

Abstract
Transgenic mice overexpressing brain-derived neurotrophic factor from the β-actin promoter were tested for behavioral, gross anatomical and physiological abnormalities. Brain-derived neurotrophic factor messenger RNA overexpression was widespread throughout brain. Overexpression declined with age, such that levels of overexpression decreased sharply by nine months. Brain-derived neurotrophic factor transgenic mice had no gross deformities or behavioral abnormalities. However, they showed a significant passive avoidance deficit. This deficit was dependent on continued overexpression, and resolved with age as brain-derived neurotrophic factor transcripts decreased. In addition, the brain-derived neurotrophic factor transgenic mice showed increased seizure severity in response to kainic acid. Hippocampal slices from brain-derived neurotrophic factor transgenic mice showed hyperexcitability in area CA3 and entorhinal cortex, but not in dentate gyrus. Finally, area CA1 long-term potentiation was disrupted, indicating abnormal plasticity.
Our data suggest that overexpression of brain-derived neurotrophic factor in the brain can interfere with normal brain function by causing learning impairments and increased excitability. The results also support the hypothesis that excess brain-derived neurotrophic factor could be pro-convulsant in the limbic system.
Keywords: passive avoidance, long-term potentiation, hippocampus, neurotrophin, epilepsy, entorhinal cortex
Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family of neurotrophic factors, is found in high concentrations throughout the adult brain. It is especially enriched in areas known to be involved in learning, memory, excitability, and plasticity such as the hippocampus and cortex.19,64,85,118 BDNF mRNA has been shown to increase after learning-related events.13,16,83 BDNF can modulate physiological plasticity in CA1 of the hippocampus.28,44,45 BDNF heterozygote knock-out mice, who have reduced levels of BDNF, show reduced long-term potentiation (LTP) in CA1 of the hippocampus.4,50,51,82 Because of its localization and the correlation with activity in learning-related structures, it is possible that BDNF plays a role in behavioral plasticity and excitability.
Mechanistic studies have shown that acute BDNF exposure enhances excitatory transmission (for reviews see Refs 10 and 62). Increases in spontaneous and evoked excitatory post-synaptic currents (EPSCs) were first demonstrated in nerve–muscle co-cultures after BDNF application63 and later shown in multiple systems, such as adult rat hippocampal and visual cortical slices.15,28,44,45,53–55,82 The effects of BDNF appear to be mediated by trk receptors, because they are blocked by a tyrosine kinase antagonist, K252a.15,44,54,95 Presumably, trkB receptors are involved, because BDNF binds trkB selectively and with high affinity.46,107,110
The exact mechanisms of BDNF's actions are unclear. Evidence that BDNF acts presynaptically to increase glutamate release comes from data showing that miniature EPSCs increase in frequency but not amplitude after exposure to BDNF,15,53,63 as well as studies of paired-pulse facilitation (PPF) and responses to trains of afferent stimulation.28,35,44 BDNF appears to phosphorylate synapsin, a presynaptic protein that is involved in synaptic transmission.43 BDNF also stimulates glutamate release from synaptosomes.111 Post-synaptic actions are indicated by the blockade of BDNF's effects by intracellular postsynaptic application of K252a.54 BDNF also phosphorylates N-methyl-d-aspartate receptors which are primarily postsynaptic.57,116 TrkB immunoreactivity is present on axons, dendrites and somata,30,76,119 indicating that trkB may reside at several sites.
Thus, BDNF may be an endogenous modulator of excitability in adult brain. In support of this hypothesis, acute application of exogenous BDNF increases evoked responses at sites of BDNF and trkB expression. In the dentate gyrus, where granule cell axons (mossy fibers) show perhaps the strongest BDNF immunoreactivity in brain,19,91,118 evoked responses to mossy fiber stimulation are extremely sensitive to BDNF.95 The entorhinal cortex, which receives BDNF-immunoreactive afferents, is also sensitive to exogenous BDNF. In both areas, repetitive stimulation of afferents leads to spontaneous activity and spreading depression after BDNF application.95 BDNF also increases excitotoxic damage in brain and cultures.48,91 These data suggest that exposure to BDNF could lead to hyperexcitability. Consistent with this hypothesis, kindling was suppressed in BDNF knockouts.49 A trkB fusion protein (“TrkB-Fc”), which scavenges BDNF, inhibits kindling.11 However, chronic infusion of BDNF has attenuated kindling elsewhere,52,81,90 perhaps owing to chronic changes in BDNF or trkB receptors.
Studies looking at the effects of BDNF on learning or excitability have, to date, relied on the administration of exogenous BDNF to the brain. Unfortunately, BDNF does not distribute well in the brain, and tissue concentrations therefore tend to be extremely high at the infusion site and negligible in more distant areas.5 Therefore, findings of no effect or detrimental effects of BDNF on learning and memory29,84 (S. D. Croll et al., unpublished observations) or excitotoxicity91 could be based on poor diffusion which can result in high local tissue concentrations of BDNF immediately adjacent to the cannula site.
One approach to enhancing tissue levels of BDNF at more physiologically relevant concentrations, but with a wide distribution, is to create animals which overexpress BDNF. These animals synthesize additional BDNF by making it not only from its own promoter, but also from another promoter. Here we report the generation and characterization of a BDNF transgenic mouse overexpressing BDNF from the β-actin promoter. We tested this mouse for learning and memory ability, as well as other behavioral changes. In addition, brains were examined for any gross abnormalities. As a behavioral reflection of increased excitability, motor seizure severity in response to the convulsant kainate was also evaluated. Finally, we examined evoked responses in area CA1, area CA3, dentate gyrus and entorhinal cortex of hippocampal slices.
We hypothesized that endogenous BDNF would increase brain excitability. This increased excitability was expected to alter learning and memory, increase seizure severity, increase neuronal excitability in hippocampal slices and alter hippocampal plasticity.
Experimental Procedures
Creation and gross characterization
Brain-derived neurotrophic factor transgenics
The human β-actin promoter construct was kindly provided by Dr Larry Kedes (Stanford University, refer to Ref. 36). This construct contains 3 kb of the promoter sequences followed by 0.078 kb of 5′UTR, 0.832 kb of the first intron, multiple cloning sites and an SV40 polyadenylation signal. Human BDNF cDNA was modified by polymerase chain reaction to include a consensus Kozak sequence just upstream of the ATG and a myc epitope immediately following the last codon, prior to the stop codon. This modified BDNF (which had been tested in vitro for activity on dorsal root ganglion neurons) was placed into the multiple cloning sites. The final construct was tested in a previously described fibroblast transformation assay,34 to ensure that BDNF was indeed being secreted, before being injected into mice.
Bacterial sequences were removed from the construct, and the DNA was extensively purified and injected into F2 CBA × C57Bl/6 embryos.
Southern analysis
Ten micrograms of tail DNA was digested with BamH1 enzyme, run on a 1% agarose gel and processed using standard techniques.65
Northern analysis
Heart, lung, liver, kidney, spleen, gonads (ovary/testis), muscle, cortex, cerebellum, midbrain, hindbrain, thalamus, striatum, hippocampus and olfactory bulbs were dissected from young adult transgenic mice (two to three months old). Tissues were immediately placed on dry ice and frozen at −80°C. They were then homogenized in 3 M LiCl and 6 M urea.6 Ten micrograms of total RNA was electrophoresed through a 1% agarose–formaldehyde gel. The gel was processed using standard techniques.65 The membrane was probed with a 32P-labeled hBDNF fragment.
In situ hybridization
Brains were collected and embedded in a mold containing OCT embedding medium. The mold containing the tissues was placed on dry ice/ethanol until frozen, and stored at −80°C until ready to cut. Brain sections were cut 10 μm thick, adhered to Probe On Plus microscope slides (Fisher Biotech, Pittsburgh, PA) and stored at −80°C.
The BDNF probe was a 35S-UTP-labeled 800 base pair (bp) fragment from the coding region of the BDNF gene as previously described.64 The trkB probe was a 35S-UTP-labeled 530 bp fragment coding the kinase domain of the full-length TrkB receptor, as previously described.4 In situ hybridization was performed on the tissue as previously described.64 In brief, the riboprobes were hydrolysed to the appropriate length and hybridized on the tissue overnight. Slides were subsequently washed, dehydrated, air dried and placed on autoradiography film. After observing appropriate signal on the autoradiography film, slides were dipped in Kodak NTB-2 emulsion (Rochester, NY) and developed after 10–14 days of exposure. Slides were then counterstained with Hematoxylin and Eosin and coverslipped from xylenes.
Brain-derived neurotrophic factor enzyme-linked immunosorbent assays
Four transgenic (varying zygosity) and four wild-type mice were assayed for BDNF protein levels by enzyme-linked immunosorbent assay (ELISA). Animals were killed by cervical dislocation, and tissue was rapidly dissected and immediately placed in liquid nitrogen. Three brain regions, the hippocampus, striatum and cortex, were collected, as well as two peripheral organs, the heart and liver. All tissues were homogenized and assayed using a modification of the BDNF two-site ELISA procedure previously described.88 Modifications from the original protocol included diluting the tissue homogenization buffer 1:1 with water, performing incubation at room temperature, using Kierkegaard and Perry Labs (KPL) 1% bovine serum albumin (BSA) Block Diluent Solution (Gaithersburg, MD) for blocking and reporting rather than a Tween solution, and substituting KPL Wash buffer for phosphate-buffered saline (PBS)/0.02% Tween in the washes. This ELISA uses a monoclonal capture antibody and a biotinylated rabbit polyclonal antibody (Dr Qiao Yan, Amgen) as the reporter.
Gross characterization
Mice were examined in their home cages by a behaviorist blind to genotype for any overt signs of behavioral abnormalities. In addition, as mice were killed, a gross subjective examination of their anatomy was performed. Finally, brains fixed with 4% paraformaldehyde were collected from transgenics and wild-types. Brains were sectioned at 30 μm horizontally and were stained with Thionin as previously reported for rats.20 An evaluator blind to genotype observed the slides to determine whether there were any gross abnormalities of brain structure.
Subjects
Subjects were BDNF overexpressing mice and their wild-type littermates or wild-type parallel-bred controls created and bred at Regeneron Pharmaceuticals and, for physiology, later transferred to Helen Hayes Hospital. Animals were group-housed with same-sex littermates in standard temperature and humidity-controlled colony conditions at Regeneron, and were individually housed at Helen Hayes Hospital. Food and water were available ad libitum. Animals were maintained on a 12:12-h light:dark cycle (lights on 06.00). All animals were allowed to acclimate to any changes in housing for at least one week before testing. All experiments were conducted in strict compliance with protocols approved by Regeneron's Institutional Animal Care and Use Committee or the New York State Department of Health. All protocols were prepared in accordance with guidelines established by the National Institutes of Health. Every effort was made to ensure the humane and ethical treatment of the animals, and to reduce animal usage.
Behavioral analyses
Eight homozygote transgenics, 12 heterozygote transgenics and 10 wild-types in two separate cohorts were tested at the age of six to eight weeks for performance on various behavioral assays. Three separate naive cohorts consisting of a total of 22 homozygote transgenics, 19 heterozygote transgenics and 16 wild-types were also tested at the age of six to eight months. Not all of the older animals were tested on all tasks, but at least two cohorts of animals were tested on each task, and all of the animals were tested for passive avoidance ability. The task order was counterbalanced to control for order effects. All behavioral tests were conducted between 10.00 and 16.00, with each animal being tested at about the same time each day for each task which ran across multiple days. Animals were placed into the behavioral testing room for approximately 1 h before testing to allow for acclimation to the testing environment. The behavioral experimenter was blind to animal genotype at the time of testing.
Passive avoidance
All animals were tested on a shuttlebox step-through passive avoidance task. During the acquisition trial, animals were placed in the less desirable bright chamber of a two-chamber shuttlebox apparatus (Coulbourn Instruments, Allentown, PA). When animals crossed to the more desirable dark side, a guillotine door between the chambers was automatically shut and the animals received a 0.8 mA, 3 s shock. The latency to cross during this trial was recorded as the latency to spontaneous crossover. Twenty-four hours, 48 h, and one week later mice were again placed in the chamber and the latency to cross to the dark side was recorded as a measure of retention. Animals received no additional shocks during the retention trials. A 3 × 3 (group × delay) mixed factorial ANOVA was used to analyse the crossover latencies. An independent groups ANOVA was used to compare latencies of spontaneous crossover between the groups.
Hot-plate test
Animals were tested on the hot-plate test to evaluate their pain thresholds. Animals were placed on a 50–52°C hot plate, and remained there for 40 s or until they attempted to escape or licked their hindpaws. The latency to respond was recorded, with the maximum latency of 40 s used to prevent tissue damage. An independent groups ANOVA was used to compare the hot-plate latencies across groups.
