of H2TPC. Thus, the two types of phenyl rings of H2TPC have their ortho-H slightly less ring-current shifted to larger chemical shift than do those of H2TPP, and one type is less ring-current shifted than the other. HMQC spectra are shown in Supporting Information Figure S2 for the pyrrole-, pyrroline- and phenyl-H, all of whose 1H and 13C chemical shifts are listed in Table 1.
Figure 1.
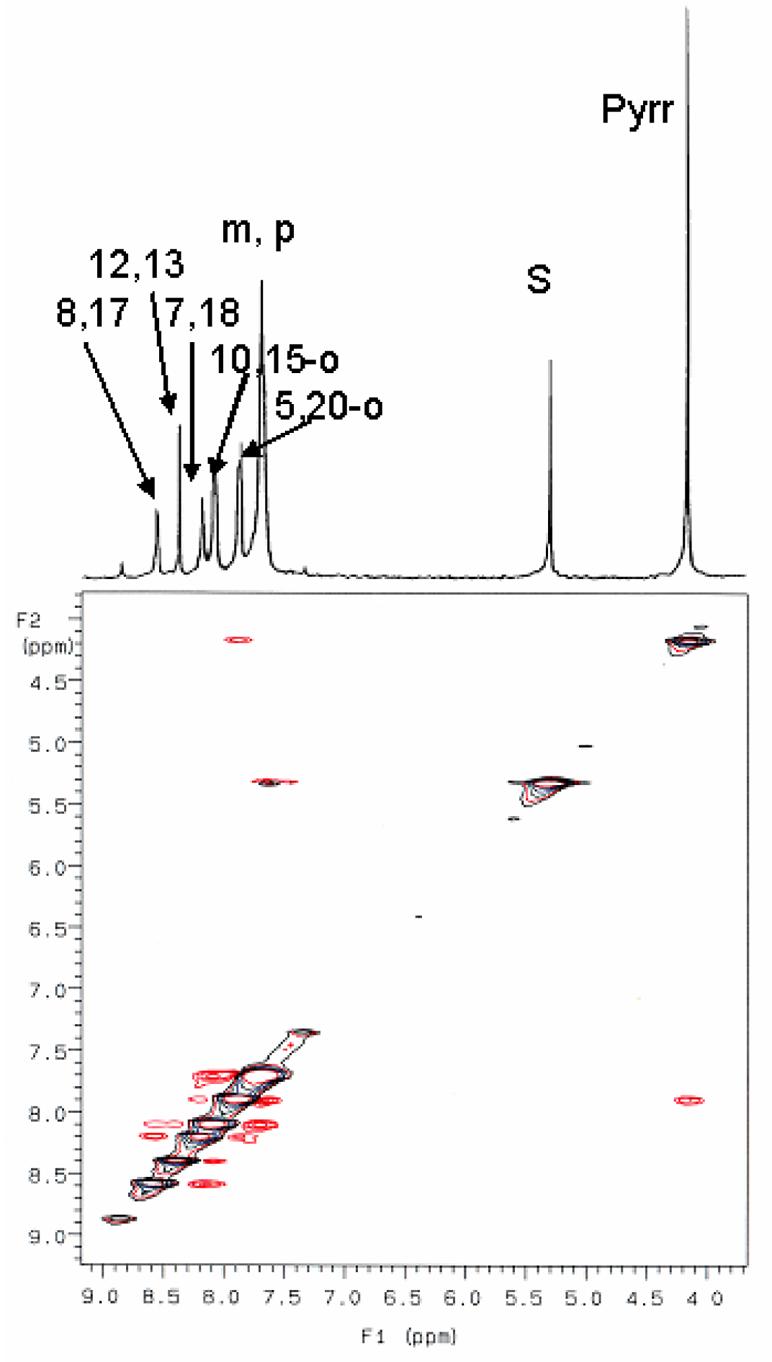
1D 1H 300 MHz NMR and 2D NOESY spectra of H2TPC in CD2Cl2 (5.32 ppm) at 10 °C. In addition to NOE and J-couplings between 7,18-H and 8,17-H, and between both 5,20-o-H and 10,15-o-H and the overlapping m,p-H (all also shown in the DQF-COSY spectrum of Supporting Information Figure S1), note the NOEs from 5,20-o-H to pyrroline-H and 7,18-H, and the NOEs from 10,15-o-H to 12,13-H and 8,17-H. S denotes solvent residual proton resonance.
Table 1.
Summary of Chemical shifts of H2TPC.
| Proton position (ppm) |
1H Chemical shift (ppm) | 3JH-H coupling (Hz) | 13C Chemical shift |
|---|---|---|---|
| 2,3-Pyrroline | 4.168 (s) | 37 | |
| 5,20-o-Phenyl | 7.892 (dd) | 7.0, v. small | 133 |
| 5,10,15,20-m,p-Phenyl | 7.683-7.719 (m) | 128, 127 | |
| 10,15-o-Phenyl | 8.100 (dd) | 7.0, v. small | 134 |
| 7,18-Pyrrole | 8.197 (d) | 4.7 | 124 |
| 12,13-Pyrrole | 8.393 (s) | 132 | |
| 8,17-Pyrrole | 8.578 (d) | 4.7 | 128 |
The NOESY spectrum of Figure 1 shows NOE cross peaks between the pyrroline-H and the ortho-H multiplet at 7.89 ppm, and also very weak NOE crosspeaks with the pyrrole-H doublet at 8.20 ppm, thus identifying that pyrrole-H doublet as arising from the 7,18-H. In like manner, the singlet pyrrole-H resonance at 8.39 ppm shows NOE cross peaks with the ortho-H resonance at 8.10 ppm, as does the pyrrole-H doublet at 8.58 ppm, thus identifying the latter as the 8,17-pyrrole-H. The chemical shifts and assignments are summarized in Table 1. The assignment of the pyrrole-H resonances of H2TPC, as a diamagnetic precursor of the iron(III) complexes of TPC, allows fitting of the temperature dependence of the pyrroline-H and each of the pyrrole-H resonances of these complexes.
NMR spectra and peak assignments of the low-spin (TPC)Fe(III) complexes
[(TPC)Fe(Im-d4)2]Cl
Upon addition of axial ligands, the high-spin complex (TPC)FeCl changes color from brown to green and becomes a low-spin, S = 1/2 (TPC)Fe(III) bis-imidazole complex. Figure 2 shows the 1D 1H NMR spectrum of low-spin [(TPC)Fe(Im-d4)2]Cl in CD2Cl2 at 0 °C. It shows four paramagnetically-shifted resonances, one at positive and three at negative chemical shifts. By integration, the resonance at +39 ppm must be that of the pyrroline protons; the same assignment of the bis-1-MeIm complex has been reported previously.27 In the COSY spectrum of [(TPC)Fe(Im-d4)2]Cl, obtained at +30 °C and shown in Supporting Information Figure S3, very weak crosspeaks were observed between the two pyrrole-H resonances at −2 and −32 ppm, which corresponds to −2 and −38 ppm at 0 °C in Figure 2. As for the free base H2TPC, these two resonances can be assigned to the adjacent pyrrole-7,18 and pyrrole-8,17 protons. Thus the third resonance with negative chemical shift is assigned to the pyrrole-12,13-H. Although from the COSY spectrum alone we can not distinguish which of the first two resonances are from protons attached to pyrrole-7,18 and which to pyrrole-8,17, ADF calculations on the corresponding [(OMC)Fe(HIm)2]+ complex (OMC = octamethylchlorin) have recently shown that the spin density at the pyrrole-8,17-carbons is much larger than that at either the 7,18- or 12,13-carbons,30 as shown in Figure 3A. (Separate ADF calculations for [(TPC)Fe(HIm)2]+ have not been carried out because of the size of the calculation (40 additional atoms).) Thus, for the spin-coupled pair of resonances the pyrrole-8,17-H must have much larger contact shift than the pyrrole-7,18-H, and the resonance near −38 ppm thus arises from the pyrrole-8,17-H. The resonances of the phenyl protons are all located in the region of 6-7.7 ppm. Their chemical shifts have not been analyzed in detail because of overlap between the expected two sets of three phenyl-H resonances.
Figure 2.

1H spectrum of low-spin [(TPC)Fe(Im-d4)2]Cl in CD2Cl2 at 0 °C. The labels “PL”, “7,18”, “12,13” and “8,17” denote pyrroline, pyrrole-7,18, pyrrole-12,13 and pyrrole-8,17 protons, respectively.
Figure 3.

Spin densities obtained from the ADF calculations for (A) [(OMC)Fe(ImH)2]+ in the planar geometry, with electron configuration (dxy)2(dxz,dyz)3 and the antisymmetric chlorin 3e(π)-like orbital contributing significantly, (B) [(OMC)Fe(MeNC)2]+ in the planar geometry, with metal electron configuration (dxy)2(dxz,dyz)3 with metal orbitals that are relatively more stable than those in (A), and (C) [(OMC)Fe(MeNC)2]+ in the ruffled geometry, with metal electron configuration (dxz,dyz)4(dxy)1 and the chlorin 3a2u(π)-like orbital contributing significantly, all shown for a generic chlorin core with the pyrroline carbons at the top. The red and blue colors represent positive and negative spin density, respectively. Adapted from Figure 8 of Reference 30.