Swim T-maze
Mice were also trained on the swim T-maze (for example see Ref 108). The swim T-maze was a clear Plexiglass T-maze submerged in opaque water. The mice could not touch the bottom of the maze. A hidden escape platform was located at one side of the T. Animals were placed at the base of the T and were allowed only one arm choice. Animals received one trial per day until they achieved criterion (three consecutive correct trials) or had been trained for 15 trials. Animals were scored by the number of trials it took them to reach criterion. Animals that did not reach criterion received a score of 15. An independent groups ANOVA was used to compare trials to criterion across groups.
Anxiety testing
Because anxiety levels can affect performance on learning tasks, BDNF overexpressing mice and their wild-type littermates were tested on two anxiety tests, the elevated plus maze61 and the staircase test.101 In brief, the elevated plus maze was a plus-shaped maze consisting of two white, open arms and two black arms with high walls. Anxious mice tend to spend more time in the closed than the open arms relative to non-anxious mice. Mice were placed on the plus maze for 5 min, and the time which the animals spent on the open versus closed arms, as well as the number of entries into each type of arm, were recorded. For the staircase test, mice were placed on a small staircase for 3 min and the number of times the animal reared relative to the number of steps climbed was measured. Step climbing is a measure of general activity, whereas rearing is a sign of decreased anxiety.101,113 An independent groups ANOVA was used to compare measures across groups.
Locomotion
Mice were tested for general activity using a gridded table as previously described for rats.20 In addition, the number of steps climbed in the stair climbing task and the total number of arms entered for the elevated plus maze are both measures of general locomotor activity. Independent groups ANOVAs were used to compare the three groups on these measures.
Kainate experiments
Seizure induction
Mice were injected with 30 mg/kg kainic acid i.p. at eight to 10 weeks of age. Animals were observed for behavioral signs of seizures by two experimenters who were blind to genotype. Animals were scored using a modification of Racine's seizure scoring system.86 Specifically, a score of 1 represented behavioral arrest or staring, 2 represented head nodding, gnawing, facial automatisms, or mild tremors, 3 represented unilateral forelimb clonus, 4 represented bilateral forelimb clonus, 5 represented severe seizures with prolonged loss of postural control or prolonged tonus, 6 represented status epilepticus defined as 10 min or more of continuous or closely spaced seizures with no return to normal behavior, 7 represented status epilepticus which included a stage 5 seizure, and 8 represented seizure-induced death. Any animal which had more than one stage 5 seizure in 5 min or reached stage 7 was immediately injected with 10 mg/kg diazepam i.p. All other animals were given diazepam after 1 h of status epilepticus or, if they never reached status, 2.5 h after the kainate injection. The maximum seizure stage attained was recorded, as well as the latency to generalized clonus (stage 4). Seizure scores and latency to clonus were analysed using Student's independent groups t-test.
Histology
One week after kainate injection, animals were killed by exsanguination after receiving an overdose of chloral hydrate/pentobarbital i.p. Animals were perfused with a brief flush of heparinized saline followed by 4% phosphate-buffered paraformaldehyde (pH 7.0), containing 7.5% saturated picric acid (v/v). Brains were removed and placed in a 30% sucrose solution for three to seven days. Once the brains had sunk in sucrose solution, they were sectioned coronally at 30 μm, mounted and stained with Thionin to look for cell damage in the hippocampus after kainate.
General electrophysiology
Slice preparation
Mice were deeply anesthetized with ether and the brain was removed immediately. It was placed in ice-cold artificial cerebrospinal fluid (ACSF) containing sucrose instead of NaCl (sucrose–ACSF, in mM, 126 sucrose, 5.0 KCl, 2.0 CaCl2, 2.0 MgSO4, 26.0 NaHCO3, 1.25 NaH2PO4 and 10.0 d-glucose, pH = 7.4). The hippocampus was cut horizontally into 400-μm-thick slices using a Vibroslice (Stoelting Instruments, Wood Dale, IL), and all slices were immediately placed in a recording chamber (modified from Fine Science Tools Model no. 21000-02, Foster City, CA), where they were semi-submerged, warmed to 32–33°C and oxygenated with 95% O2/5% CO2. After 30 min, perfusion was switched from sucrose–ACSF to ACSF containing NaCl, substituted equimolar for sucrose (NaCl–ACSF). NaCl–ACSF was used to perfuse slices for the rest of the experiment. Recordings were started 30–45 min after the change from sucrose–ACSF to NaCl–ACSF. In the Results section, the number of slices is large because five to 10 slices (both hippocampi were sliced) could be maintained in the recording chamber at once.
Recording and stimulation
Extracellular recordings were made with a 2–4 MΩ resistance glass microelectrode filled with NaCl–ACSF. Monopolar stimulating electrodes were made from Teflon-coated stainless steel wire (75 μm diameter). The stimulating electrode was placed at the slice surface and the recording electrode was 50 μm below the surface. Rectangular current pulses were used for stimulation (10–200 μA, 10 μs) and were triggered with a stimulus isolator (Model A340, World Precision Instruments, Sarasota, FL) and an interval generator (Pulsemaster, World Precision Instruments, Sarasota, FL). Recordings were made with an Axoclamp 2B Amplifier (Axon Instruments, Foster City, CA) and saved on diskette using Nicolet programming (Nicolet Instruments, Model 410, Madison, WI).
Recording and stimulating parameters in different regions
Area CA3
The response to hilar stimulation was recorded in the area CA3 pyramidal cell layer on the border between area CA3b and c. The hilar stimulating electrode was placed just below the granule cell layer at the junction between the upper and lower blades (the “crest”). A half-maximal stimulus strength was used to test responses to repetitive stimulation, which consisted of 10 pairs of stimuli at 1 Hz. Each pair of stimuli was 40 ms apart. If multiple population spikes were evoked by any of the stimuli during this repetitive train, area CA3 was considered to be abnormally excitable. The frequency of hyperexcitable slices in each group was analysed using a chi-square test for 2 × 2 contingency tables. Area CA3 data were not used if a single maximal stimulus, tested prior to repetitive stimulation, did not evoke a response that exceeded 3 mV in peak amplitude, because intracellular recordings from such tissue demonstrated that most neurons were unhealthy.
Entorhinal cortex
The white matter input to the medial entorhinal cortex was tested using a stimulating electrode placed at the white matter/layer VI border of the medial entorhinal cortex. Recordings were made in the deep (layer V, VI) and superficial (layer II, layer III) layers of the adjacent cortex, along a tangent to the pia that intersected the stimulation site.94 In adult rat slices, a single stimulus to the white matter evokes a stereotypical response in each of the layers and contains no more than two components (orthodromic and antidromic) unless a convulsant is added.41,42,89,94 A hyperexcitable response was defined as a field potential with more than two components that was evoked by a single stimulus. The stimulus strength chosen was that which evoked a maximal response in layer VI. The entorhinal cortex was defined as abnormally excitable if multiple field potentials were evoked to this stimulus in both superficial and deep layers. The frequency of hyperexcitable slices in each group was analysed using a chi-square test for 2 × 2 contingency tables. Slices with a maximal response which included an antidromic component less than 5 mV in amplitude and an orthodromic component less than 1 mV were excluded from the study, because intracellular recordings of neurons in such slices showed that most neurons were unhealthy.
Dentate gyrus
To examine the perforant path input to dentate gyrus granule cells, the outer molecular layer was stimulated near the hippocampal fissure, just below the subiculum, while recording at the granule cell layer/hilar border at the crest or center of the dorsal blade. Paired-pulse inhibition was tested using a maximal stimulus intensity and a 20- or 40-ms interstimulus interval. To address paired-pulse inhibition or facilitation, population spike amplitude of the first and second response to two identical stimuli was measured. Population spike amplitude was defined as the average of both rapid deflections that comprise the spike. Repetitive stimulation was tested by triggering pairs of stimuli (40 ms interval) at 1 Hz for a total of 10 pairs of stimuli. Hyperexcitability was defined as a response to a single stimulus with more than one population spike. The dentate gyrus was studied only in those slices in which the response to a maximal stimulus included a “population excitatory postsynaptic potential (EPSP)” (defined as the positive potential upon which the population spike is superimposed) greater than 4 mV.
Area CA1
Recordings were made in area CA1b at a site 50–100 μm below the surface of the slice. The same recording site was used to test responses to Schaffer collateral and stratum lacunosum-moleculare stimulation. In all experiments, responses to single and paired stimuli of both pathways were assessed prior to tetanization (100 Hz, 1 s). Data included in the Results section were from slices that had robust responses to Schaffer collateral stimulation (a maximal population spike > 5 mV). Slices that did not meet this criterion also failed to show robust responses to other stimuli, such as stimulation of stratum lacunosum-moleculare or stratum oriens, and were judged to be unhealthy. Population spike amplitude was defined as the average of the two rapid deflections that comprise the population spike.
To stimulate the Schaffer collaterals, the stimulating electrode was placed on the surface of the slice, approximately 150 μm from the border of stratum pyramidale and stratum radiatum in area CA2. An opaque band was present in this area, reflecting the Schaffer collateral axons, and was used to guide the electrode position. To stimulate stratum lacunosum-moleculare, the stimulating electrode was placed in the center of this layer, approximately 200 μm from the hippocampal fissure, and near the border of CA1 and the subiculum.
Paired-pulse inhibition/facilitation was based on responses to two identical stimuli triggered 20 or 40 ms apart. Paired pulse inhibition was defined by a population spike evoked by a second stimulus that was smaller than the spike evoked by the first stimulus, and PPF was defined by a larger second response. These tests were made using several stimulus intensities and results were consistent regardless of intensity. To compare paired pulse responses in different slices statistically, a half-maximal stimulus was used.
Input–output slope was defined as the maximal increase in population spike amplitude per 10 μA change in stimulus strength. Input–output data were obtained using a 10-μs stimulus duration and current was increased in 10-μA steps between the minimum and maximum responses.
For tetanic stimulation, a 100-Hz train was triggered for 1 s, using a stimulus intensity that was approximately half-maximal. Stimuli were tested before tetanization using < 0.05 Hz frequency for 5–10 min. Potentiation was defined as an increase in amplitude of the population spike greater than two standard deviations of the baseline average. Short-term potentiation (STP) was defined by measurements made 1 min after tetanization. LTP was defined on the basis of amplitude measurements 30 min after tetanization.
Results
Creation and gross characterization of brain-derived neurotrophic factor transgenic mice
Generation of the transgenics
DNA was injected into F2CBA × C57Bl/6 embryos. Thirty-five pups were born, out of which nine were found to be transgenic by Southern analysis. Out of the nine founders, three had multiple integrations and were therefore not used to establish lines. Homozygote mice were generated with two lines that had the transgene integrated at only one site. Both lines had two copies of the transgene. Pilot studies were conducted using animals from both lineages (northern analysis, Southern analysis, behavioral tasks). Both lineages showed overexpression of BDNF in a similar ratio from tissue to tissue, and both lineages had similar behavioral abnormalities (i.e. passive avoidance, activity, etc.) relative to wild-types (data not shown). Because of limited space and resources, the decision was made to continue breeding only one line. All data in this report were collected using animals from the selected lineage. For the behavioral studies, most animals were littermates, although some of the animals were drawn from a first-generation parallel breeding of wild-type-only and homozygote-only breeding pairs in order to increase the sample size in the wild-type and homozygote groups. No difference was noted in the behavior of wild-type or homozygote animals bred in this manner relative to those bred from heterozygote–heterozygote breeding pairs to obtain littermates of mixed genotype. For the physiology experiments, all animals were tested with littermate controls, but the majority of transgenic animals tested was heterozygotes owing to the increased availability of these animals. Both heterozygotes and homozygotes showed the physiological abnormalities reported, although the homozygotes had a more striking phenotype.
Northern analysis
Northern analysis of tissues collected from young adult BDNF transgenic mice showed that the BDNF transgene was expressed in most tissues (Fig. 1). Neither the BDNF transgene nor the endogenous BDNF transcript was observed in liver. The BDNF transgene was observed in all other tissues, with the lowest levels in the kidney, and the highest levels in the heart, muscle and gonads. In the brain, the lowest levels of the transgene were observed in the thalamus, with fairly high levels observed across all other brain regions examined. The greatest increases in BDNF (i.e. transgene relative to endogenous BDNF) appeared to occur in the gonads and muscle in the periphery, and the striatum, cerebellum and hindbrain in the brain. The striatum was the most striking of the brain regions with almost no endogenous BDNF message, but fairly high expression of the BDNF transgene.
Fig. 1.
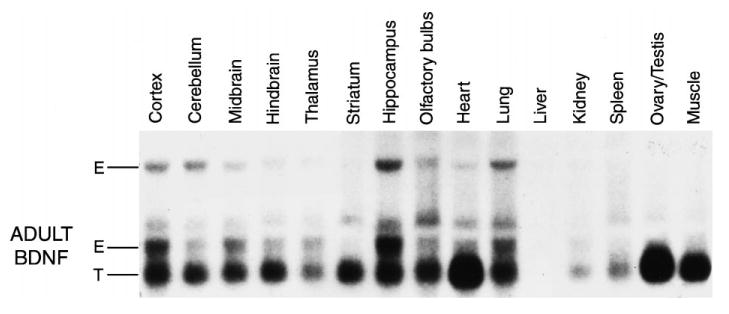
Northern analysis showing the expression of the endogenous BDNF transcript (E) and the BDNF transgene (T) in various tissues collected from two- to three-month-old transgenic mice.
In situ hybridization
In situ hybridization studies were performed on brains from eight-week-old and nine-month-old transgenic and wild-type mice for both BDNF and trkB. The in situ hybridization analysis for BDNF in the brain agreed with the results found by northern analysis. That is, BDNF mRNA in eight-week-old mice was increased fairly ubiquitously, with the least overexpression occurring in the thalamus and the most dramatic overexpression occurring in the striatum (Fig. 2B, compared with wild-type, Fig. 2A). Interestingly, the in situ hybridization results revealed that the expression of the BDNF transgene dropped as the animals went from young adulthood (Fig. 2B) to middle adulthood (Fig. 2C). The trkB in situ hybridization revealed no obvious differences between the wild-type and transgenic mice in any region of the brain (data not shown).
Fig. 2.
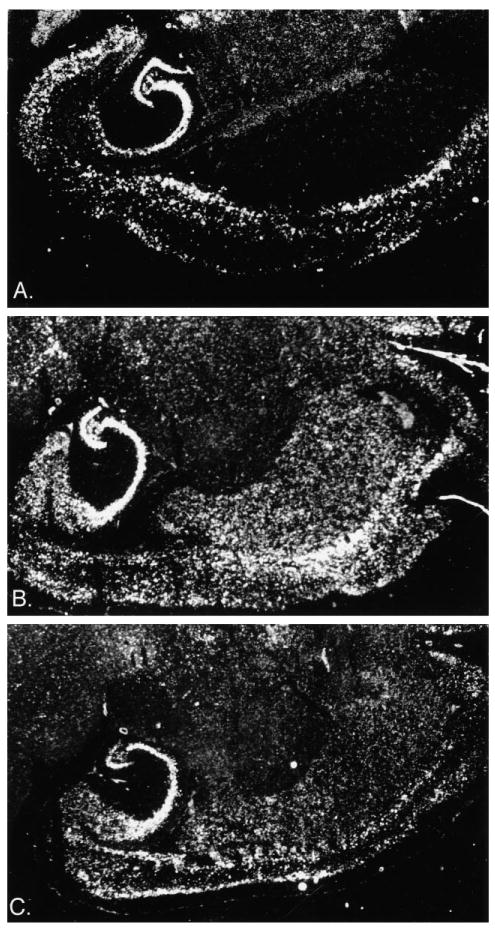
Dark-field photomicrographs of the BDNF in situ hybridization emulsion autoradiography showing BDNF mRNA in the brains of (A) a wild-type, (B) an eight-week-old BDNF transgenic, and (C) a nine-month-old BDNF transgenic.
Brain-derived neurotrophic factor enzyme-linked immunosorbent assays
BDNF ELISAs were performed on four wild-types and four transgenics at approximately eight weeks of age. Tissue was collected from the heart, liver and three brain regions: the striatum, cortex and hippocampus. Mean tissue levels of BDNF are provided in Table 1. Levels of up-regulation were modest and averaged 28%. The striatum and hippocampus showed the most up-regulation at 39% and 32%, respectively. A two-way ANOVA revealed an overall significant BDNF elevation in the transgenics (F1,24 = 7.145, P < 0.037).
Table 1.
Brain-derived neurotrophic factor enzyme-linked immunosorbent assay data for two- to three-month-old brain-derived neurotrophic factor transgenic mice (mixed zygosity) and their wild-type littermates
| Region | Wild-types | Transgenics | % increase |
|---|---|---|---|
| Hippocampus | 44.75 ± 3.82 | 59.00 ± 7.63 | 32% |
| Striatum | 19.75 ± 2.67 | 27.40 ± 5.81 | 39% |
| Cortex | 26.25 ± 3.76 | 28.50 ± 2.51 | 9% |
| Heart | 32.52 ± 7.35 | 41.40 ± 6.28 | 27% |
| Liver | 101.58 ± 10.13 | 130.55 ± 9.49 | 28% |
| Overall mean | 44.97 ± 7.20 | 57.37 ± 9.19 | 28% |
Values are given in ng BDNF/g tissue. Means ± S.E.M. are given for each region. Four BDNF transgenics and four wild-type littermates were used to measure these values. The statistical analysis is included in the Results section.
Gross characterization
BDNF overexpressing mice could not be distinguished from their wild-type littermates on the basis of size, physical appearance or overt behavior. They were viable and fertile. BDNF transgenics had no gross abnormalities in peripheral anatomy, nor did they show any gross abnormalities in brain structure by Thionin stain (data not shown; for hippocampus, see Fig. 4).
Fig. 4.
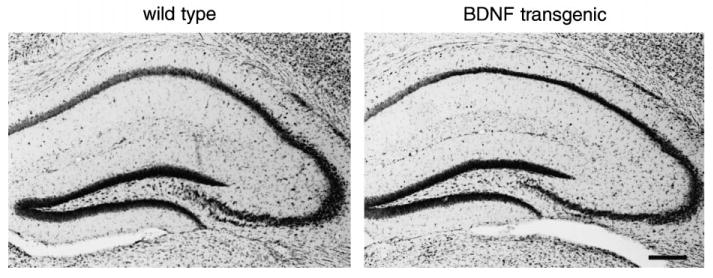
Nissl-stained sections of hippocampus from wild-type and transgenic mice. (Left) A Nissl-stained coronal section from a wild-type mouse. (Right) A Nissl-stained section from a comparable level of a transgenic mouse. Scale bar = 200 μm.
Behavioral characterization
To determine whether the BDNF transgenics were behaviorally different from their wild-type littermates we tested them on tasks evaluating learning, anxiety, pain and locomotion. These behaviors were selected partly on the basis of observations in rats of the effects of exogenous BDNF on pain thresholds (increased; Refs 102 and 103), learning (decreased, unpublished observations) and reactivity (increased, unpublished observations). Because BDNF was increased most substantially in the striatum, we started by testing the animals on a task in which the striatum plays a role, passive avoidance.24,79,92
Passive avoidance
Six- to eight-week-old BDNF homozygote transgenic mice were significantly impaired on passive avoidance retention 24 h, 48 h and one week after acquisition compared with both wild-type and heterozygote mice (group F2,27 = 9.077, P < 0.001, Fig. 3A). The impairment remained consistent and did not change between the delays (delay F2,54 = 0.462, P > 0.64). Heterozygote mice had a latency intermediate to that of the wild-types and homozygotes (Fig. 3A). This deficit is not likely to be accounted for by a tendency to cross more quickly into the more desirable dark chamber, because there was no difference between the groups in spontaneous crossover during the acquisition trial (F2,27 = 0.003, P > 0.99, Table 2). To determine whether the passive avoidance deficit observed at six to eight weeks was dependent on continued high levels of BDNF overexpression, six- to eight-month-old transgenics were also tested on passive avoidance. In contrast to the effect observed in six- to eight-week-old transgenics, six- to eight-month-old transgenics showed no significant passive avoidance deficit (F2,54 = 1.506, P > 0.23, Fig. 3B; spontaneous crossover F2,54 = 0.622, P > 0.54, Table 2).
Fig. 3.
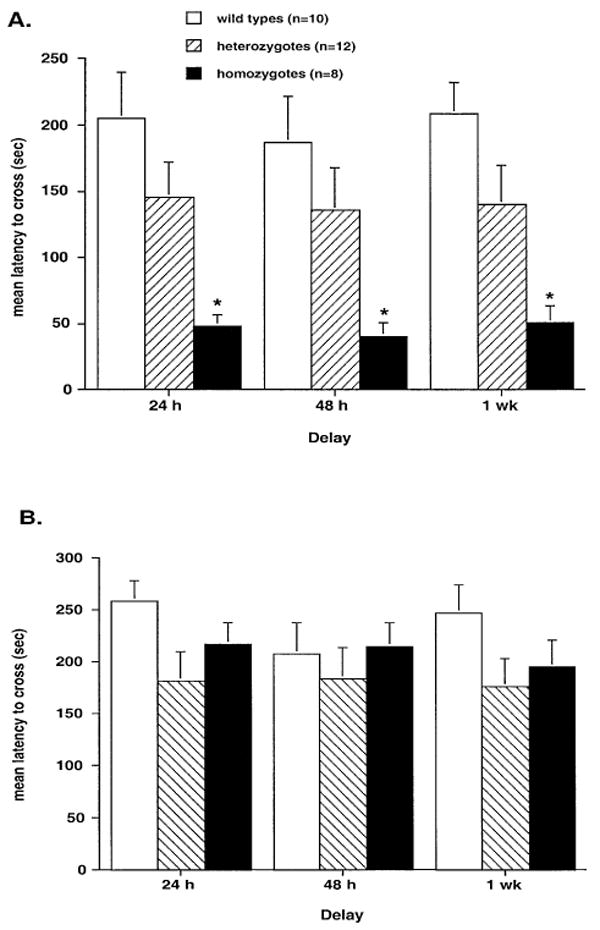
Bar graphs showing passive avoidance retention, expressed as latency to cross, for (A) six- to eight-week-old mice (n = 10 wild-types, 12 heterozygotes, and 8 homozygotes) and (B) six- to eight-month-old mice (n = 16 wild-types, 19 heterozygotes, and 22 homozygotes). *P < 0.05 compared with wild-types, Tukey–Kramer post hoc test.
Table 2.
Means ± S.E.M. for wild-type, heterozygote brain-derived neurotrophic factor overexpressing mice and homozygote brain-derived neurotrophic factor overexpressing mice on all behavioral measures except for passive avoidance retention
| Task | Wild-types | Heterozygote | Homozygote |
|---|---|---|---|
| Passive avoidance spontaneous crossover (s) | 21.8 ± 3.2 | 21.3 ± 5.1 | 21.6 ± 5.6 |
| 48.5 ± 10.7 | 35.0 ± 6.8 | 41.3 ± 7.5 | |
| Hot-plate test: latency (s) | 18.8 ± 3.2 | 18.0 ± 1.8 | 17.6 ± 24 |
| 16.4 ± 2.4 | 17.9 ± 2.0 | 18.1 ± l.4 | |
| Swim T-maze: trials to criterion | 7.7 ± 1.7 | 7.2 ± 0.8 | 9.2 ± 1.8 |
| 5.9 ± 0.5 | 7.1 ± 0.9 | 6.6 ± 0.7 | |
| Grid locomotor: no. of grid crosses | 19.8 ± 3.9 | 22.6 ± 4.5 | 31.5 ± 2.6 |
| 27.2 ± 2.7 | 26.5 ± 2.9 | 26.8 ± 3.6 | |
| Elevated plus maze: | |||
| Proportion of entries into open arms | 0.31 ± 0.08 | 0.25 ± 0.04 | 0.30 ± 0.03 |
| 0.19 ± 0.04 | 0.17 ± 0.04 | 0.25 ± 0.02 | |
| Proportion of time in open arms | 0.19 ± 0.09 | 0.12 ± 0.03 | 0.21 ± 0.04 |
| 0.15 ± 0.05 | 0.10 ± 0.02 | 0.18 ± 0.03 | |
| Total arm entries | 11.1 ± 2.18 | 10.75 ± 1.61 | 19.88 ± 3.12* |
| 9.1 ± 1.63 | 10.93 ± 2.43 | 15.22 ± 1.63 | |
| Stair climbing task: | |||
| Number of rears | 10.7 ± 2.2 | 8.0 ± 1.6 | 9.9 ± 1.3 |
| 17.5 ± 2.1 | 14.8 ± 1.5 | 12.7 ± 1.4 | |
| Number of steps | 24.3 ± 4.1 | 19.2 ± 3.2 | 32.5 ± 4.3 |
| 39.6 ± 6.7 | 40.2 ± 4.4 | 45.1 ± 4.3 | |
| Rears per step | 0.403 ± 0.045 | 0.438 ± 0.073 | 0.319 ± 0.033 |
| Behavioral motor seizures after kainic acid injections: | 0.502 ± 0.065 | 0.398 ± 0.054 | 0.301 ± 0.027 |
| Wild-types | Transgenics | ||
| (n = 8/7†) | (n = 9) | ||
| Seizure score | 5.00 ± 0.66 | 7.11 ± 0.35* | |
| Latency to stage 4 (s) | 19.71 ± 2.29 | 15.00 ± 3.55 |
Six- to eight-week-old data are provided in the first row and six- to eight-month-old data are provided in the second, bold, row (when available).
Significantly different from wild-types, P < 0.05. For seizure measurements, transqenics are mixed zygosity.
Eight animals are included in the seizure score, but only seven in the latency because one animal did not achieve stage 4 seizures in the wild-type group.