[(TPC)Fe(2-MeHIm)2]Cl
[(TPC)Fe(2-MeHIm)2]Cl shows a similar 1D spectrum (Figure 4) to that of [(TPC)Fe(Im-d4)2]Cl, except that the magnitude of the chemical shifts is smaller at the same temperature, indicating smaller spin delocalization to the α-pyrroline and β-pyrrole carbons in this complex. In the COSY spectrum shown in Supporting Information Figure S4, as seen for [(TPC)Fe(Im-d4)2]Cl, Figure S3, there are also weak crosspeaks between the pyrrole-7,18-H and pyrrole-8,17-H. In a similar fashion, resonance assignments of the pyrroline and all the pyrrole protons can be made. Although [(TPC)Fe(2-MeHIm)2]Cl and [(TPC)Fe(Im-d4)2]Cl have different chemical shifts, the order of the chemical shifts of the three pyrrole-H resonances is same for the two (from the least to the most negative chemical shifts: in magnitude, 7,18 > 12,13 > 8,17). This suggests that these two complexes have similar electronic structures but with less spin density delocalized to the chlorin ring in the case of the 2-MeHIm ligands.
Figure 4.

1D 1H NMR spectrum of low-spin [(TPC)Fe(2-MeHIm)2]Cl in CD2Cl2 at 0 °C. “FL” denotes free axial ligand in the solution; “BL” denotes bound axial ligand. The N-H of the axial ligands are not observed, probably due to their fast proton exchange rate with traces of water in the solution, which leads to very broad peaks. Asterisks denote resonances arising from the minor amount of [(TPP)Fe(2-MeHIm)2]Cl present in the sample due to re-oxidation of TPC to TPP upon iron insertion.
The assignments of the bound axial 2-MeHIm ligands can be made from chemical exchange cross peaks observed between the bound and free axial ligands in the NOESY/EXSY spectrum shown in Supporting Information Figure S5. As for [(TPC)Fe(Im-d4)2]Cl, the phenyl proton peaks are tightly grouped and strongly overlapping between +6 and +7.7 ppm. Thus, their chemical shifts have not been analyzed in detail.
[(TPC)Fe(4-Me2NPy2)Cl
This complex has a similar 1H NMR spectrum to those of the former two complexes, with chemical shifts intermediate between the two, as shown in Supporting Information Figure S6. As for the former two complexes, the assignments of [(TPC)Fe(4-Me2NPy)2]Cl were also made from the COSY (Supporting Information Figure S7) and NOESY (Figure S8) spectra. The intermediate chemical shifts of the pyrroline and pyrrole-8,17 protons of [(TPC)Fe(4-Me2NPy)2]Cl between those of [(TPC)Fe(2-MeHIm)2]Cl and [(TPC)Fe(Im-d4)2]Cl is a point that will be considered further in the discussion section.
[(TPC)Fe(Py)2]+
Pyridine is a relatively weak base that binds to (TPC)FeCl only when chloride ion has been replaced by CF3SO3− by addition of AgOSO2CF3. Figure 5a shows the 1D 1H spectrum of [(TPC)Fe(Py)2]+. To simplify the spectrum and help make assignments, the complex with deuterated pyridine as the axial ligands, [(TPC)Fe(Py-d5)2]+, was also studied by 1H NMR spectroscopy (Figure 5b). The peak assignments were made from COSY (Supporting Information Figure S9) and NOESY (Figure S10) spectra. The order of the chemical shifts of the pyrroline and pyrrole protons is still the same as those of the former three complexes, although the magnitude of the chemical shifts of the pyrroline and pyrrole-8,17 protons is much smaller. The temperature dependence of the chemical shifts of the three types of pyrrole-H and the pyrroline-H do not follow the Curie law, as will be discussed in detail below.
Figure 5.

1D 1H NMR spectrum of low-spin (a) [(TPC)Fe(Py)2]Cl and (b) [(TPC)Fe(Py-d5)2]Cl in CD2Cl2 at 0 °C. “BL-m”. “BL-p” and “BL-o” denote meta, para and ortho protons of the bound axial ligands, respectively; “m”, “p” and “o” denote meta, para and ortho protons of the meso phenyl groups, respectively. Asterisks denote resonances arising from the minor amount of [(TPP)Fe(Py)2]Cl and [(TPP)Fe(Py-d5)2]Cl present in the samples due to re-oxidation of TPC to TPP upon iron insertion. X represents residual H2O.
Unlike the former three complexes, for [(TPC)Fe(Py)2]+ the phenyl protons are well-resolved and can be assigned from the COSY spectrum, as shown in Supporting Information Figure S9. Since there are two kinds of meso carbons, in total six resonances were observed from the phenyl protons. The COSY spectrum shows four pairs of crosspeaks, two of which are between meta- and ortho-H, the other two from meta- and para-H. The two resonances with half intensity as compared to the other four can be assigned to para-H; the two resonances which have crosspeaks with both ortho- and para-H are the meta-H, and the other two are the ortho-H. The resonances of the phenyl para- and meta-H in Figure 5a appear to be smaller than they should be. This is because the phenyl para- and meta-H are farther away from the iron center as compared to the ortho-H, leading to longer relaxation times (for para-H, T1≈ 250 ms). Under the conditions of the 1D NMR experiments shown in Figure 5 (total time period for one transient is 100 ms), the relaxation delay was not long enough for the magnetization of m- and p-H to relax to equilibrium. The relative resonance intensity of phenyl ortho-, meta- and para-H is 2:2:1 when a long delay time (1 s) is used.
It should be noted that the phenyl-H resonances are shifted from their diamagnetic positions in an alternating fashion, with the meta-H appearing at more positive chemical shift than its diamagnetic counterpart, while the ortho- and para-H appear at more negative chemical shifts than their diamagnetic counterparts. Thus, the phenyl-H shift differences δm – δo and δm – δp are positive, which indicates positive spin density at the meso-carbons,41 as discussed in greater detail in the Discussion section.
[(TPC)Fe(4-CNPy)2]+
4-CNPy is the weakest-binding of the axial ligands studied in this work. Even in the presence of AgOSO2CF3, it binds to (TPC)Fe(III) well only below −20 °C. The 1D 1H NMR spectrum recorded at −40 °C shown in Figure 6 for [(TPC)Fe(4-CNPy)2]+ is quite different from those of other complexes discussed above. Although the order of the chemical shifts of the three pyrrole-H peaks from positive to negative is still 7,18 >12,13 > 8,17, it is found that the 7,18- and 12,13-H of this complex have positive chemical shifts and the 8,17-H chemical shift is much less negative than for all other complexes. Furthermore, the pyrroline-H of this complex have a negative chemical shift; thus the order of pyrrole-H resonances remains the same as that of the other complexes of this study, while the pyrroline-H resonance has moved from most positive to most negative chemical shift at −40 °C. The peak assignments were also accomplished by COSY (Supporting Information Figure S11) and NOESY experiments (Figure S12). As for [(TPC)Fe(Py)2]+, the resonances from the phenyl protons are also well resolved, which enables full peak assignments. The temperature dependence of the three pyrrole-H and the pyrroline-H chemical shifts is anti-Curie, as will be discussed further below. Again, the pattern of shifts is that of the phenyl-H shift differences δm – δo and δm – δp being positive, which indicates positive spin density at the meso-carbons. This finding supports the assignment of the ground state electron configuration in this case as (dxz,dyz)4(dxy)1 (see discussion section).
Figure 6.

1D 1H NMR spectrum of low-spin [(TPC)Fe(4-CNPy)2]Cl in CD2Cl2 at −40 °C. The asterisk marks an impurity, probably traces of water.
EPR spectra of the low-spin (TPC)Fe(III) complexes
The rhombic signal in the EPR spectrum of [(TPC)Fe(HIm)2]Cl shown in Figure 8 is typical of low-spin Fe(III) porphyrinates and chlorinates with (dxy)2(dxzdyz)3 ground states.30,42 And although broader, the [(TPC)Fe(2-MeHIm)2]+ and [(TPC)Fe(4-Me2NPy)2]+ complexes have very similar observed g-values to those of [(TPC)Fe(HIm)2]+, suggesting that they also have the same ground state. However, for [(TPC)Fe(Py)2]+ and [(TPC)Fe(4-CNPy)2]+, the EPR signals are broad and poorly resolved. As reported recently for the corresponding complexes of (TMP)Fe(III),33-35 these two complexes may not have a pure (dxy)2(dxzdyz)3 or (dxy)1(dxzdyz)4 ground state, even though their ambient-temperature 1H NMR shifts, discussed above, are consistent with an S = ½ ground state. The same broad EPR signals were found for not only the (TMP(Fe(III) complexes of the same ligands,33,35 but also for (TPP)Fe(III) complexes having 2,6-substituents on the four phenyl rings.34,35
Figure 8.