Hot-plate test
There was no significant difference between the wild-type and BDNF transgenic mice from either age group in hot-plate latency (six to eight week F2,27 = 0.06, P > 0.94; six to eight month F2,41 = 0.016, P > 0.98, Table 2). Therefore, the difference in passive avoidance retention observed in the younger animals was not likely to be accounted for by differences in pain threshold.
Swim T-maze
Neither six- to eight-week-old nor six- to eight-month-old BDNF transgenic mice differed from their wild-type littermates in swim T-maze performance, measured by trials to criterion (six to eight weeks F2,27 = 0.490, P > 0.61; six to eight months F2,42 = 0.607, P > 0.55, Table 2). Therefore, the passive avoidance deficit is not likely to reflect a non-specific, generalized learning impairment.
Anxiety tests
Young BDNF transgenics did not show significantly different anxiety levels from their wild-type controls on either the staircase test (rears F2,27 = 0.678, P > 0.51; rears per step F2,26 = 0.991, P > 0.38, Table 2) or the elevated plus maze (proportion of open entries F2,27 = 0.324, P > 0.72; proportion of open time F2,27 = 0.643, P > 0.53, Table 2). In contrast, the older BDNF transgenics showed significantly increased rears/step in the staircase test (F2,45 = 5.622, P < 0.007; rears F2,45 = 2.05, P > 0.14, Table 2). They did not, however, show a significant decrease in anxiety in the elevated plus maze (proportion of open entries F2,42 = 1.890, P > 0.16; proportion of open time F2,42 = 1.517, P > 0.23, Table 2).
Locomotion
In neither age group did transgenics show significantly different locomotor activity on a gridded table from wild-type controls (six to eight week F2,27 = 1.975, P > 0.158; six to eight month F2,42 = 0.009, P > 0.99, Table 2), although the younger transgenic animals did have a slight tendency to spend more time mobile. This slight tendency to spend more time mobile also was apparent in the step climbing measure of locomotion, in which the homozygote mice showed an increased tendency to climb stairs which approached significance (F2,27 = 2.976, P > 0.06, Table 2). In addition, the young transgenics showed a significant elevation in the number of total elevated plus maze arm entries, an indication of increased general locomotor activity (F2,27 = 4.852, P < 0.016, Table 2). This tendency decreased in the older transgenics such that the increased arms entered only approached significance (F2,42 = 2.856, P > 0.06, Table 2) and the number of steps climbed on the staircase was no different from wild-types (step climbing F2,45 = 0.383, P > 0.68, Table 2).
Kainate experiments
Seizure behavior
Initial observations of transgenic animals indicated that they were hyperexcitable because some of the transgenics, but none of the wild-type littermates had spontaneous motor seizures that could reach stage 5. To examine seizure susceptibility systematically, nine BDNF transgenics and eight wild-types were injected with kainic acid. Animals were observed by two experimenters blind to genotype for behavioral signs of seizures. BDNF transgenics had significantly more severe seizures than their wild-type littermates (t15 = 1.042, P < 0.011, Table 2). The latencies to reach stage 4 for the two groups were not significantly different (t14 = 1.042, P > 0.31, Table 2).
Histology
No hippocampal damage was observed in the brains of either the transgenic or wild-type animals after kainic acid treatment (Fig. 4). This result was not entirely surprising, because the mice were made on a C57BL/6 background, and these mice have been shown to be less susceptible to seizure-induced cell death than other strains.96
Hippocampal physiology
In the first series of experiments from transgenic and wild-type mice, slices from six transgenics were compared with slices from eight wild-type mice. All mice were approximately the same age (two to three months). There were no differences in the behavior or phenotype of the mice to indicate differences to the experimenter, who was blind to the genotype in five cases (n = 3 transgenics; n = 2 wild-type). The blind experimenter who recorded physiological responses was able to predict the genotype of each animal solely on the basis of its evoked responses. Thus, slices from the transgenics exhibited more hyperexcitable responses than slices from wild-type animals (described below).
Area CA3
Area CA3 responses to single stimuli of the dentate hilus were qualitatively similar in slices from transgenics and wild-types (Fig. 5). In contrast, responses to repetitive stimulation of the hilus differed between the two types of mice. Ten pairs of stimuli at 1 Hz, using a half-maximal stimulus, led to multiple population spikes in 24/39 (62%) transgenic animal slices vs 4/20 wild-type slices (20%), a statistically significant difference (χ2 = 9.18, P < 0.01; Table 3, Fig. 5). In at least one slice of every transgenic animal, repetitive stimulation led to spreading depression, whereas spreading depression was not evoked in any slice from a wild-type mouse. Each spreading depression episode was heralded by spontaneous population spikes, which were immediately followed by a rapid and large direct current (d.c.) negative shift (−12 to −26 mV). During the d.c. shift, all spontaneous activity ceased, and stimulus-evoked responses could not be evoked. This was followed by a slow recovery of the d.c. potential and evoked responses, taking approximately 3–5 min. These events are similar to spreading depression episodes that have been reported in studies of rat cortex or hippocampus.106 The hyperexcitability in area CA3 of the transgenics, occurring in the absence of exogenous BDNF, was similar to the responses of control rat slices after exposure to synthetic BDNF, in which 1 Hz stimulation for less than 10 s induced spreading depression after exposure, but not before.95
Fig. 5.
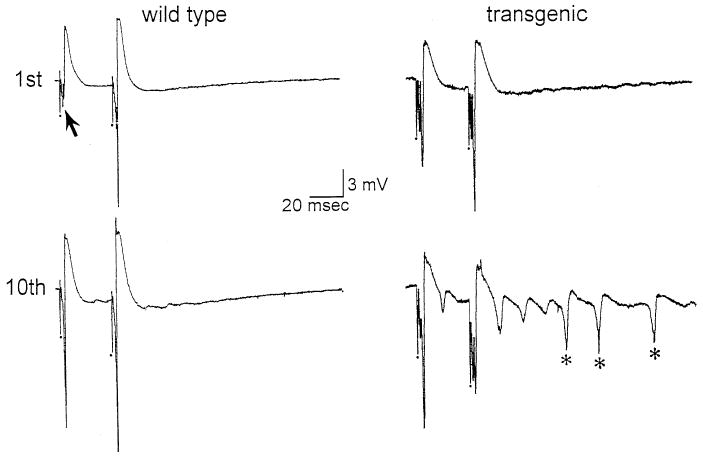
Hyperexcitability in area CA3 in transgenic mice. (Left) Top: Responses to two stimuli of the hilus were recorded in the area CA3 pyramidal cell layer. These stimuli were identical in intensity and the population spike (arrow) demonstrated strong facilitation. Stimulus artifacts in this figure, and other figures, are indicated by the dots and are truncated. Bottom: 10 pairs of stimuli were triggered at 1 Hz (1 pair every second) and the 10th pair is illustrated. Note that in this example there was an increase in the population spike amplitudes, but this did not happen in all cases. (Right) Top: Responses to hilar stimulation are shown for a transgenic mouse slice. Intensity of stimulation was adjusted so that it was half-maximal, as in “wild type”. Note that the extent of facilitation is less than in “wild type”, but this was not observed in all transgenic slices. Bottom: Responses to the 10th pair of stimuli demonstrated multiple population spikes (asterisks).
Table 3.
Hyperexcitability in brain-derived neurotrophic factor transgenics vs wild-type mice
| Area CA3 | Entorhinal cortex | ||
|---|---|---|---|
| Hyperexcitability | Spreading depression | Hyperexcitability | |
| Transgenics (n = 8 mice) | 23/39 (62%) | 6/33 (15%) (one slice per animal*) | 13/33 (39%) |
| Wild-types (n = 7 mice) | 4/20 (20%) | 0 | 3/37 (8%) |
| P-Value: | < 0.01 | > 0.05 | < 0.01 |
The number of slices in which hyperexcitable responses or spreading depression were evoked by stimulation is shown as a fraction of the total number of slices tested, and is also listed as a percentage. Hyperexcitability was defined as multiple population spikes and judged in area CA3 by the response to 10 half-maximal stimuli at 1 Hz. Hyperexcitability was assessed in the entorhinal cortex by the response to a single maximal stimulus to the white matter. For stimulation and recording sites, see Experimental Procedures.
One slice from every animal exhibited spreading depression; 15% of the total number of slices tested.
Entorhinal cortex
Responses to white matter stimulation were tested in deep and superficial layers of all slices that demonstrated a robust response in layer VI to a single stimulus. In wild-type mice, as in control rat slices, a maximal white matter stimulus evoked a short latency (less than 2 ms to peak) antidromic population spike (amplitude >5 mV), followed by a longer latency (2–4 ms to peak) orthodromic population spike (amplitude, 1–4 mV; Fig. 6). Responses to the same stimulus that were recorded in the superficial layers (i.e. layer III) consisted of a slow negative wave94(Fig. 6).
Fig. 6.
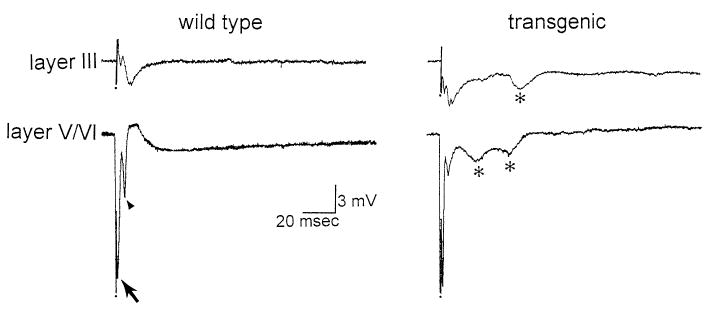
Hyperexcitability in the entorhinal cortex of transgenic slices. (Left) A typical response to a maximal stimulus to the white matter, recorded in layer III (Top) or layer V/VI (Bottom) of a slice from a wild-type mouse. The response recorded in layer VI consists of a short-latency antidromic population spike (arrow) followed by an orthodromic population spike (arrowhead). (Right) A representative response from a transgenic slice. Note that a maximal stimulus evoked similar field potentials immediately after the stimulus as in the wild-type mouse, and in addition secondary field potentials occurred (asterisks).
In slices from transgenics and wild-types, initial responses to white matter stimulation were similar, but in the transgenics they were followed in many cases by additional field potentials (Fig. 6). After a single stimulus, 13/33 (39%) of transgenic slices exhibited more than two field potentials in both deep and superficial layers after a single stimulus, whereas this occurred in only 3/37 (8%) of wild-type slices, a statistically significant difference (χ2 = 9.84, P < 0.01; Table 3).
Age dependence of hyperexcitability in transgenics
To determine whether physiological abnormalities were related to age-related BDNF expression, similar to the behavioral alterations, the results from young animals were compared with those from older animals. Three older animals were examined, two that were eight months old and one that was 12 months old. In the animals that were eight months old, hyperexcitability in area CA3 and the entorhinal cortex (as defined previously) was present in comparable frequency to young transgenics (area CA3: 2/8 slices or 25%, entorhinal cortex 3/9 slices or 33%). In the animal that was 12 months old, hyperexcitability was absent in all but one slice in area CA3 (1/4 slices or 25%) and absent in all slices of entorhinal cortex (0/4 slices or 0%). Spreading depression after hilar stimulation did not occur in the tissue from the 12-month-old mouse at all, and occurred once in the slices from the eight-month-old mice. Thus, for all three animals, spreading depression occurred in 1/12 slices (8.3%), whereas it occurred in 5/24 slices (20.8%) of the younger group. This difference in frequency was not statistically significant (χ2 = 0.95, P > 0.20).
Dentate gyrus
There was no detectable difference between transgenic and wild-type tissue in the responses to single stimuli of the outer molecular layer that were recorded in the granule cell layer. Large population EPSPs were recorded at all stimulus intensities, with small population spikes superimposed upon them, similar to adult rat slices.22,93 The maximal population EPSP amplitudes were not different (Table 4). The maximal population spike amplitudes were also not different (Table 4). Upon repetitive 1 Hz stimulation there was no evidence of multiple population spikes in the granule cell layer in any slice tested.
Table 4.
Dentate gyrus evoked responses in transgenics and wild-type mice
| Parameter | Wild-types | Transgenics | P-Value |
|---|---|---|---|
| Mean population EPSP amplitude | 7.2 ± 0.87 | 7.5 ± 1.00 | > 0.802 |
| mV ± S.E.M. | |||
| (wt n = 8; trsg n = 10) | |||
| Mean population spike amplitude | 1.4 ± 0.7 | 3.2 ± 1.5 | > 0.265 |
| mV ± S.E.M. | |||
| (wt n = 16; trsg n = 15) | |||
| Paired pulse ratio (20 ms ISI) | 378 ± 142 | 205 ± 44 | > 0.201 |
| (pS2/pS1)% ± S.E.M. | |||
| (wt n = 7; trsg n = 6) | |||
| Paired pulse ratio (40 ms ISI) | 643 ± 190 | 189 ± 83 | < 0.044* |
| (pS2/pS1)% ± S.E.M. | |||
| (wt n = 5; trsg n = 6) |
Responses to stimulation of the outer molecular layer were recorded in the granule cell layer as described in the Experimental Procedures. Population EPSP amplitude was measured from baseline to peak. Population spike amplitude was defined as the average of the two fast deflections that comprise the population spike. Paired pulse ratios were based on responses to an identical pair of stimuli with a 20 or 40 ms interstimulus interval (ISI) and are expressed as the ratio of the second population spike amplitude (pS2) to the first (pS1). trsg, transgenic; wt, wild-type.
Statistically significant (Student's t-test, P < 0.05).
In contrast to normal rat slices,93 paired-pulse stimulation using a short (20 ms) interstimulus interval often elicited facilitation in mouse slices, regardless of stimulus strength. The percentage of slices with facilitation was similar among transgenic and wild-type tissue. For paired pulses with a 20-ms interval and a maximal stimulus strength, 4/6 slices from transgenics exhibited PPF, and 4/7 of wild-type slices exhibited PPF (χ2 = 0.00, not significant, P < 0.99). For paired pulses with a 40-ms interval, transgenic slices facilitated in 5/6 slices, whereas 5/5 slices from wild-types facilitated. However, the paired pulse ratio, defined as the population spike amplitude evoked by the second stimulus as a function of the first, was significantly different in transgenics when the 40-ms interval was analysed. There was less facilitation using the 40-ms interval in transgenics (t9 = 2.33, P < 0.045; Table 4). There was no significant difference for the 20 ms interval (t8= 1.394, P > 0.201; Table 4).
Area CA1
Responses to single stimuli. Slices from nine BDNF transgenics were compared with slices from seven wild-type littermates. The transgenics were two to three months old (n = 6) or nine to 11 months old (n = 3). The wild-types were two to three months old (n = 4) or nine to 11 months old (n = 3). Data from the two age groups were combined, because the results were equivalent. A total of 37 slices was examined from transgenic and 28 from wild-type mice. There were no differences in the behavior or phenotype of the mice to indicate differences to the experimenter, who was blind to the genotype in many of the experiments (transgenics, n = 3; wild-types, n = 3, all two to three months old). Results from blinded experiments were equivalent to those of non-blinded experiments.
Area CA1 responses to single stimulation of both Schaffer collaterals and stratum lacunosum-moleculare in transgenics did not differ significantly from wild-types in any experiments. The general morphology of the response was qualitatively similar. The stimulus strength required to produce a maximal response to Schaffer collateral stimulation (t32 = 0.60, P > 0.55, Table 5) or a minimal response (t32 = 0.58, P > 0.56, Table 5) was not different. The maximal population spike amplitude was not different (t33 = 0.54, P > 0.58, Table 5). In contrast, the input–output curves, while not significantly different, showed a statistical trend (t30 = 1.996, P > 0.05, Table 5) towards a steeper response in the transgenics than in their wild-type controls. In four slices from transgenics, but in none from the wild-types, more than one population spike was evoked in response to a single stimulus.
Table 5.
Physiological data for CA1 responses to Schaffer collateral stimulation
| Parameter | Wild-types | Transgenics |
|---|---|---|
| Maximal input–output slope (mV/μA) | 3.2 ± 0.4 | 4.7 ± 0.6 |
| (wt n = 15; trsg n = 17) | ||
| Current for maximum response (μA) | 112.7 ± 9.6 | 104.2 ± 10.0 |
| (wt n = 15; trsg n = 19) | ||
| Current for minimum response (μA) | 30.7 ± 3.6 | 28.3 ± 2.4 |
| (wt n = 15; trsg n = 19) | ||
| Maximum spike amplitude (mV) | 13.0 ± 1.0 | 12.0 ± 1.1 |
| (wt n = 15; trsg n = 20) | ||
| % change, 20 ms paired pulse | 91.7 ± 12.4 | 159.7 ± 19.0* |
| (wt n = 13; trsg n = 18) | ||
| % change, 40 ms paired pulse | 162.6 ± 15.6 | 204.3 ± 32.0 |
| (wt n = 14; trsg n = 16) | ||
| % change, STP† | 332.1 ± 51.3 | 269.6 ± 37.0 |
| (wt n = 7; trsg n = 11) | ||
| % change, LTP† | 178.7 ± 9.7 | 213.6 ± 19.8 |
| (wt n = 7; trsg n = 6) | ||
| Potentiation in BDNF transgenic mice
(wild-type mice all exhibited LTP and no spreading depression) | ||
| STP | LTP | |
| All experiments | 228.2 ± 41.9 | 162.1 ± 19.6 |
| (n = 13) | ||
| Experiments with no spreading depression | 269.6 ± 37.1 | 160.6 ± 18.4 |
| (n = 11) | ||
| Experiments with LTP | 316.1 ± 51.5 | 214.1 ± 20.1 |
| (n = 7) | ||
| Experiments with LTP and no spreading depression | 322.8 ± 160.4 | 203.2 ± 20.0 |
| (n = 6) | ||
Data expressed as means ± S.E.M. Transgenic values which are significantly different than wild-type values are indicated by *. Not all slices were measured for all parameters; therefore the number of slices used for wild-types (wt) and transgenics (trsg) is included. Full statistics are included in the Results section, and a description of the measurements is included in the Experimental Procedures.
Experiments with no spreading depression.
Responses to paired stimulation. Responses to paired stimuli revealed similar PPF at a 40-ms interval in both wild-type and transgenic mice to Schaffer collateral stimulation (t28 = 1.117, P > 0.27, Table 5). In contrast, wild-type mice rarely exhibited PPF when a 20-ms inter-stimulus interval was used, while transgenic mice showed significantly more facilitation at this interval (t29 = 2.740, P < 0.01, Table 5).
Responses to tetanic stimulation. STP and LTP of the population spike were studied in BDNF transgenics and wild-type mice in vitro using Schaffer collateral stimulation. STP was measured 1 min post-tetanus and LTP was measured 30 min post-tetanus.
There were no significant differences between wild-types and transgenics in the amplitude of potentiation for either STP (t16 = 1.011, P > 0.32, Table 5) or LTP (t12 = 1.579, P > 0.14, Table 5). However, LTP was induced in 7/7 (100%) wild-type slices, but only 6/13 (43%) transgenic slices (a significant difference; χ2(1) = 5.8, P < 0.02, Fig. 7). In addition, the transgenics showed significantly more population spikes during tetanus than the wild-types [13/19 (68%) transgenics vs 2/11 (18%) wild-types, χ2(1) = 7.04, P < 0.01, Fig. 8]. Furthermore, two of the transgenic slices showed spreading depression after tetanus, whereas none of the wild-types showed this effect (Fig. 8).
Fig. 7.
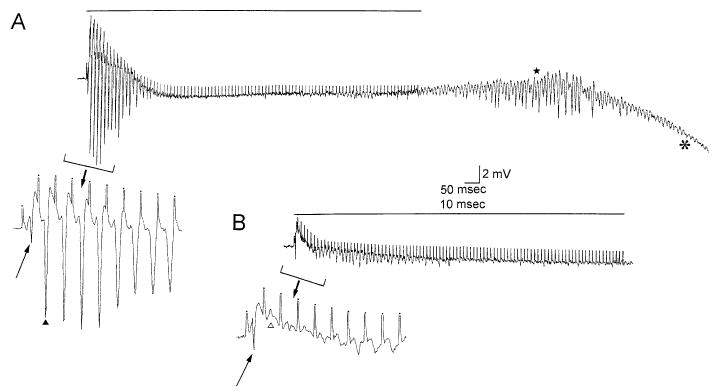
Responses of CA1 pyramidal cells to tetanic stimulation of the Schaffer collateral axons in slices from transgenic and wild-type mice. (A) Recordings from a transgenic mouse slice. The response to a 100 Hz train of stimuli for 1 s to the Schaffer collaterals, recorded in the CA1 cell body layer extracellularly, is shown. The horizontal line marks the tetanus. Note that after tetanic stimulation there were spontaneous population spikes (star) that led to a spreading depression episode. The onset of spreading depression is indicated by the asterick. Spreading depression began with a large d.c. negative shift in the extracellular potential, and during spreading depression all spontaneous evoked activity ceased, as is typical of these events. The extracellular potential recovered within 1–2 min and the evoked responses recovered in 10–15 min. Stimulus artifacts are truncated. (Calibration = 2 mV, 50 ms). Inset: The beginning of the tetanus is shown with a different time base (calibration at right, 2 mV, 10 ms). Stimulus artifacts are clipped and indicated by dots. The arrow points to the population spike evoked by the first stimulus, and the arrowhead indicates the population spike following the second stimulus. Note the large size of population spikes occurring after the first stimulus. This tetanus led to STP and LTP, as did all tetanic trains in wild-type mice (see text). This was not the case in transgenics (see text). (B) The response to tetanic stimulation is shown, recorded from a slice of a wild-type mouse. Calibration as for A. This tetanus, as well as all others recorded from wild-type tissue, was not followed by spontaneous activity or changes in the extracellular potential. Inset: The beginning of the tetanus is shown. The arrow points to the population spike evoked by the first stimulus. Note that the response to the second stimulus (open arrowhead) is small relative to the analogous stimulus in part A. Successive stimuli also evoked minimal responses.
Fig. 8.
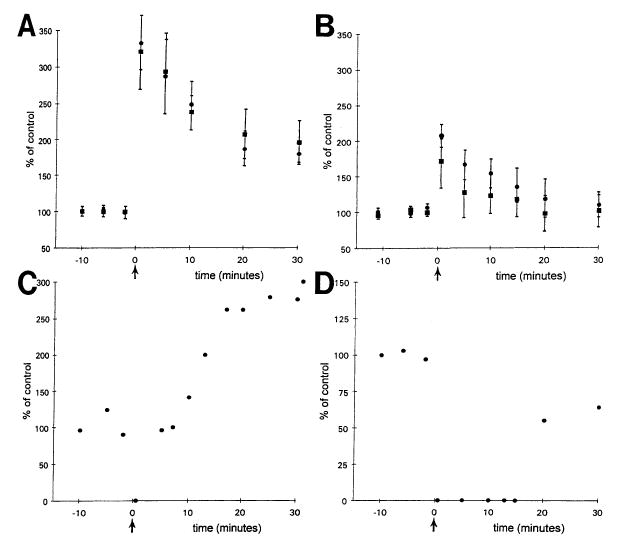
LTP in BDNF transgenic and wild-type mice. (A) Population spike amplitude is expressed as per cent of the mean amplitude (± S.E.M.) prior to tetanic stimulation (at time zero, marked by the arrow) for transgenics (circles, n = 6) and wild-type mice (squares, n = 7). Excluded are experiments that demonstrated no LTP and experiments in which spreading depression occurred immediately after the tetanus. (B) Analogous to part A, population spike amplitude is plotted as a function of time in relation to tetanic stimulation. Only those experiments in which LTP did not occur are included. Because this only occurred in transgenic mice, all data are from transgenics. Circles are data from experiments in which LTP and spreading depression did not occur (n = 5); squares include data from one experiment in which spreading depression occurred (n = 6). (C–D) The two experiments from transgenic mice, in which spreading depression followed tetanic stimulation, are shown. In C, LTP followed spreading depression, and in D, LTP did not.
Discussion
Summary
We generated BDNF transgenic mice that overexpressed BDNF from the β-actin promoter. These mice showed no gross brain abnormalities or overt behavioral abnormalities. Behavioral testing revealed a significant passive avoidance deficit which was dependent on continued high levels of BDNF expression, and did not reflect a non-specific learning impairment. In addition, BDNF transgenic mice were more active than their wild-type littermates overall, and this activity also diminished when BDNF levels dropped.
This study also demonstrated markedly increased excitability in BDNF transgenics compared with wild-type littermates. Specifically, motor seizure severity was greater in transgenics after kainic acid injection. In vitro, area CA3 and entorhinal cortex field potentials evoked in transgenics demonstrated increased excitability. In addition, repetitive stimulation of the mossy fiber axons of granule cells produced spreading depression episodes in CA3 pyramidal cells in transgenic but not wild-type mice. Evoked responses in CA1 showed differences which suggested that the abnormal excitability in the transgenics may have interfered with normal LTP induction.
These data support the hypothesis that BDNF predisposes the hippocampal region to become epileptiform. In addition, the hyperexcitability in these animals may contribute to deficits in learning and plasticity. Thus, although BDNF is neurotrophic and neuroprotective in many areas, particularly during development, in the adult animal it may disrupt learning, enhance excitability and even be proconvulsant.