Temperature-dependent fitting plots of the chemical shifts of the pyrroline- and three pyrrole-H to Equation (1) for (A) [(TPC)Fe(4-Me2NPy)2]+, (B) [(TPC)Fe(Im-d4)2]+, (C) [(TPC)Fe(Py)2]+, and (D) [(TPC)Fe(4-CNPy)2]+. In all plots, ■ represents pyrroline-H, ▼ represents 7,18-pyrrole-H, ▲ represents 12,13-pyrrole-H, and ● represents 8,17-pyrrole-H. The best-fit spin densities and ΔE values are summarized in Table 3.
Temperature dependence of the chemical shifts of low-spin (TPC)Fe(III) complexes
The non- and anti-Curie behavior observed for [(TPC)Fe(Py)2]+ and [(TPC)Fe(4-CNPy)2]+, as well as the deviations of the intercepts of the Curie plots of even the bis-imidazole (this work) and -1-methylimidazole27 complexes, indicate the existence of a thermally-accessible excited state for all of these (TPC)Fe(III) complexes. Two S = 1/2 electron configurations are possible for iron(III) porphyrinates and chlorinates, the more commonly-observed (dxy)2(dxzdyz)3 and the now not-so-uncommon (dxy)1(dxzdyz)4 electron configuration.41,48 In addition, a thermally-accessible excited state having S = 3/2 or S = 5/2 is also possible. As we have shown previously,35-38,43-45 a number of ferriheme complexes have a thermally-accessible excited state that causes at least some curvature of the Curie plot, and can sometimes show extremely curved chemical shift dependence.35,36,44 Expansion of the Curie law to include the contribution from the Boltzmann population of this thermally-accessible excited state yields the following expression,43 if the 1/T2 contribution to the intermediate- or high-spin excited state is neglected:46-49
| (1) |
In this equation Cn1 is the coefficient for the position of interest for level 1, Cn2 is the corresponding coefficient for level 2, W1 and W2 are the statistical weights for each level (= 2S + 1 for each), and ΔE is the energy separation between the two levels. The coefficients Cn1 and Cn2 are approximately equal to the Curie coefficients of each level (except for the small contribution of the pseudocontact contribution to the paramagnetic shifts47,48), and they can be determined by fitting the temperature dependence of the proton chemical shifts, assuming that the diamagnetic shift of each proton type is known. The coefficients Cn1 and Cn2 can be further subdivided into the McConnell constant QC and the variable ρC, where QC = −496.8 and 591.4 ppm K for protons attached to aromatic and aliphatic carbons, respectively,47,50-52 and ρC is the spin density at the carbon of interest; in general ρC is not larger than about 0.03 for the systems of this study. A program that carries out this fitting procedure, with least-squares minimization of the errors between experimental and calculated shifts, has been created in our laboratory and is available on the Internet.53 By entering the diamagnetic shifts of the free-base ligand for each proton type (Table 1) and the contact shift versus inverse temperature data for each complex into the two-level fitting program, specifying the ground state spin as S = 1/2, and testing each possible spin S for the excited state, ρC1, ρC2 and ΔE for each spin choice were calculated. Choosing S = 1/2 for the excited state as well as the ground state (to cover the possibility that the ground state electron configuration was (dxy)2(dxz,dyz)3 while the excited state electron configuration was (dxy)1(dxz,dyz)4 or vice versa) did not lead to convergence of the fits, whereas choosing S = 3/2 or S = 5/2 for the excited state led to convergence in all cases. However, upon viewing the spin densities calculated from the fits, those ρC2 for the excited state S = 5/2 were found to be unreasonably small. Thus, the reasonable spin state was found to be S = 3/2 in each case. The same result was found as in previous studies of most octaalkyltetraphenylporphyrinatoiron(III),36-39 (OEC)Fe(III) and OEPFe(III),30 (TMP)Fe(III)35 and four ((2,6-X2)4TPP)Fe(III)35 series of complexes with axial ligands of varying basicities. The results of the S = 3/2 excited state fits for ρC1, ρC2 and ΔE are summarized in Table 3. Checks for contributions to the temperature dependence from ligand on-off equilibria at near ambient temperatures were made by not including higher-temperature chemical shift data in the fitting. It was found that including data points from the entire temperature range (T = 303 – 183 K) yielded the same ΔE and spin densities ρC1 and ρC2 as if only the T < 253 K data were used for Im-d4 and 4-Me2NPy, but that only T < 253 K chemical shifts could be used for the other three complexes. And because the pseudocontact shifts are small and have not been accurately determined, yet are expected to have the same temperature dependence as the contact shift, the paramagnetic shift instead of contact shift was used for fitting the temperature dependence.
Table 3.
Summary of Results Obtained from the TDFw53 Fits of the Temperature Dependence of the three δ(pyrrole-H) and δ(pyrroline-H) of the [(TPC)Fe(L)2]+ Complexes of this Study.
| System | GSa | ESb | ΔE,c cm−1 |
MSDd | pyrroline-α and β-pyrrole carbon spin densities, ρC,e comments on quality or nature of fit |
|---|---|---|---|---|---|
| L = Im-d4 | 1/2 | 3/2 | 238 | 0.054 | 1: 0.0210, 0.0143 av (0.0064, 0.0308, 0.0057). OK for S = 1/2 dπ/dxy. |
| 2: 0.0117, 0.0127 av (0.0102, 0.0215, 0.0066). OK for S = 3/2. | |||||
| L = 4-Me2NPy | 1/2 | 3/2 | 132 | 0.050 | 1: 0.0152, 0.0121 av (0.0071, 0.0275, 0.0017). OK for S = 1/2 dπ/dxy. |
| 2: 0.0103, 0.0115 av (0.0083, 0.0206, 0.0057). Av. ρC too small for S = 3/2. | |||||
| L = 2-MeHIm | 1/2 | 3/2 | 639 | 0.083 | 1: 0.0091, 0.0098 av (0.0081, 0.0182, 0.0030). OK for S = 1/2 dπ/dxy. |
| 2: 0.0032, 0.0080 av (0.0061, 0.0130, 0.0049). Av. ρC too small for S = 3/2. | |||||
| L = Py | 1/2 | 3/2 | 700 | 0.149 | 1: 0.0032, 0.0042 av (0.0043, 0.0115, −0.0032). OK for S = 1/2 dxy/dπ. |
| 2: 0.0205, 0.0206 av (0.0048, 0.0402, 0.0169). OK for S = 3/2. | |||||
| L = 4-CNPy | 1/2 | 3/2 | 563 | 0.174 | 1: −0.0101, −0.0010 av (0.0011, 0.0011, −0.0050). OK for S = 1/2 dxy. |
| 2: 0.0335, 0.0133 av (0.0060, 0.0386, 0.0029). OK for S = 3/2. |
Ground state spin (S); electron configuration may be (dxy)2(dxz,dyz)3 (abbreviated dπ), (dxz,dyz)4(dxy)1 (abbreviated dxy), or a mixture of the two, as marked in the comments at the far right.
Excited state spin (S); all systems appear to have the electron configuration (dxz,dyz)3(dxy)1(dz2)1.
Best fit energy difference between ground and excited state (±30%).
MSD = mean-square deviation of the data points from the best fit.
Spin densities, ρC, obtained from fits, using the program TDFw, to Equation (1); 1 = ground state, 2 = excited state. Spin densities are given in the order pyrroline-H, average of three pyrrole-H, with individual pyrrole-H spin densities in parentheses in the order 7,18; 8,17; 12,13. Comments indicate how the spin density distribution is interpreted in terms of the sizes of the coefficients for each pyrrole position.