Plasticity and learning
Hippocampal CA1 physiology
Although the BDNF transgenic mice showed similar CA1 evoked responses to single stimuli as their wild-type littermates, their responses to multiple stimuli were different. Specifically, the transgenics showed what appeared to be an increase in excitability because there was increased PPF, increased excitatory activity during tetanic stimulation and spreading depression after tetanic stimuli. The observation of increased excitatory activity during tetanic stimulation is interesting in light of the studies of Figurov et al.,28 who showed that, in normal rats, exposure to BDNF had a similar effect. In addition, BDNF transgenics did not exhibit LTP as consistently as wild-types, although their LTP, when induced, appeared normal in magnitude. Although the mechanisms of impaired LTP induction are uncertain, the most parsimonious explanation is that the hyperexcitability in the transgenics interfered with normal CA1 plasticity.
Learning and memory
Our most striking behavioral finding was a reliable and dramatic impairment of passive avoidance performance. This deficit is unlikely to be a consequence of the slight tendency towards increased motor activity, both because of the magnitude of the impairment, and because of the finding of absolutely no difference between transgenics and wild-types in tendency to spontaneously cross over in the shuttlebox apparatus. Our discovery that the passive avoidance impairment resolves as the BDNF mRNA levels begin to drop in middle adulthood is reassuring, because it suggests that the impairment is due to the elevated BDNF and not to permanent developmental changes in circuitry. Because we have found a physiological abnormality in CA1 of the hippocampus, it is possible that abnormalities in this region of the brain underlie the passive avoidance deficit measured in the BDNF transgenic mice. This possibility seems unlikely, however, because the passive avoidance deficit resolves when the BDNF levels drop with age, but the disruptions in CA1 physiology remain. Therefore, it is possible that some other structure in the brain underlies the passive avoidance deficit. The striatum is a likely candidate, because it shows the greatest relative up-regulation of BDNF in these mice, and has also been shown to mediate the successful performance of passive avoidance tasks.24,79,92
The role of brain-derived neurotrophic factor in learning, memory and plasticity
Learning and learning-related processes have previously been shown to be associated with increases in BDNF,13,16,25,27,83 although the effects of decreased amounts of BDNF on learning have been controversial.60,72 However, the addition of exogenous BDNF has resulted in either no change or impairments in learning and memory29,84 (S. D. Croll et al., unpublished observations). These findings, combined with the present data, suggest that although it is possible that BDNF is involved in the modulation of normal learning and memory, too much BDNF may have adverse effects on these processes. That is, abnormally high levels of BDNF may result in learning impairments. These impairments may result from too much excitability in the circuits involved in learning, or from too much plasticity, leading to a loss of synaptic refinement. Indeed, our finding of increased excitability in CA1 provides support for this explanation. It is also consistent with other studies showing that excess activation of hippocampal circuits involved in plasticity interferes with learning in “LTP saturation” studies.12,17,69,74
Age dependence of brain-derived neurotrophic factor overexpression and transgenic phenotype
BDNF overexpression was initially very high and fairly ubiquitous in the adult mouse brain, but began to decrease as the animals approached eight to nine months of age. Because the behavioral deficits resolved as BDNF transcript levels dropped with age, the data suggest that BDNF overexpression could underlie the behavioral deficits. The results argue against the possibility that BDNF overexpression during development caused permanent alterations in the brain that led to abnormal behavior. However, the changes in physiology were less clearly correlated with the age of the transgenic mice that were examined, although they did suggest some decreases in excitability in the older animals. One possibility is that changes in excitability require lower levels of BDNF overexpression than behavioral alterations. Further studies are necessary to determine whether the changes in physiology were due directly to overexpression of BDNF, or whether there were also developmental changes or alterations in circuitry caused by seizures that contributed to the observed hyperexcitability and disruption of LTP.
Seizures and excitability
Brain-derived neurotrophic factor and hyperexcitability
Recordings in area CA3 and entorhinal cortex from BDNF transgenics were similar to those observed after recombinant BDNF exposure in normal rat hippocampal slices,95 suggesting that there could be a similar underlying mechanism of hyperexcitability. Thus, in transgenics, we hypothesize that repetitive stimulation of afferents containing increased BDNF led to BDNF release at high concentrations that enhanced excitatory transmission. Because of the robust paired-pulse inhibition in the dentate gyrus in transgenics, and the lack of change in paired-pulse responses in area CA3 (see Fig. 5), it is unlikely that a reduction in inhibition led to the hyperexcitability. However, there was a reduction in paired-pulse inhibition in area CA1, and Tanaka et al.112 showed that BDNF can decrease IPSCs (inhibitory postsynaptic currents) of CA1 pyramidal cells. Therefore, changes in GABAergic neurons or GABA receptors in transgenics cannot be ruled out.
The restriction of hyperexcitability to areas with the most endogenous BDNF is consistent with BDNF release mediating hyperexcitability in transgenics. However, it has not been proved that BDNF is released upon afferent stimulation, although it is known that BDNF is anterogradely transported.3,19,120 Therefore, other reasons for transgenic hyperexcitability are important to consider. Increased BDNF during development could have played a role because BDNF has effects on synaptogenesis, morphological development of neurons,40,47,56,59,68,105 and the morphology and phenotype of GABA neurons.71,77,115 These effects could lead to hyperexcitability independent of released BDNF during recordings. Spontaneous seizures in transgenic mice are also potentially relevant, because they could influence excitability. Even brief excitatory activity in rats leads to alterations in gene expression.31,100 Repetitive activity, if sufficient, can lead to neuronal damage or loss, consequent reactive gliosis, synaptic reorganization and further alterations in function. An argument against the possibility that severe cell loss occurred is that transgenic animals, even after kainic acid injection, did not exhibit obvious hippocampal cell loss. Still, alterations in gene expression and subtle anatomical changes could have had effects that led to a hyperexcitable state.
Comparing rat and mouse excitability
Caution is appropriate concerning the term “hyperexcitability.” Hyperexcitability was defined, for the purposes of this study, on the basis of rat experiments, in which decades of hippocampal slice analyses have distinguished normal and abnormal synaptic responses to afferent inputs.23,98,99 Multiple field potentials and spreading depression are both indications of hyperexcitability because such events occur in response to application of high concentrations of potassium or convulsants. However, what is true for the rat may not be true for the mouse.
It is important to bear in mind that there is some strain-dependent hyperexcitability in wild-type mice.96 For the purposes of the present study, this issue is perhaps less of a problem because the strain used (C57BL/6) is relatively seizure resistant.96 Therefore, it was unexpected that hyperexcitability occurred in transgenics of this strain at all. Because of the strain used, the data more strongly support the hypothesis that BDNF may be an important regulator of neuronal excitability than if the strain was seizure sensitive.
Changes in dentate gyrus
There was more facilitation in paired pulse tests (40-ms interval) of the dentate gyrus in slices from wild-type mice than slices from transgenics. This finding is different from the case in CA1, where there was more PPF in the transgenics. However, these results are actually consistent because paired-pulse inhibition in the dentate gyrus appears to increase, paradoxically, in animal models of epilepsy with chronic seizures. Thus, in the dentate gyrus of rats that have been treated with the convulsant kainic acid, there is a chronic increase in paired-pulse inhibition.14,104 However, in the presence of GABA antagonists, underlying hyperexcitability is revealed in slices at the chronic stage.21 In the kindling model of epilepsy, paired-pulse inhibition in dentate gyrus also increases.80,87,114 In Oliver and Miller's80 studies, increased inhibition occurs only at long interstimulus intervals, similar to the BDNF transgenics. Thus, there are similarities between transgenic mice overexpressing BDNF and rat models of epilepsy, not just in area CA3 and entorhinal cortex, but also in the dentate gyrus and CA1. This supports the hypothesis that BDNF may have some actions that are similar to convulsants, or produce long-lasting changes that are similar to the end result of convulsant treatment and chronic seizures.
Implications for epilepsy
If endogenous BDNF enhances limbic excitation, its actions may be relevant to the etiology of neurological diseases such as epilepsy. Indeed, anatomical studies in human tissue resected from medically intractable temporal lobe epileptics show increased mRNA for BDNF in granule cells.67 Thus, human granule cells are likely to synthesize BDNF, and increased BDNF may be present after seizures, similar to rat.7,9,26,38,39,75,78,91,97 Because mice overexpressing BDNF were hyperexcitable in hippocampus, and normal rat hippocampus treated with BDNF becomes hyperexcitable,95 increased hippocampal excitability may also occur in humans after seizures due to increased BDNF.
Based on these observations, we hypothesize that, in the normal animal, endogenous BDNF may not increase excitability dramatically. Synaptic effects that do occur could actually be beneficial rather than pathological, as suggested by CA144,50,82 and dentate gyrus studies in vivo70 showing that BDNF application induces LTP, and hence could be involved in learning and memory. After seizures, when one would predict that BDNF expression is enhanced, effects of BDNF might increase excitability, and possibly lead to spontaneous seizures. Since each seizure may further increase BDNF expression, one seizure could lead to another. Enhanced excitation, specifically of mossy fiber targets and neurons in superficial layers of entorhinal cortex, may eventually lead to damage or cell loss. Perhaps it is no coincidence that these areas are vulnerable regions observed upon autopsy of temporal lobe epileptics.32,66
Conclusions
BDNF may have beneficial effects in developing systems, and could be neuroprotective or neurorestorative after some injuries to CNS structures,1,2,8,18,33,37,58,73,109,117 but in the adult hippocampus and entorhinal cortex, high levels of BDNF might have very different, potentially detrimental, effects on learning, plasticity and excitability.
Acknowledgments
The authors would like to thank Ning Cai, Joyce McClain, Nefertiti Greene, Michelle Russell, Cathy Chesnutt and Annmarie Curcio for technical and administrative assistance, Dr Ann Acheson for the BDNF ELISA and Dr Jeffrey Goodman for comments on the manuscript. In addition, we would like to thank Evan Burrows and Claudia Murphy for assistance with the figures. Finally, we would like to thank Dr Qiao Yan from Amgen for providing the rabbit polyclonal BDNF antibody and Dr Larry Kedes from Stanford University for providing the human β-actin promoter construct. This work was supported in part by NS 37562 to H.E.S. from the National Institutes of Health.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- BDNF
brain-derived neurotrophic factor
- bp
base pairs
- d.c.
direct current
- ELISA
enzyme-linked immunosorbent assay
- EPSC
excitatory postsynaptic current
- EPSP
excitatory postsynaptic potential
- LTP
long-term potentiation
- PPF
paired-pulse facilitation
- STP
short-term potentiation
References
- 1.Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, Thadani A, Squinto SP, Yancopoulos GD, Lindsay RM. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–452. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- 2.Alderson RF, Alterman AL, Barde YA, Lindsay RM. Brain-derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron. 1990;5:297–306. doi: 10.1016/0896-6273(90)90166-d. [DOI] [PubMed] [Google Scholar]
- 3.Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- 4.Altar CA, Siuciak JA, Wright P, Ip NY, Lindsay RM, Wiegand SJ. In situ hybridization of trkB and trkC receptor mRNA in rat forebrain and association with [125I]NT-3, [125I]BDNF, and [125I]NT-4/5 binding autoradiography. Eur J Neurosci. 1994;6:1389–1405. doi: 10.1111/j.1460-9568.1994.tb01001.x. [DOI] [PubMed] [Google Scholar]
- 5.Anderson KD, Alderson RF, Altar CA, DiStefano PS, Corcoran TL, Lindsay RM, Wiegand SJ. Differential distribution of exogenous BDNF, NGF, and NT-3 in the brain corresponds to the relative abundance and distribution of high-affinity and low-affinity neurotrophin receptors. J comp Neurol. 1995;357:296–317. doi: 10.1002/cne.903570209. [DOI] [PubMed] [Google Scholar]
- 6.Auffray C, Rougeon F. Purification of mouse immunoglobulin heavy-chain messenger RNAs from total myeloma tumor RNA. Eur J Biochem. 1980;107:303–314. doi: 10.1111/j.1432-1033.1980.tb06030.x. [DOI] [PubMed] [Google Scholar]
- 7.Ballarín M, Ernfors P, Lindefors N, Persson H. Hippocampal damage and kainic acid injection induce a rapid increase in mRNA for BDNF and NGF in the rat brain. Expl Neurol. 1991;114:35–43. doi: 10.1016/0014-4886(91)90082-n. [DOI] [PubMed] [Google Scholar]
- 8.Beck T, Lindholm D, Castrén E, Wree A. Brain-derived neurotrophic factor protects against ischemic cell damage in rat hippocampus. J cerebr Blood Flow Metab. 1994;14:689–692. doi: 10.1038/jcbfm.1994.86. [DOI] [PubMed] [Google Scholar]
- 9.Bengzon J, Kokaia Z, Ernfors P, Kokaia M, Leanza G, Nilsson OG, Persson H, Lindvall O. Regulation of neurotrophin and trkA, trkB and trkC tyrosine kinase receptor messenger RNA expression in kindling. Neuroscience. 1993;53:433–446. doi: 10.1016/0306-4522(93)90207-v. [DOI] [PubMed] [Google Scholar]
- 10.Berninger B, Poo MM. Fast actions of neurotrophic factors. Curr Opin Neurobiol. 1996;6:324–330. doi: 10.1016/s0959-4388(96)80115-2. [DOI] [PubMed] [Google Scholar]
- 11.Binder DK, Routbort MJ, Ryan TE, Yancopoulos GD, McNamara JO. Selective inhibition of kindling development by intraventricular administration of trkB receptor body. J Neurosci. 1996;19:1424–1436. doi: 10.1523/JNEUROSCI.19-04-01424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bliss TVP. The saturation debate. Science. 1998;281:1975–1976. doi: 10.1126/science.281.5385.1975. [DOI] [PubMed] [Google Scholar]
- 13.Bramham CR, Southard T, Sarvey JM, Herkenham M, Brady LS. Unilateral LTP triggers bilateral increases in hippocampal neurotrophin and trk receptor mRNA expression in behaving rats: evidence for interhemispheric communication. J comp Neurol. 1996;368:371–382. doi: 10.1002/(SICI)1096-9861(19960506)368:3<371::AID-CNE4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 14.Buckmaster PS, Dudek FE. Network properties of the dentate gyrus in epileptic rats with neuron loss and granule cell axon reorganization. J Neurophysiol. 1997;77:2685–2696. doi: 10.1152/jn.1997.77.5.2685. [DOI] [PubMed] [Google Scholar]
- 15.Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. J Physiol. 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castrén E, Pitkänen M, Sirviö J, Parsadanian A, Lindholm D, Thoenen H, Riekkinen PJ. The induction of LTP increases BDNF and NGF mRNA but decreases NT-3 mRNA in the dentate gyrus. NeuroReport. 1993;4:895–898. doi: 10.1097/00001756-199307000-00014. [DOI] [PubMed] [Google Scholar]
- 17.Castro CA, Silbert LH, McNaughton BL, Barnes CA. Recovery of spatial learning deficits after decay of electrically induced synaptic enhancement in the hippocampus. Nature. 1989;342:545–548. doi: 10.1038/342545a0. [DOI] [PubMed] [Google Scholar]
- 18.Cheng B, Mattson MP. NT-3 and BDNF protect CNS neurons against metabolic/excitotoxic insults. Brain Res. 1994;640:56–67. doi: 10.1016/0006-8993(94)91857-0. [DOI] [PubMed] [Google Scholar]
- 19.Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Croll SD, Greene NA, Lindsay RM, Wiegand SJ. Sodium azide-induced learning deficits in rats: time course and corresponding pathology. Psychobiology. 1997;25:34–47. [Google Scholar]
- 21.Cronin J, Obenaus A, Houser CR, Dudek FE. Electrophysiology of dentate granule cells after kainate-induced synaptic reorganization. Brain Res. 1992;573:305–310. doi: 10.1016/0006-8993(92)90777-7. [DOI] [PubMed] [Google Scholar]
- 22.Dahl D, Burgard EC, Sarvey JM. NMDA receptor antagonists reduce medial but not lateral perforant path-evoked EPSPs in dentate gyrus of rat hippocampal slice. Expl Brain Res. 1990;83:172–177. doi: 10.1007/BF00232206. [DOI] [PubMed] [Google Scholar]
- 23.Delgado-Escueta AV, Ward AA, Woodbury DM, Porter RJ. Basic Mechanisms of the Epilepsies. Raven; New York: 1986. [Google Scholar]
- 24.Diaz del Guante MA, Cruz-Morales SE, Prado-Alcal RA. Time-dependent effects of cholinergic blockade of the striatum on memory. Neurosci Lett. 1991;122:79–82. doi: 10.1016/0304-3940(91)90198-3. [DOI] [PubMed] [Google Scholar]
- 25.Dragunow M, Beilharz E, Mason B, Lawlor P, Abraham W, Gluckman P. Brain-derived neurotrophic factor expression after long-term potentiation. Neurosci Lett. 1993;160:232–236. doi: 10.1016/0304-3940(93)90420-p. [DOI] [PubMed] [Google Scholar]
- 26.Ernfors P, Bengzon J, Kokaia Z, Persson H, Lindvall O. Increased levels of mRNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron. 1991;7:165–176. doi: 10.1016/0896-6273(91)90084-d. [DOI] [PubMed] [Google Scholar]
- 27.Falkenberg T, Mohammed AK, Henriksson B, Persson H, Winblad B, Lindefors N. Increased expression of brain-derived neurotrophic factor mRNA in rat hippocampus is associated with improved spatial memory and enriched environment. Neurosci Lett. 1992;138:153–156. doi: 10.1016/0304-3940(92)90494-r. [DOI] [PubMed] [Google Scholar]
- 28.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 29.Fischer W, Sirevaag A, Wiegand SJ, Lindsay RM, Björklund A. Reversal of spatial memory impairments in aged rats by nerve growth factor and neurotrophins 3 and 4/5 but not by brain-derived neurotrophic factor. Proc natn Acad Sci USA. 1994;91(18):8607–8611. doi: 10.1073/pnas.91.18.8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Grayson DR, Kromer LF. Developmental and mature expression of full-length and truncated trkB receptors in the rat forebrain. J comp Neurol. 1996;374:21–40. doi: 10.1002/(SICI)1096-9861(19961007)374:1<21::AID-CNE2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 31.Gall CM. Seizure-induced changes in neurotrophin expression: implications for epilepsy. Expl Neurol. 1993;124:150–166. doi: 10.1006/exnr.1993.1186. [DOI] [PubMed] [Google Scholar]
- 32.Gastaut H, Toga M, Roger J, Gibson WC. A correlation of clinical electroencephalographic and anatomical findings in nine autopsied cases of “temporal lobe epilepsy”. Epilepsia. 1959;1:59–85. [Google Scholar]
- 33.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 34.Glass DJ, Nye SH, Hantzopoulos P, Macchi MJ, Squinto SP, Goldfarb M, Yancopoulos GD. TrkB mediates BDNF/NT-3-dependent survival and proliferation in fibroblasts lacking the low affinity NGF receptor. Cell. 1991;66:405–413. doi: 10.1016/0092-8674(91)90629-d. [DOI] [PubMed] [Google Scholar]
- 35.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6836. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gunning P, Leavitt J, Muscat G, Ng SY, Kedes L. A human beta-actin expression vector system directs high-level accumulation of antisense transcripts. Proc natn Acad Sci USA. 1987;84:4831–4835. doi: 10.1073/pnas.84.14.4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hofer MM, Barde YA. Brain-derived neurotrophic factor prevents neuronal death in vivo. Nature. 1988;331:261–262. doi: 10.1038/331261a0. [DOI] [PubMed] [Google Scholar]
- 38.Humpel C, Wetmore C, Olson L. Regulation of brain-derived neurotrophic factor messenger RNA and protein at the cellular level in pentylenetetrazol-induced epileptic seizures. Neuroscience. 1993;53:909–918. doi: 10.1016/0306-4522(93)90476-v. [DOI] [PubMed] [Google Scholar]
- 39.Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- 40.Jelsma TN, Aguayo AJ. Trophic factors. Curr Opin Neurobiol. 1994;4:717–725. doi: 10.1016/0959-4388(94)90015-9. [DOI] [PubMed] [Google Scholar]
- 41.Jones RS. Complex synaptic responses of entorhinal cortical cells in the rat to subicular stimulation in vitro: demonstration of an NMDA receptor-mediated component. Neurosci Lett. 1987;81:209–214. doi: 10.1016/0304-3940(87)90000-0. [DOI] [PubMed] [Google Scholar]
- 42.Jones RS, Heinemann U. Spontaneous and evoked NMDA-receptor mediated potentials in the entorhinal cortex of the neonate rat in vitro. Adv exp med Biol. 1990;268:181–186. doi: 10.1007/978-1-4684-5769-8_21. [DOI] [PubMed] [Google Scholar]
- 43.Jovanic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I actin interactions. Proc natn Acad Sci USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 45.Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 46.Klein R, Nanduri V, Jing S, Lamballe F, Tapley P, Bryant S, Cordon-Cardo C, Jones KR, Reichardt LF, Barbacid M. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66:395–402. doi: 10.1016/0092-8674(91)90628-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knüsel B, Winslow JW, Rosenthal A, Burton LE, Seid DP, Nikolics K, Hefti F. Promotion of central cholinergic and dopaminergic neuron differentiation by brain-derived neurotrophic factor but not neurotrophin 3. Proc natn Acad Sci USA. 1991;88:961–965. doi: 10.1073/pnas.88.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koh JY, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–576. doi: 10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- 49.Kokaia M, Ernfors P, Kokaia Z, Elmer E, Jaenisch R, Lindvall O. Suppressed epileptogenesis in BDNF mutant mice. Expl Neurol. 1995;133:215–224. doi: 10.1006/exnr.1995.1024. [DOI] [PubMed] [Google Scholar]
- 50.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc natn Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korte M, Staiger V, Griesbeck O, Thoenen H, Bonhoeffer T. The involvement of brain-derived neurotrophic factor in hippocampal long-term potentiation revealed by gene targeting experiments. J Physiol, Paris. 1996;90:157–164. doi: 10.1016/s0928-4257(97)81415-5. [DOI] [PubMed] [Google Scholar]
- 52.Larmet Y, Reibel S, Carnahan J, Nawa H, Marescaux C, Depaulis A. Protective effects of brain derived neurotrophic factor on the development of hippocampal kindling in the rat. NeuroReport. 1995;6:1937–1941. doi: 10.1097/00001756-199510020-00027. [DOI] [PubMed] [Google Scholar]
- 53.Leβmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 54.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc natn Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levine ES, Dreyfus CF, Black IB, Plummer MR. Selective role for trkB neurotrophin receptors in rapid modulation of hippocampal synaptic transmission. Molec Brain Res. 1996;38:300–303. doi: 10.1016/0169-328x(96)00025-3. [DOI] [PubMed] [Google Scholar]
- 56.Lewin GR, Barde YA. Physiology of neurotrophins. A Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- 57.Lin SY, Wu K, Mount HTJ, Suen PC, Black IB. BDNF rapidly enhances tyrosine phosphorylation of NMDA receptor subunit 2B in postsynaptic densities of rat cerebral cortex and hippocampus. Soc Neurosci Abstr. 1997;23:59. [Google Scholar]
- 58.Lindholm D, Dechant G, Heisenberg CP, Thoenen H. Brain-derived neurotrophic factor is a survival factor for cultured rat cerebellar granule neurons and protects them against glutamate-induced neurotoxicity. Eur J Neurosci. 1993;5:1455–1464. doi: 10.1111/j.1460-9568.1993.tb00213.x. [DOI] [PubMed] [Google Scholar]
- 59.Lindsay RM, Wiegand SJ, Altar CA, DiStefano PS. Neurotrophic factors: from molecule to man. Trends Neurosci. 1994;17:182–190. doi: 10.1016/0166-2236(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 60.Linnarsson S, Bjorklund A, Ernfors P. Learning deficit in BDNF mutant mice. Eur J Neurosci. 1997;9:2581–2587. doi: 10.1111/j.1460-9568.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- 61.Listar RG. The use of a plus-maze to measure anxiety in the mouse. Psychopharmacology. 1987;92:180–185. doi: 10.1007/BF00177912. [DOI] [PubMed] [Google Scholar]