In Figure 8 are shown the Curie plots of the pyrroline and three pyrrole-H of the (TPC)Fe(III) complexes with 4-Me2NPy, Im-d4, Py and 4-CNPy ligands. In Figure 8A and B, respectively, the proton resonances of [(TPC)Fe(4-Me2NPy)2]+ and [(TPC)Fe(Im-d4)2]+ exhibit approximate Curie behavior, which, before the advent of the 2-level fitting procedure43 would have been passed off as simple Curie behavior, even though the intercepts of such linear plots were not quite the same as the diamagnetic shifts. In contrast, in Figure 8D, the proton resonances of [(TPC)Fe(4-CNPy)2]+ show anti-Curie behavior. [(TPC)Fe(Py-d5)2]+ shows behavior in between these extremes. Table 3 summarizes the fitting results for all complexes of this study. Both [(TPC)Fe(4-CNPy)2]+ and [(TPC)Fe(Py)2]+ are seen to have a ground state with much smaller spin densities on all pyrrole and pyrroline carbons, but most especially on the 12,13- and 1,4-carbons than for [(TPC)Fe(4-Me2NPy)2]+ and [(TPC)Fe(Im-d4)2]+, and an excited state with large spin densities on these two sets of carbons. This is consistent with the ground state of these two complexes having a largely (dxzdyz)4(dxy)1 electron configuration, probably a mixture of (dxy)2(dxz,dyz)3 and (dxz,dyz)4(dxy)1 for the former and quite pure (dxz,dyz)4(dxy)1 for the latter. The excited state has S = 3/2 with a probable (dxzdyz)3(dxy)1(dz2)1 electron configuration. As mentioned above, the spin densities obtained for the excited states of all complexes clearly are not consistent with S = 5/2---the ρC values are much too small for the high-spin configuration. An average spin density of 0.014-0.018 has been found for the S = 3/2 excited states of a series of (TMP)Fe(III) and four 2,6-disubstituted (TPP)Fe(III) complexes;35 thus, especially for the bis-2-methylimidazole complex of (TPC)Fe(III), the spin density obtained for the β-pyrrole-carbons is much too small (Table 3), and the fit may be considered unreliable; this is not surprising, considering the dynamics of ring inversion that are probably occurring over the same temperature range,54,55 and may affect the temperature dependence of the resonances.56 The bis-Im-d4 and -4-Me2NPy complexes have (dxy)2(dxzdyz)3 ground state electron configurations and the same S = 3/2 excited state with the same electron configuration, while the pyridine complex has much smaller spin density coefficients than the two just mentioned, yet larger than the 4-CNPy complex. It thus appears to have a mixed ground state configuration of (dxz,dyz)4(dxy)1/(dxy)2-(dxz,dyz)3. Therefore, the fits of the NMR spectra to the expanded version of the Curie law, equation (1), provide more definitive information about the electronic ground state of the low-basicity pyridine complexes than do the EPR spectra of the same complexes discussed above.
NMR spectra and peak assignments of high-spin (TPC)FeCl
Figure 9A shows the 1D 1H NMR spectrum of high-spin (TPC)FeCl in CD2Cl2 at 303 K. Partial peak assignments of pyrroline-H and phenyl-H have been published previously.16 However, the assignments of the three pyrrole-H resonances have not been reported before. The average pyrrole-H chemical shift is close to that of the pyrrole-H in (TPP)FeCl57 (seen as a minor impurity in Figure 9A and 9B(a) because of the ease of re-oxidation of (TPC)FeCl to (TPP)FeCl), indicating that these two S = 5/2 complexes have the same electronic structure.
Figure 9.

(A) 1D 1H NMR spectrum of high-spin (TPC)FeCl in CD2Cl2 at 303 K. The small peak marked with an asterisk arises from the impurity (TPP)FeCl produced during iron insertion. The “m” stands for the meta-H of the meso phenyl groups. (B) Saturation transfer experiments on the mixture of high-spin (TPC)FeCl and low-spin of [(TPC)Fe(4-Me2NPy)2]Cl at 303 K. (a) is the control spectrum. (b), (c) and (d) are difference spectra.
It is difficult to assign the three pyrrole proton peaks of high-spin (TPC)FeCl using normal 2D NMR techniques because of their very short relaxation times (T1 ≈ 1∼2 ms). Since full peak assignments (with the help of ADF calculations) have been made on the low-spin species, we can make full assignments for the high-spin species from the information provided by chemical exchange between the low- and high-spin species30,58 by saturation transfer experiments. As reported previously,30,58 the method is to make a mixture of low-spin and high-spin complexes by adding less axial ligand than necessary for full conversion of all of the high-spin complex to low-spin, 2 equivalents if the equilibrium constant for ligand binding is very large. As a result, only part of the (TPC)FeCl will be converted to the low-spin species (e.g., if (TPC)FeCl:axial ligand ≈ 1:1, at best, the ratio of concentrations of (TPC)FeCl:[(TPC)Fe(L)2]+ ≈ 1:1). Fortunately, as for other Fe(III) porphyrinates and chlorinates,30 no evidence of mono-axial ligand complexes in the solution is detected in the NMR spectra. By irradiating, one by one, the pyrrole-H resonances of (TPC)FeCl and observing the corresponding resonances of the low-spin species in the difference spectrum, which arise from chemical exchange, the peaks of (TPC)FeCl can be assigned.
The axial ligand used for saturation transfer studies cannot bind too strongly (as does imidazole) or too weakly (as does 4-CNPy), because chemical exchange between high- and low-spin complexes will either not be observed or will lead to severe broadening of the low-spin complex resonances, respectively, in these cases. We found that 4-Me2NPy is an appropriate ligand for the saturation transfer experiments; pyridine would also work for this purpose. Figure 9B shows the control and all difference spectra from saturation transfer experiments on the mixture of (TPC)FeCl and [(TPC)Fe(4-Me2NPy)2]Cl. The assignments of the three pyrrole-H peaks are given on the control spectrum.
In Figure 9B, trace (b), besides the peak irradiated (pyrrole-7,18) and the corresponding pyrrole-7,18 proton peak of the low-spin complex, there is another negative peak observed, which is at the position of the ortho-H of the bound axial ligand 4-Me2NPy. Actually, if the pyridine ortho-H resonance of the complex is irradiated, the difference spectrum also contains a negative peak appearing at the position of the pyrrole-7,18 protons of (TPC)FeCl. These signals cannot arise from chemical exchange, nor from an NOE (the NOE difference signal usually has the same sign as the irradiated peak at temperatures above −50 °C for intermediate-sized molecules such as (TPC)FeCl, and its magnitude is much smaller). In fact, even when we used pure (TPC)FeCl and irradiated at the blank position of the chemical shift of the ortho-H peak of the complex (−16 ppm), where there was then no peak, we still obtained a negative peak at 84 ppm (the chemical shift of the pyrrole-7,18-H). This peak disappeared when the same experiment was carried out on the DRX-500, and we have shown conclusively that it is an instrumental artifact of the Unity-300.59 Thus the extra negative-phase peak in saturation transfer difference spectrum (b) of Figure 9B located at −16 ppm is an artifact.
Discussion
Electronic Structure of the Low-Spin (TPC)Fe(III) Complexes
The large negative chemical shifts of the pyrrole-8,17-H and the large positive chemical shifts of the pyrroline-H of [(TPC)Fe(Im-d4)2]Cl indicate that there is an unpaired electron in the predominantly dyz orbital with partial delocalization into the appropriate pi orbitals of the chlorin ring. In this discussion we define a coordinate system centered at the metal atom, with the z axis perpendicular to the mean plane of the chlorin ring and the x axis coinciding with the approximate C2 axis of the molecule that runs from the metal to bisect the two pyrroline carbon atoms (Scheme 1). As found for the pyrrole protons of low-spin [(TPP)Fe(HIm)2]C1,60,61 the chemical shifts of the pyrrole proton resonances are dominated by the contact interaction. This is supported by the ADF calculations reported previously30 and shown in Figures 3, where it is seen that the overlap between the 3e(π)-like and 1a1u(π)-like orbitals of the chlorin ring with the dyz orbital of the iron center30 leads to large spin density on the 8,17-pyrrole and the pyrroline carbons, and smaller spin densities at the pyrrole-7,18 and pyrrole-12,13 positions. The spin densities at the meso carbons are also very small, consistent with the NMR results (the closely-spaced phenyl-H resonances between 6 and 7.7 ppm, Figure 2). Figure 3B shows the spin density distribution calculated for metal orbitals that are relatively more stable than those in Figure 3A. Note that the 7,18-carbon positions have negative spin density, while the 8,17 and 12,13-carbon positions have more positive spin density in comparison to Figure 3A. A relative stability of the metal orbitals intermediate between that obtained in 3A and that obtained in 3B reproduces the order of the chemical shifts of the 8,17-pyrrole, 12,13-pyrrole, and 7,18-pyrrole positions of (TPC)Fe(III) with imidazole ligands.
The ground state and peak assignments of [(TPC)Fe(Im-d4)2]Cl and [(TPC)Fe(2-MeHIm)2]Cl made here are also supported by the ENDOR and ESEEM study42 on the bisimidazole complexes of (TPC)FeCl and (TPP)FeCl. In that work it was found that both [(TPP)Fe(HIm)2]Cl and [(TPC)Fe(HIm)2]Cl have a (dxy)2(dxzdyz)3 ground state, even though this disagrees with the definitions of Taylor based upon g-values.42,62 The spin delocalization pattern obtained in those systems is also consistent with the 1H chemical shifts found in this work.
The decrease in the magnitude of the chemical shifts from [(TPC)Fe(Im-d4)2]Cl to [(TPC)Fe(2-MeHIm)2]Cl might be explained on the basis of the expected trend of the stability of the metal orbitals according to the donor abilities of the axial ligands. The observation that the magnitude of the chemical shifts is smaller in this case is consistent with 2-MeHIm being a stronger donor than HIm or Im-d4. A stronger σ donor destabilizes the metal and places more metal d character and less chlorin character in the singly-occupied molecular orbital (SOMO), as illustrated by comparison of the spin densities of the stronger ligand donor case, Figure 3A, with the weaker σ donor case, Figure 3B. However, although a stronger σ donor toward the proton, 2-MeHIm is “hindered” in binding to iron porphyrinates, as is evidenced by the slightly longer FeNax bonds found for [TPPFe(2-MeHIm)2]+ (2.015(4), 2.010(4) Å),63 [TMPFe(1,2-Me2Im)2]+ (2.004(5), 2.004(5) Å),64 [OMTPPFe(2-MeHIm)2]+ (2.007(7), 2.010(7) Å in one structure, 2.006(5), 2.032(5) Å in another),65 and [OETPPFe(2-MeHIm)2]+ (2.09(2), 2.09(2) Å)66 as compared to those for “non-hindered” imidazole complexes of iron porphyrinates such as [TMPFe(1-MeIm)2]+ (1.975(3) Å for both ligands in one structure and 1.965(3) Å for both in the other),67 paral-[TMPFe(5-MeHIm)2]+ (1.978(6), 1.961(5) Å in one molecule, 1.980(5), 1.985(5) Å in the other),68 perp-[TMPFe(5-MeHIm)2]+ (1.957(6), 1.973(6) Å),68 an average of 0.037 Å longer bond for the 2-MeHIm ligand. While only a small difference, it may be sufficient to weaken the σ-donor strength of 2-MeHIm toward Fe(III) somewhat when the metal is in a highly aromatic macrocyclic ring such as a chlorin or a porphyrin where the Fe-Nax bond lengths must be increased to accommodate the steric requirements of the 2-MeHIm ligands, such that 2-MeHIm is only a slightly stronger σ-donor than Im-d4.
The pyridines present a somewhat different situation from the imidazoles. The intermediate magnitude of the chemical shifts of [(TPC)Fe(4-Me2NPy)2]Cl compared to [(TPC)Fe(Im-d4)2]Cl and [(TPC)Fe(2-MeHIm)2]Cl does not follow from the expected greater donor strength of 4-Me2NPy compared to the imidazoles, and the trend of decreasing magnitude of the chemical shifts with decreasing donor strength of the substituted pyridines is opposite to the trend for the substituted imidazoles, if only σ-basicity is considered.
Considering first the donor strength of 4-Me2NPy as compared to the two imidazoles of this study, four different sets of physical data indicate that 4-Me2NPy is the stronger donor: (1) The pKa of the conjugate acid of the ligand, pKa(BH+), for which 4-Me2NPy is the most basic (pKaBH+) = 9.7, 6.65, 7.56,69 respectively, for 4-Me2NPy, HIm and 2-MeHIm.70,71 (2) The equilibrium constant for binding of two ligands to a given Fe(III) porphyrinate in a common solvent, usually expressed as log β2III, for which 4-Me2NPy has the largest values: For (TPP)Fe(III) in DMF, log β2III = 6.8,72 5.5,73 5.2,72 for 4-Me2NPy, HIm and 2-MeHIm, respectively, although it should be noted that a different anion was used for the HIm measurement (Cl− vs. ClO4− for the other two, even though this should not be an important factor in DMF solution, where the anion is already dissociated and 2 DMF molecules are bound before N-donor axial ligands are added). (3) The energies of the axial ligand σ orbitals, for which that of 4-Me2NPy is less stable (9.1 eV ionization energy from gas-phase photoelectron spectroscopy)74 than those of HIm or 2-MeHIm (∼10.30 and ∼10.08 eV, respectively).75 (4) The energies of the axial ligand π-donor orbitals, for which the symmetric π-donor orbital of 4-Me2NPy is less stable in energy (7.82 eV)74 than the comparable orbitals of HIm and 2-MeHIm (the π orbitals with large electron density on the nitrogen that bears the lone pair ionize in the same region as the lone pair at ∼10.30 and ∼10.08 eV, respectively).75 These four sets of data are consistent in indicating that 4-Me2NPy is both a stronger sigma and a stronger pi donor than either of the two imidazoles, in contrast to the intermediate magnitude of the 1H NMR chemical shifts of the 4-Me2NPy complex relative to the two imidazole complexes.
The lack of correlation between the sigma or pi donor strengths of these ligands and the magnitudes of the chemical shifts may suggest that (a) 4-Me2NPy has greater π acceptor ability than the imidazoles to stabilize the metal orbitals, which counters the effect of the σ donor ability, that (b) the π-donor ability of 4-Me2NPy is greater than that of either of the imidazoles, thus increasing the spin delocalization to the pyridine ligand through ligand to metal pi donation at the expense of spin delocalization to the chlorin ring, or that (c) the pyridines induce a slight geometry change in the chlorin ring that alters the electronic interactions and energies. These three factors may not be independent. With regard to π-acceptor abilities, photoelectron spectroscopy studies in our laboratories of CpMn(CO)2L complexes, where L is a variety of substituted pyridines, show that the π-acceptor abilities of a series of pyridines have a significant effect in stabilizing the metal orbitals.76 The π-acceptor abilities of pyridines and imidazoles have been mentioned in other contexts also,77-91 but there is no clear evidence of the relative π-acceptor abilities of these ligands to stabilize metal orbitals. The π-acceptor abilities of 4-Me2NPy and the imidazoles with the Fe(III) center of these molecules are expected to be much smaller than observed in the CpMn(CO)2L molecules, and probably insufficient to explain the trends, although further investigation is needed.
As for geometry changes of the chlorin ring, the energy required to ruffle the chlorin ring is very small (22 cm−1 calculated for (OMC)Fe(MeNC)2, where OMC = octamethylchlorin).30 The low frequency of the ruffle vibrational mode and the similar energies of the (dxy)2(dxzdyz)3 and (dxzdyz)4(dxy)1 configurations assist vibronic coupling of these configurations in the ground state wave function.30 The steric tendency of axial ligands to cause ruffling of the TPC ring is expected to increase in the order Im-d4 < 4-Me2NPy < 2-MeHIm, since 5-membered ring imidazoles have their α-hydrogens moved considerably away from interference with the porphyrin π system than do 6-membered ring pyridine ligands. This is the same order as the decreasing magnitude of the chemical shifts among these three strong-field ligands. Furthermore, the contribution of the (dxzdyz)4(dxy)1 electron configuration to the ground state is promoted not only by the ruffling of the chlorin ring, but also by π stabilization of the dxzdyz orbitals relative to the dxy orbital by the axial ligands. In previous studies of low-spin Fe(III) tetraphenylporphyrin (TPP) and tetramesitylporphyrin (TMP) complexes33-35 with 4-CNPy ligands, the ground state of the complexes was found to be (dxzdyz)4(dxy)1, as shown for the chlorin complex in Figure 3C for the ruffled geometry. In this case, the spin density at the pyrrole positions is much smaller than expected for the (dxy)2(dxzdyz)3 configuration. The trends for the pyrrole-H chemical shifts of the substituted pyridine complexes in this study are consistent with increasing contribution of the spin density represented by the (dxzdyz)4(dxy)1 configuration, as shown in Figure 3C, in the ground state as the σ-donor ability of the pyridine decreases and the π-acceptor ability correspondingly increases.
For [(TPC)Fe(4-CNPy)2]+, which is the weakest sigma donor and the strongest pi acceptor of the ligands in this study, the chemical shifts indicate that the ground state wave function is now dominated by the spin density distribution in Figure 3C, with the (dxzdyz)4(dxy)1 configuration and the ruffled geometry. The most negative pyrrole-H resonance (also the 8,17-H) at −40 °C is at −4.5 ppm, whereas it is at −46.5 ppm for [(TPC)Fe(Im-d4)2]Cl at the same temperature. There are two additional differences between the 1H NMR spectra of these two complexes: (i) while the six expected phenyl-H resonances of [(TPC)Fe(Im-d4)2]+ are clustered between 6 and 7.7 ppm, those of [(TPC)Fe(4-CNPy)2]+ are well-resolved, with the two types of m-H (13.2, 12.2 ppm) having more positive chemical shifts than those of the o-H (3.0, 2.1 ppm) and p-H (4.9, 4.8 ppm), which thus yields δm − δo and δm − δp chemical shift differences that are positive; and (ii) while the pyrroline-H of [(TPC)Fe(Im-d4)2]+ have a very positive chemical shift (+44.9 ppm at −40 °C), those of [(TPC)Fe(4-CNPy)2]+ have a negative chemical shift (−6.5 ppm at −40 °C). A negative chemical shift for the pyrroline-H is opposite the expected sign of the contact shift for the protons attached to an aliphatic carbon that is attached to a carbon that is part of the π system of the macrocycle.47,48
With regard to (i), the positive chemical shift differences δm − δo and δm − δp indicate that there is significant positive spin density at the meso-carbons,41 as is consistent with spin delocalization from the metal dxy orbital to the 3a2u(π)-like orbital of a ruffled TPC ring, Figure 3C. And with regard to (ii), the negative chemical shift of the pyrroline-H of [(TPC)Fe(4-CNPy)2]+ indicates negative spin density at the α-carbons of the pyrroline ring.30,48 As shown in Figure 3C, ADF calculations have shown that for a ruffled macrocycle conformation, there is negative spin density at the α-carbons of the pyrroline ring.30 The magnitude of this negative spin density is smaller than that reported for [(TPC)Fe(t-BuNC)2]+, which has a chemical shift of −36 ppm for the pyrroline-H at +25 °C, or about −42 ppm at −40 °C.26 Thus, the “purity” of the (dxz,dyz)4(dxy)1 ground state of [(TPC)Fe(4-CNPy)2]+ is considerably less than that of [(TPC)Fe(t-BuNC)2]+. This is also consistent with the EPR spectra of the two, very broad for [(TPC)Fe(4-CNPy)2]+, with g ∼ 2.68, 2.36, 1.7-1.6 or smaller (Figure 7), but very sharp for [(TPC)Fe(t-BuNC)2]+, with g⊥ = 2.15, g∥ = 1.97.26
Figure 7.