- 62.Lo DC. Neurotrophic factors and synaptic plasticity. Neuron. 1995;15:979–981. doi: 10.1016/0896-6273(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 63.Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 64.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 65.Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. CSH Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 66.Margerison JH, Corsellis JAN. Epilepsy and the temporal lobes: a clinical encephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain. 1966;89:499–530. doi: 10.1093/brain/89.3.499. [DOI] [PubMed] [Google Scholar]
- 67.Mathern GW, Babb TL, Micevych PE, Blanco CE, Pretorius JK. Granule cell mRNA levels for BDNF, NGF, and NT-3 correlate with neuron lossess or supragranular mossy fiber sprouting in the chronically damaged and epileptic human hippocampus. Molec chem Neuropath. 1997;30:53–76. doi: 10.1007/BF02815150. [DOI] [PubMed] [Google Scholar]
- 68.McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 69.McNaughton BL, Barnes CA, Rao G, Baldwin J, Rasmussen M. Long-term enhancement of hippocampal synaptic transmission and the acquisition of spatial information. J Neurosci. 1986;6:563–571. doi: 10.1523/JNEUROSCI.06-02-00563.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Messaoudi E, Bårdsen K, Srebro B, Bramham CR. Acute intrahippocampal infusion of brain-derived neurotrophic factor induces lasting potentiation of synaptic transmission in the rat dentate gyrus. J Neurophysiol. 1998;79:496–499. doi: 10.1152/jn.1998.79.1.496. [DOI] [PubMed] [Google Scholar]
- 71.Mizuno K, Carnahan J, Nawa H. Brain-derived neurotrophic factor promotes differentiation of striatal GABAergic neurons. Devl Biol. 1994;165:243–256. doi: 10.1006/dbio.1994.1250. [DOI] [PubMed] [Google Scholar]
- 72.Montkowski A, Holsboer F. Intact spatial learning and memory in transgenic mice with reduced BDNF. NeuroReport. 1997;8:779–782. doi: 10.1097/00001756-199702100-00040. [DOI] [PubMed] [Google Scholar]
- 73.Morse JK, Wiegand SJ, Anderson K, You Y, Cai N, Carnahan J, Miller J, DiStefano PS, Altar CA, Lindsay RM, Alderson RF. Brain-derived neurotrophic factor (BDNF) prevents the degeneration of medial septal cholinergic neurons following fimbria transection. J Neurosci. 1993;13:4146–4156. doi: 10.1523/JNEUROSCI.13-10-04146.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moser EI, Krobert KA, Moser MB, Morris RGM. Impaired spatial learning after saturation of long-term potentiation. Science. 1998;281:2038–2042. doi: 10.1126/science.281.5385.2038. [DOI] [PubMed] [Google Scholar]
- 75.Mudò G, Jiang XH, Timmuske T, Bindoni M, Belluardo N. Change in neurotrophins and their receptor mRNAs in the rat forebrain after status epilepticus induced by pilocarpine. Epilepsia. 1996;37:198–207. doi: 10.1111/j.1528-1157.1996.tb00012.x. [DOI] [PubMed] [Google Scholar]
- 76.Muragaki Y, Timothy N, Leight S, Hempstead BL, Chao MV, Trojanowski JQ, Lee VMY. Expression of trk receptors in the developing and adult human central and peripheral nervous system. J comp Neurol. 1995;356:387–397. doi: 10.1002/cne.903560306. [DOI] [PubMed] [Google Scholar]
- 77.Nawa H, Bessho Y, Carnahan J, Nakanishi S, Keiko M. Regulation of neuropeptide expression in cultured cerebral cortical neurons by brain-derived neurotrophic factor. J Neurochem. 1993;60:772–775. doi: 10.1111/j.1471-4159.1993.tb03216.x. [DOI] [PubMed] [Google Scholar]
- 78.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nishino H, Koide K, Aihara N, Kumazaki M, Sakurai T, Nagai H. Striatal grafts in the ischemic striatum improve pallidal GABA release and passive avoidance. Brain Res Bull. 1993;32:517–520. doi: 10.1016/0361-9230(93)90300-z. [DOI] [PubMed] [Google Scholar]
- 80.Oliver MW, Miller JJ. Alterations of inhibitory processes in the dentate gyrus following kindling-induced epilepsy. Expl Brain Res. 1985;57:443–447. doi: 10.1007/BF00237830. [DOI] [PubMed] [Google Scholar]
- 81.Osehobo P, Adams B, Sazgar M, Xu Y, Racine R, Fahnestock M. Brain-derived neurotrophic factor infusion delays amygdala and perforant path kindling without affecting paired pulse measures of neuronal inhibition in adult rats. Neuroscience. 1999 doi: 10.1016/s0306-4522(99)00048-2. in press. [DOI] [PubMed] [Google Scholar]
- 82.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 83.Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- 84.Pelleymounter MA, Cullen MJ, Baker MB, Gollub M, Wellman C. The effects of intrahippocampal BDNF and NGF on spatial learning in aged Long Evans rats. Molec chem Neuropath. 1996;29:211–226. doi: 10.1007/BF02815003. [DOI] [PubMed] [Google Scholar]
- 85.Phillips HS, Hains JM, Laramee GR, Rosenthal A, Winslow JW. Widespread expression of BDNF but not NT3 by target areas of basal forebrain cholinergic neurons. Science. 1990;250:290–294. doi: 10.1126/science.1688328. [DOI] [PubMed] [Google Scholar]
- 86.Racine RJ. Modification of seizure activity by electrical stimulation II. Motor seizure. Electroenceph clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 87.Racine RJ, Burnham WM, Gilbert M, Kairiss EW. Kindling mechanisms: electrophysiological studies. In: Wada JA, editor. Kindling Three. Raven; New York: 1986. pp. 263–279. [Google Scholar]
- 88.Radka SF, Holst PA, Fritsche M, Altar CA. Presence of brain-derived neurotrophic factor in brain and human and rat but not mouse serum detected by a sensitive and specific immunoassay. Brain Res. 1996;709:122–130. doi: 10.1016/0006-8993(95)01321-0. [DOI] [PubMed] [Google Scholar]
- 89.Rafiq A, DeLorenzo RJ, Coulter DA. Generation and propagation of epileptiform discharges in a combined entorhinal cortex/hippocampal slice. J Neurophysiol. 1993;70:1962–1974. doi: 10.1152/jn.1993.70.5.1962. [DOI] [PubMed] [Google Scholar]
- 90.Reibel S, Larmet Y, Lê BT, Carnahan J, Nawa H, Marescaux C, Depaulis A. Protective effects of BDNF in two models of epilepsy in the rat. Soc Neurosci Abstr. 1996;22:996. [Google Scholar]
- 91.Rudge JS, Mather PE, Pasnikowski EM, Cai N, Corcoran T, Acheson A, Anderson K, Lindsay RM, Wiegand SJ. Endogenous BDNF protein is increased in adult rat hippocampus after a kainic acid induced excitotoxic insult but exogenous BDNF is not protective. Expl Neurol. 1998;149:398–410. doi: 10.1006/exnr.1997.6737. [DOI] [PubMed] [Google Scholar]
- 92.Sandberg K, Sanberg PR, Hanin I, Fisher A, Coyle JT. Cholinergic lesion of the striatum impairs acquisition and retention of a passive avoidance response. Behav Neurosci. 1984;98:162–165. doi: 10.1037//0735-7044.98.1.162. [DOI] [PubMed] [Google Scholar]
- 93.Scharfman HE. Dentate hilar cells with dendrites in the molecular layer have lower thresholds for synaptic activation by perforant path stimulation than granule cells. J Neurosci. 1991;11:1660–1673. doi: 10.1523/JNEUROSCI.11-06-01660.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Scharfman HE. Hyperexcitability of entorhinal cortex and hippocampus after application of aminooxyacetic acid (AOAA) to layer III of the rat medial entorhinal cortex in vitro. J Neurophysiol. 1996;76:2986–3001. doi: 10.1152/jn.1996.76.5.2986. [DOI] [PubMed] [Google Scholar]
- 95.Scharfman HE. Hyperexcitability in combined entorhinal/hippocampal slices of adult rat after exposure to brain-derived neurotrophic factor. J Neurophysiol. 1997;78:1082–1095. doi: 10.1152/jn.1997.78.2.1082. [DOI] [PubMed] [Google Scholar]
- 96.Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc natn Acad Sci USA. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schmidt-Kastner R, Humpel C, Wetmore C, Olson L. Cellular hybridization for BDNF, trkB, and NGF mRNAs and BDNF-immunoreactivity in rat forebrain after pilocarpine-induced status epilepticus. Expl Brain Res. 1996;107:331–347. doi: 10.1007/BF00230416. [DOI] [PubMed] [Google Scholar]
- 98.Schwartzkroin PA. Epilepsy: Models, Mechanisms, and Concepts. Cambridge University Press; Cambridge: 1993. [Google Scholar]
- 99.Schwartzkroin PA, Wheal HV. Electrophysiology of Epilepsy. Academic; New York: 1984. [Google Scholar]
- 100.Sheng M, Greenberg ME. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron. 1990;4:477–485. doi: 10.1016/0896-6273(90)90106-p. [DOI] [PubMed] [Google Scholar]
- 101.Simiand J, Keane PE, Morre M. The staircase test in mice: a simple and efficient procedure for primary screening of anxiolytic agents. Psychopharmacology. 1984;84:48–53. doi: 10.1007/BF00432023. [DOI] [PubMed] [Google Scholar]
- 102.Siuciak JA, Altar CA, Wiegand SJ, Lindsay RM. Antinociceptive effect of brain-derived neurotrophic factor and neurotrophin-3. Brain Res. 1994;633:326–330. doi: 10.1016/0006-8993(94)91556-3. [DOI] [PubMed] [Google Scholar]
- 103.Siuciak JA, Wong V, Pearsall D, Wiegand SJ, Lindsay RM. BDNF produces analgesia in the formalin test and modifies neuropeptide levels in rat brain and spinal cord areas associated with nociception. Eur J Neurosci. 1995;7:663–670. doi: 10.1111/j.1460-9568.1995.tb00670.x. [DOI] [PubMed] [Google Scholar]
- 104.Sloviter RS. Possible functional consequences of synaptic reorganization in the dentate gyrus of kainate-treated rats. Neurosci Lett. 1992;137:91–96. doi: 10.1016/0304-3940(92)90306-r. [DOI] [PubMed] [Google Scholar]
- 105.Snider WD, Lichtman JW. Are neurotrophins synaptotrophins. Molec cell Neurosci. 1996;7:433–442. doi: 10.1006/mcne.1996.0031. [DOI] [PubMed] [Google Scholar]
- 106.Somjen GG, Aitken PG, Czéh GL, Herreras JJ, Young JN. Mechanisms of spreading depression: a review of recent findings and a hypothesis. Can J Physiol Pharmac. 1992;70:S248–254. doi: 10.1139/y92-268. [DOI] [PubMed] [Google Scholar]
- 107.Soppet D, Escandon E, Maragos J, Middlemas DS, Reid SW, Blair J, Burton LE, Stanton BR, Kaplan DR, Hunter T, Nikolics K, Parada LF. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell. 1991;65:895–903. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- 108.Spangler EL, Ingram DK. Effects of inescapable shock on maze performance as a function of age in mice. Expl Aging Res. 1986;12:39–42. doi: 10.1080/03610738608259433. [DOI] [PubMed] [Google Scholar]
- 109.Spina MB, Squinto SP, Miller J, Lindsay RM, Hyman C. Brain-derived neurotrophic factor protects dopamine neurons against 6-hydroxydopamine and N-methyl-d-phenylpyridinium ion toxicity: involvement of the glutathione system. J Neurochem. 1992;59:99–106. doi: 10.1111/j.1471-4159.1992.tb08880.x. [DOI] [PubMed] [Google Scholar]
- 110.Squinto SP, Stitt TN, Aldrich TH, Davis S, Bianco SM, Radziejewski C, Glass DJ, Masiakowski P, Furth ME, Valenzuela DM, DiStefano PS, Yancopoulos GD. trkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell. 1991;65:885–893. doi: 10.1016/0092-8674(91)90395-F. [DOI] [PubMed] [Google Scholar]
- 111.Takei N, Sasaoka K, Inoue K, Takahashi M, Endo Y, Hatanaka H. Brain-derived neurotrophic factor increases the stimulation-evoked release of glutamate and the levels of exocytosis-associated proteins in cultured cortical neurons from embryonic rats. J Neurochem. 1997;68:370–375. doi: 10.1046/j.1471-4159.1997.68010370.x. [DOI] [PubMed] [Google Scholar]
- 112.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thiébot MH, Soubri P, Simon P, Boissier JR. Dissociation de deux composantes du comportement chez le rat sous l'effet de psychotropes. Application á l'étude des anxiolytiques. Psychopharmacologia. 1973;31:77–90. doi: 10.1007/BF00429300. [DOI] [PubMed] [Google Scholar]
- 114.Tuff LP, Racine RJ, Adamec R. The effects of kindling on GABA-mediated inhibition in the dentate gyrus of the rat I. Paired pulse depression. Brain Res. 1983;27:79–90. doi: 10.1016/0006-8993(83)90909-5. [DOI] [PubMed] [Google Scholar]
- 115.Ventimiglia R, Mather PE, Jones BE, Lindsay RM. The neurotrophins BDNF, NT-3 and NT-4/5 promote survival and morphological and biochemical differentiation of striatal neurons in vitro. Eur J Neurosci. 1995;7:213–222. doi: 10.1111/j.1460-9568.1995.tb01057.x. [DOI] [PubMed] [Google Scholar]
- 116.Wu K, Suen PC, Levine ES, Xu JL, Mount HRJ, Lin SY, Black IB. BDNF rapidly enhances phosphorylation of the postsynaptic NMDA receptor subunit 1. Soc Neurosci Abstr. 1997;23:59. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yan Q, Elliott J, Snider WJ. Brain-derived neurotrophic factor rescues spinal motor neurons from axotomy-induced cell death. Nature. 1992;360:753–755. doi: 10.1038/360753a0. [DOI] [PubMed] [Google Scholar]
- 118.Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA. Expression of brain-derived neurotrophic factor (BDNF) protein in the adult rat central nervous system. Neuroscience. 1997;78:431–448. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]
- 119.Zhou XF, Parada LF, Soppet D, Rush RA. Distribution of trkB tyrosine kinase immunoreactivity in the rat central nervous system. Brain Res. 1993;622:63–70. doi: 10.1016/0006-8993(93)90802-t. [DOI] [PubMed] [Google Scholar]
- 120.Zhou XF, Rush RA. Endogenous brain-derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience. 1996;74:945–951. doi: 10.1016/0306-4522(96)00237-0. [DOI] [PubMed] [Google Scholar]