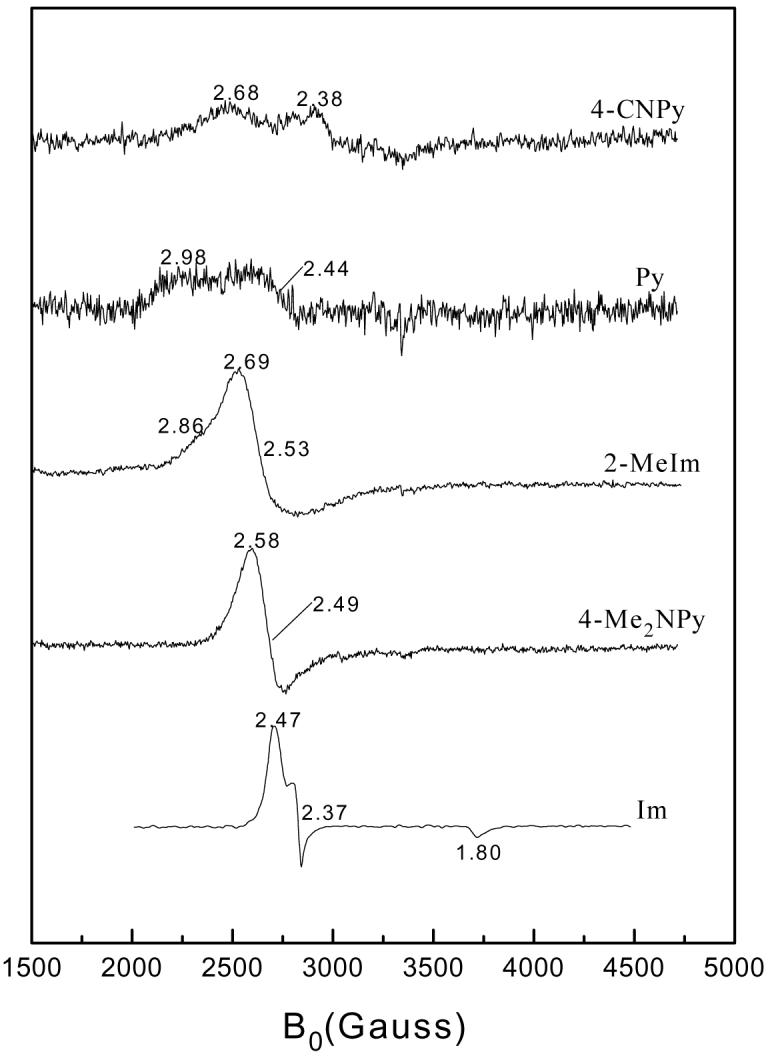
EPR spectra of low-spin (TPC)Fe(III) complexes with the axial ligands of this study.
It should be noted that the trends observed in this work for the magnitudes of the chemical shifts of the pyrroline- and pyrrole-H of the [(TPC)Fe(L)2]+ complexes as a function of ligand L (Im-d4 > 4-Me2NPy > 2-MeHIm > Py > 4-CNPy) are opposite those observed earlier for the corresponding [(OEC)Fe(L)2]+ complexes, where the pyrrole-CH2 chemical shifts increase in the order Im-d4 < 4-Me2NPy < Py < 4-CNPy and the pyrroline-H chemical shifts increase in the same order.30 In the case of the octaethylchlorin complexes of pyridine and 4-cyanopyridine, it was found in that study that the spin state over the NMR temperature range was not S = 1/2, but rather either S = 3/2 or S = 5/2.30 That alkyl-substituted porphyrin complexes of lower-basicity pyridine ligands are often not low-spin at either ambient or low temperatures has been shown previously.36,92-95 However, all of the TPC complexes of this study and those reported previously25-28 have been found to have S = 1/2 over the temperature range of the NMR measurements, albeit with a thermally-accessible excited state of S = 3/2 for the complexes of this study, and probably the others as well.
Electronic Structure of High-Spin (TPC)FeCl
(TPC)FeCl and (TPP)FeCl are similar in electronic structure; in both the chemical shifts of the pyrrole protons are dominated by the (positive) contact shifts that result from σ spin delocalization, with a smaller contact shift contribution (negative) arising from π spin delocalization. The average chemical shift of the three pyrrole-H resonances of high-spin (TPC)FeCl (+73 ppm) is fairly similar to the chemical shift of the (TPP)FeCl pyrrole-H (+79 ppm). This suggests that the amount of spin density delocalized from the iron center to the chlorin ring is nearly the same as that delocalized to the porphyrin ring in (TPP)FeCl, but that it is distributed quite asymmetrically among the three pyrrole positions of the chlorin ring.16 The order of the three pyrrole-H chemical shifts of (TPC)FeCl is 7,18 > 12,13 > 8,17, with the pyrrole-7,18 the most positive. This is the same as the order of the three pyrrole-H chemical shifts of [(TPC)Fe(Im-d4)2]Cl, with pyrrole-8,17 the most negative. This indicates that the π spin delocalization mechanism is the same for [(TPC)Fe(Im-d4)2]Cl and (TPC)FeCl, since the σ-delocalization contribution that results from the dx2-y2 electron should be equal for all pyrrole-carbons. From the order of the pyrrole-H chemical shifts we can conclude that high-spin (TPC)FeCl also has larger π spin density on the pyrrole-8,17 carbons, leading to larger (in magnitude) negative π contact shift and thus smaller overall chemical shift. For the pyrrole-7,18-H the observed chemical shift is the largest of the three, and thus the π spin density and the contact shift (in magnitude) from this unpaired π spin are the smallest of the three pyrrole-H resonances. As in [(TPC)Fe(Im-d4)2]Cl, it is also the 3e(π)-like orbital that is involved in the interaction with the appropriate dπ orbital of the metal center and thus contribute to the π spin distribution.
The small negative chemical shift of the pyrroline-H is probably completely a result of the pseudocontact contribution to the paramagnetic shift, which is expected to be negative,46-49 indicating essentially zero contact shift for these protons. This suggests essentially zero π spin density at the pyrroline α-carbons.
Conclusions
The electronic structure of the low-spin [(TPC)Fe(L)2]+ complexes with L = imidazoles is predominantly the (dxy)2(dxzdyz)3 configuration, and the extent of spin delocalization to the TPC ring depends mostly on the σ- and π-donor ability of the axial ligand. This is a result of σ- and π-donor effects that modulate the d-orbital energies of the metal, such that 4-Me2NPy and the non-hindered imidazole that are strong donors increase the metal character and decrease the ligand character of the spin density. Steric effects (most important for 2-MeHIm, somewhat important for the pyridines, and relatively unimportant for HIm or Im-d4) and distortions of the TPC ring may also play a role. The chemical shifts of the pyrroline and pyrrole-8,17-H resonances of these complexes are dominated by the contact shifts from π spin delocalization which arises from the interaction between the 3e(π)-like orbital of the chlorin ring and the appropriate dπ orbital of the metal center. For L = lower-basicity pyridines the ground state of the metal center tends to change from (dxy)2(dxzdyz)3 to (dxzdyz)4(dxy)1 with weakening of the σ- and π-donor ability and strengthening of the π-acceptor ability of the ligands. The 4-CNPy complex, which represents the weakest sigma donor and strongest pi acceptor, is believed to be ruffled and thus has negative spin density at the α-pyrroline carbons, due to mixing of the a2u-like π orbital of the chlorin in the (dxzdyz)4(dxy)1 configuration.
(TPC)FeCl is a high-spin Fe(III) complex. The chemical shifts of the pyrrole-H are dominated by the contact contribution from σ spin delocalization. The 3e(π)-like orbitals of the chlorin ring are involved in π interaction with the iron center.
Supplementary Material
TOC Graphic. Synopsis.

NMR and EPR spectra of five [(TPC)Fe(L)2]+ complexes (L = Im-d4, 4-NMe2Py, 2-MeHIm, Py-d5 and 4-CNPy) are reported. The 1H NMR shifts indicate a (dxy)2(dxz,dyz)3 ground state for the first two complexes, a mixed ground state for the next two, and a pure (dxz,dyz)4(dxy)1 ground state for the 4-CNPy complex, with significant ruffling of the chlorin ring. The temperature dependence of all complexes is consistent with a (dxz,dyz)3(dxy)1(dz2)1 excited state.
Table 2.
1H Chemical shifts of the [(TPC)Fe(L)2]+ Complexes of This Study in CD2Cl2 at 233 K.
| Ligand L | Pyrroline-H | 8,17-H | 12,13-H | 7,18-H | o-H | m-H | p-H |
|---|---|---|---|---|---|---|---|
| Imidazole-d4 | 44.9 | −46.5 | −9.8 | −4.8 | ∼+7.7 | ∼+6.7 | ∼+6.0 |
| 4-Dimethylaminopyridine | 34.0 | −39.8 | −8.7 | −1.0 | ∼+7.7 | ∼+6.7 | ∼+6.0 |
| 2-Methylimidazole | 26.2 | −29.2 | −8.7 | +1.6 | ∼+7.7 | ∼+6.7 | ∼+6.0 |
| Pyridine | 15.1 | −19.8 | −1.0 | +12.4 | +5.2, +5.7 | +9.8, +9.6 | +6.5, +6.7 |
| 4-Cyanopyridine | −6.5 | −4.5 | +4.5 | +16.7 | +3.3, +2.2 | +13.3, +12.3 | +4.9, +4.8 |
| t-Butylisocyanidea | ∼−42a | ∼+7a,b | ∼+5a,b | ∼+12a,b | ∼−3a | ∼+17a | ∼+1a |
Acknowledgments
The National Institutes of Health, grant DK-31038 (FAW), and National Science Foundation, grant CHE0416004 (DLL) are gratefully acknowledged.
Footnotes
Supporting Information Available: Figures S1-S12 are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jünemann S. Biochim. Biophys. Acta. 1997;1321:107–127. doi: 10.1016/s0005-2728(97)00046-7. [DOI] [PubMed] [Google Scholar]
- 2.Jasaitis A, Borisov VB, Belevich NP, Morgan JE, Konstantinov AA, Verhovsky MI. Biochemistry. 2000;39:13800–13809. doi: 10.1021/bi001165n. [DOI] [PubMed] [Google Scholar]
- 3.Lorence RM, Koland JG, Gennis RB. Biochemistry. 1986;25:2314–2321. doi: 10.1021/bi00357a003. [DOI] [PubMed] [Google Scholar]
- 4.Sotiriou C, Chang CK. J. Am. Chem. Soc. 1988;110:2264–2270. [Google Scholar]
- 5.Bravo J, Mate MJ, Schneider T, Switala J, Wilson K, Loewen PC, Fita I. Proteins, Struct. Funct. Genet. 1999;34:155–166. doi: 10.1002/(sici)1097-0134(19990201)34:2<155::aid-prot1>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 6.Loewen P. Gene. 1996;179:39–44. doi: 10.1016/s0378-1119(96)00321-6. [DOI] [PubMed] [Google Scholar]
- 7.Chiu JT, Loewen PC, Switala J, Gennis RB, Timkovich R. J. Am. Chem. Soc. 1989;111:7046–7050. [Google Scholar]
- 8.Berzofsky JA, Peisach J, Horecker BL. J. Biol. Chem. 1972;247:3783–3791. [PubMed] [Google Scholar]
- 9.Chatfield MJ, La Mar GN, Kauten RJ. Biochemistry. 1987;26:6939–6950. doi: 10.1021/bi00396a013. [DOI] [PubMed] [Google Scholar]
- 10.Chang CK, Wu W. J. Biol. Chem. 1986;261:8593–8596. [PubMed] [Google Scholar]
- 11.Crane BR, Siegel LM, Getzoff ED. Biochemistry. 1997;36:12101–12119. doi: 10.1021/bi971065q. [DOI] [PubMed] [Google Scholar]
- 12.Stroupe ME, Leech HK, Daniels DS, Warren MJ, Getzoff ED. Nature Struct. Biol. 2003;10:1064–1073. doi: 10.1038/nsb1007. [DOI] [PubMed] [Google Scholar]
- 13.Stolzenberg AM, Strauss SH, Holm RH. J. Am. Chem. Soc. 1981;103:4763–4778. [Google Scholar]
- 14.Strauss SH, Pawlik MJ. Inorg. Chem. 1986;25:1921–1923. [Google Scholar]
- 15.Dixon DW, Woehler S, Hong X, Stolzenberg AM. Inorg. Chem. 1988;27:3682–3685. [Google Scholar]
- 16.Pawlik MJ, Miller PK, Sullivan EP, Levstik MA, Almond DA, Strauss SH. J. Am. Chem. Soc. 1988;110:3007–3012. [Google Scholar]
- 17.Sullivan EP, Jr., Grantham JD, Thomas CS, Strauss SH. J. Am. Chem. Soc. 1991;113:5264–5270. [Google Scholar]
- 18.Holm RH, Kennepohl P, Solomon EI. Chem. Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 19.Licoccia S, Chatfield MJ, La Mar GN, Smith KM, Mansfield KE, Anderson RR. J. Am. Chem. Soc. 1989;111:6087–6093. [Google Scholar]
- 20.Feng D, Ting Y-S, Ryan MS. Inorg. Chem. 1985;24:612–617. [Google Scholar]
- 21.Ozawa S, Watanabe Y, Morishima I. Inorg. Chem. 1992;31:4042–4043. [Google Scholar]
- 22.Ozawa S, Watanabe Y, Morishima I. Inorg. Chem. 1994;33:306–313. [Google Scholar]
- 23.Ozawa S, Watanabe Y, Nakashima S, Kitagawa T, Morishima I. J. Am. Chem. Soc. 1994;116:634–641. [Google Scholar]
- 24.Ozawa S, Watanabe Y, Morishima I. J. Am. Chem. Soc. 1994;116:5832–5838. [Google Scholar]
- 25.Kobeissi M, Toupet L, Simonneaux G. Inorg. Chem. 2001;40:4494–4499. doi: 10.1021/ic0013328. [DOI] [PubMed] [Google Scholar]
- 26.Simonneaux G, Kobeissi M. Dalton Trans. 2001:1587–1592. [Google Scholar]
- 27.Simonneaux G, Kobeissi M, Toupet L. Dalton Trans. 2002:4011–4016. [Google Scholar]
- 28.Kobeissi M, Simonneaux G. Inorg. Chim. Acta. 2003;343:18–26. [Google Scholar]
- 29.Simonneaux G, Kobeissi M, Toupet L. Inorg. Chem. 2003;42:1644–1651. doi: 10.1021/ic026039h. [DOI] [PubMed] [Google Scholar]
- 30.Cai S, Lichtenberger DL, Walker FA. Inorg. Chem. 2005;44:1890–1903. doi: 10.1021/ic0490876. [DOI] [PubMed] [Google Scholar]
- 31.Cai S, Belikova E, Yatsunyk L, Stolzenberg AM, Walker FA. Inorg. Chem. 2005;44:1882–1889. doi: 10.1021/ic049088y. [DOI] [PubMed] [Google Scholar]
- 32.La Mar GN, Del Gaudio J, Frye JS. Biochim. Biophys. Acta. 1977;498:422–435. doi: 10.1016/0304-4165(77)90280-x. [DOI] [PubMed] [Google Scholar]
- 33.Safo MK, Gupta GP, Watson CT, Simonis U, Walker FA, Scheidt WR. J. Am. Chem. Soc. 1992;114:7066–7075. [Google Scholar]
- 34.Watson CT. University of Arizona; 1996. Ph. D. Dissertation. [Google Scholar]
- 35.Watson CT, Cai S, Shokhirev NV, Walker FA. Inorg. Chem. 2005;44:7468–7484. doi: 10.1021/ic0507316. [DOI] [PubMed] [Google Scholar]
- 36.Yatsunyk LA, Walker FA. Inorg. Chem. 2004;43:757–777. doi: 10.1021/ic035010q. [DOI] [PubMed] [Google Scholar]
- 37.Yatsunyk LA, Walker FA. Inorg. Chem. 2004;43:4341–4352. doi: 10.1021/ic035242p. [DOI] [PubMed] [Google Scholar]
- 38.Yatsunyk LA, Shokhirev NV, Walker FA. Inorg. Chem. 2005;44:2848–2866. doi: 10.1021/ic049089q. [DOI] [PubMed] [Google Scholar]
- 39.Yatsunyk LA, Walker FA. J. Porphyrins Phthalocyanines. 2005;9:214–228. [Google Scholar]
- 40.Nesset MJM. University of Arizona; 1994. pp. 83–88. Ph. D. Dissertation. [Google Scholar]
- 41.Walker FA. Inorg. Chem. 2003;42:4526–4544. doi: 10.1021/ic026245p. [DOI] [PubMed] [Google Scholar]
- 42.Astashkin AV, Raitsimring AM, Walker FA. J. Am. Chem. Soc. 2001;123:1905. doi: 10.1021/ja002777y. [DOI] [PubMed] [Google Scholar]
- 43.Shokhirev NV, Walker FA. J. Phys. Chem. 1995;99:17795–17804. [Google Scholar]
- 44.Nesset MJM, Cai S, Shokhireva T. Kh., Shokhirev NV, Jacobson SE, Jayaraj K, Gold A, Walker FA. Inorg. Chem. 2000;39:532–540. doi: 10.1021/ic9907866. [DOI] [PubMed] [Google Scholar]
- 45.Banci L, Bertini I, Luchinat C, Pierattelli R, Shokhirev NV, Walker FA. J. Am. Chem. Soc. 1998;120:8472–8479. [Google Scholar]
- 46.Kurland RJ, McGarvey BR. J. Magn. Reson. 1970;2:286–301. [Google Scholar]
- 47.La Mar GN, Walker FA. NMR Studies of Paramagnetic Metalloporphyrins. In: Dolphin D, editor. The Porphyrins. IV. Academic Press; N.Y.: 1979. pp. 61–157. [Google Scholar]
- 48.Walker FA. In: The Porphyrin Handbook. Kadish KM, Smith KM, Guilard R, editors. V. Academic Press; Burlington, MA: 1999. pp. 81–183. [Google Scholar]
- 49.When S > 1/2 and there is a relatively large zero-field splitting, the pseudocontact term has a C'/T2 dependence and the contact term has a C/T dependence.46-48
- 50.Carrington A, McLachlan AD. Introduction to Magnetic Resonance. Harper and Row; New York: 1967. p. 80. [Google Scholar]
- 51.McLachlan AD. Mol. Phys. 1958;1:233. [Google Scholar]
- 52.Chestnut DB. J. Chem. Phys. 1958;29:43–47. [Google Scholar]
- 53.Shokhirev NV, Walker FA. http://www.shokhirev.com/nikolai/programs/prgsciedu.html.
- 54.Shokhirev NV, Shokhireva T. Kh., Polam JR, Watson CT, Raffii K, Simonis U, Walker FA. J. Phys. Chem. A. 1997;101:2778–2886. [Google Scholar]
- 55.Polam JR, Shokhireva T. Kh., Raffii K, Simonis U, Walker FA. Inorg. Chim. Acta. 1997;263/1-2:109–117. [Google Scholar]
- 56.Yatsunyk LA, Ogura H, Walker FA. Inorg. Chem. 2005;44:2867–2881. doi: 10.1021/ic049090p. [DOI] [PubMed] [Google Scholar]
- 57.La Mar GN, Eaton GR, Holm RH, Walker FA. J. Am. Chem. Soc. 1973;95:63–75. [Google Scholar]
- 58.Shokhireva T. Kh., Shokhirev NV, Walker FA. Biochemistry. 2003;42:679–693. doi: 10.1021/bi026765w. [DOI] [PubMed] [Google Scholar]
- 59.Cai S. University of Arizona; 2001. Ph.D. Dissertation. [Google Scholar]
- 60.Satterlee JD, La Mar GN. J. Am. Chem. Soc. 1976;98:2804–2808. doi: 10.1021/ja00439a027. [DOI] [PubMed] [Google Scholar]
- 61.La Mar GN, Walker FA. J. Am. Chem. Soc. 1973;95:1782–90. doi: 10.1021/ja00787a016. [DOI] [PubMed] [Google Scholar]
- 62.Taylor CPS. Biochim. Biophys. Acta. 1977;491:137–149. doi: 10.1016/0005-2795(77)90049-6. [DOI] [PubMed] [Google Scholar]
- 63.Scheidt WR, Kirner JF, Hoard JL, Reed CA. J. Am. Chem. Soc. 1987;109:1963–1968. [Google Scholar]
- 64.Munro OQ, Marques HM, Debrunner PG, Mohanrao K, Scheidt WR. J. Am. Chem. Soc. 1995;117:935–954. [Google Scholar]
- 65.Yatsunyk LA, Carducci MD, Walker FA. J. Am. Chem. Soc. 2003;125:15986–16005. doi: 10.1021/ja036398r. [DOI] [PubMed] [Google Scholar]
- 66.Ogura H, Yatsunyk L, Medforth CJ, Smith KM, Barkigia KM, Renner MW, Melamed D, Walker FA. J. Am. Chem. Soc. 2001;123:6564–6578. doi: 10.1021/ja004053s. [DOI] [PubMed] [Google Scholar]
- 67.Safo MK, Gupta GP, Walker FA, Scheidt WR. J. Am. Chem. Soc. 1991;113:5497–5510. [Google Scholar]
- 68.Munro OQ, Serth-Guzzo JA, Turowska-Tyrk I, Mohanrao K, Walker FA, Debrunner PG, Scheidt WR. J. Am. Chem. Soc. 1999;121:11144–11155. [Google Scholar]
- 69.The two imidazole pKa(BH+) values have been corrected for the statistical factor that results from the two protons present in the protonated form.
- 70.Albert A. In: Physical Methods in Heterocyclic Chemistry. Katritzky AR, editor. I. Academic Press; New York: 1971. pp. 1–108. [Google Scholar]
- 71.Albert A. In: Physical Methods in Heterocyclic Chemistry. Katritzky AR, editor. III. Academic Press; New York: 1971. pp. 1–26. [Google Scholar]
- 72.Nesset MJM, Shokhirev NV, Enemark PD, Jacobson SE, Walker FA. Inorg. Chem. 1996;35:5188–5200. [Google Scholar]
- 73.Walker FA, Lo MW, Ree MT. J. Am. Chem. Soc. 1976;98:5552–5560. doi: 10.1021/ja00434a024. [DOI] [PubMed] [Google Scholar]
- 74.Ramsey BG, Walker FA. J. Am. Chem. Soc. 1974;96:3314–3316. [Google Scholar]
- 75.Ramsey BG. J. Org. Chem. 1979;44:2093–2097. [Google Scholar]
- 76.Weisser JT, Gruhn NE, Walker FA, Lichtenberger DL. Abstracts of Papers, 221st ACS National Meeting; San Diego, CA, United States. 2001; Apr 1-5, INOR-508. CODEN: 69FZD4 Journal; Meeting Abstract. [Google Scholar]
- 77.Nelson SM, Shepherd TM. Inorg. Chem. 1965;4:813–817. [Google Scholar]
- 78.Herberhold M, Alt H. Justus Liebigs Ann. der Chemie. 1976:292–299. [Google Scholar]
- 79.Sutton JE, Taube H. Inorg. Chem. 1981;20:4021–4023. [Google Scholar]
- 80.Ries W, Bernal I, Quast M, Albright TA. Inorg. Chim. Acta. 1984;83:5–15. [Google Scholar]
- 81.Johnson CR, Shepherd RE. Synth. React. Inorg.. Metal-Org. Chem. 1984;14:339–353. [Google Scholar]
- 82.Shepherd RE, Proctor A, Henderson WW, Myser TK. Inorg. Chem. 1987;26:2440–2444. [Google Scholar]
- 83.Aroney MJ, Clarkson RM, Hambley TW, Pierens RK. J. Organomet. Chem. 1992;426:331–342. [Google Scholar]
- 84.Schoenherr T, Degen J. Z. Naturforsch. A. 1990;45:161–168. [Google Scholar]
- 85.Clarke MJ, Bailey V, Doan P, Hiller C, LaChance-Galang KJ, Daghlian H, Mandal S, Bastos CM, Lang D. Inorg. Chem. 1996;35:4896–4903. doi: 10.1021/ic960355c. [DOI] [PubMed] [Google Scholar]
- 86.Asperger S, Cetina-Cizmek B. Croat. Chem. Acta. 1996;69:1305–1328. [Google Scholar]
- 87.Sidorov AA. Zh. Neorgan. Khim. 1997;42:243–246. [Google Scholar]
- 88.Choi J-H. Bull. Korean Chem. Soc. 1999;20:81–84. [Google Scholar]
- 89.Hameed A, Rybarczyk-Pirek A, Zakrzewski J. J. Organomet. Chem. 2002;656:102–107. [Google Scholar]
- 90.Zakrzewski J, Delaire JA, Daniel C, et al. New J. Chem. 2004;28:1514–1519. [Google Scholar]
- 91.Ost TWB, Clark JP, Anderson JLR, Yellowlees LJ, Daff S, Chapman SK. J. Biol. Chem. 2004;279:48876–48882. doi: 10.1074/jbc.M408601200. [DOI] [PubMed] [Google Scholar]
- 92.Scheidt WR, Geiger DK, Hayes RG, Lang G. J. Am. Chem. Soc. 1983;105:2625–2632. [Google Scholar]
- 93.Kintner ET, Dawson JH. Inorg. Chem. 1991;30:4892–4897. [Google Scholar]
- 94.Scheidt WR, Osvath SR, Lee YJ, Reed CA, Shavez B, Gupta GP. Inorg. Chem. 1989;28:1591–1595. [Google Scholar]
- 95.Geiger DK, Scheidt WR. Inorg. Chem. 1984;23:1970. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.