

Abstract
Two myosin light chain (MLC) kinase (MLCK) proteins, smooth muscle (encoded by mylk1 gene) and skeletal (encoded by mylk2 gene) MLCK, have been shown to be expressed in mammals. Even though phosphorylation of its putative substrate, MLC2, is recognized as a key regulator of cardiac contraction, a MLCK that is preferentially expressed in cardiac muscle has not yet been identified. In this study, we characterized a new kinase encoded by a gene homologous to mylk1 and -2, named cardiac MLCK, which is specifically expressed in the heart in both atrium and ventricle. In fact, expression of cardiac MLCK is highly regulated by the cardiac homeobox protein Nkx2-5 in neonatal cardiomyocytes. The overall structure of cardiac MLCK protein is conserved with skeletal and smooth muscle MLCK; however, the amino terminus is quite unique, without significant homology to other known proteins, and its catalytic activity does not appear to be regulated by Ca2+/calmodulin in vitro. Cardiac MLCK is phosphorylated and the level of phosphorylation is increased by phenylephrine stimulation accompanied by increased level of MLC2v phosphorylation. Both overexpression and knockdown of cardiac MLCK in cultured cardiomyocytes revealed that cardiac MLCK is likely a new regulator of MLC2 phosphorylation, sarcomere organization, and cardiomyocyte contraction.
Keywords: kinase, transcription, contraction
Phosphorylation of both myosin heavy chain and myosin light chain (MLC) affects motor activity and thick filament assembly.1 In smooth muscle cells, phosphorylation of MLC2 by smooth muscle MLCK is thought to be responsible for the initiation of contraction.2 In skeletal and cardiac muscles, however, initiation of muscle contraction depends on voltage-gated L-type Ca2+ channels in the plasma membrane and T-tubules. Increased local Ca2+ concentrations allow the sarcoplasmic reticulum to release large amounts of Ca2+, which bind to troponin C followed by myosin–actin cross-bridge formation. During this process, MLCK potentiates peak tension in skeletal muscle1,3 and the force and rate of cross-bridge recruitment in cardiac myocytes.4,5
To date, smooth muscle (encoded by mylk1 gene) and skeletal (encoded by mylk2 gene) MLCKs have been characterized.3 Mouse skeletal muscle MLCK is predominantly expressed in skeletal muscle, and mouse smooth muscle MLCK is expressed in several tissues but predominantly in smooth muscle.6,7 Mutations in human skeletal MLCK on human chromosome 20 have been mapped to a disease locus for familial cardiac hypertrophy (Online Mendelian Inheritance in Man no. 606566), suggesting that abnormal function of skeletal MLCK stimulates cardiac hypertrophy.8 However, the abundance of skeletal MLCK expression in the heart is controversial,8–10 and gene-targeted mice for skeletal MLCK appear to have normal cardiac function.10 Short-form (130-kDa) smooth muscle MLCK is expressed in the heart at lower levels than those detected in smooth muscle–rich organs such as gut, uterus, and lung.6,7 Mice with ablation of long-form smooth muscle MLCK appear to have normal cardiac function,11 and those with short-form ablation remain to be studied. These results suggest that an additional MLCK is preferentially expressed and functional in the heart because MLC2 phosphorylation in cardiac muscle is a key regulator of heart contraction.5
In the process of identifying genes regulated by the cardiac homeobox transcription factor Nkx2-5, we identified a gene product highly homologous to the previously characterized skeletal and smooth muscle MLCK. The sequence of this MLCK homolog has been available (National Center for Biotechnology Information [NCBI] UniGene no. Rn.43838 [rat], Mm.32804 [mouse]), yet limited information regarding this MLCK gene has sometimes confounded its identity with the previously characterized skeletal and smooth muscle MLCKs. In this study, we report the initial characterization of this MLCK regarding its cardiac-specific expression, intracellular localization, catalytic activity, and potential functions in sarcomere organization and cardiac contraction.
Materials and Methods
The following materials and methods used for this study are described in detail in the online data supplement, available at http://circres.ahajournals.org.
Cardiomyocyte preparation
Animal models
Cloning of cardiac, skeletal, and smooth muscle MLCK
Production and infection of adenovirus
Glutathione S-transferase (GST) fusion proteins (cardiac MLCK, MLC2v, MLC2a)
Production of anti–cardiac MLCK antibody
Northern and Western blotting
Immunostaining
Real-time PCR
Phosphorylation assays in vitro and in cardiomyocytes
Two-dimensional gel electrophoresis
Simultaneous measurements of cell shortening and intracellular free calcium
Statistical analyses
Results
Identification of a Cardiac-Specific MLCK as a Downstream Target of Cardiac Homeobox Protein, Nkx2-5
In Nkx2-5 knockdown neonatal rat cardiomyocytes (Figure 1A) and inducible Nkx2-5 knockout hearts (Figure 1B), reduced expression of Nkx2-5 dramatically decreased mRNA expression of the MLCK homolog (NCBI UniGene no. Rn.43838 [rat], Mm.32804 [mouse]). Because Nkx2-5 expression is nearly restricted to cardiac myocytes at the postnatal stage,12–14 we tested whether expression of the MLCK homolog is also cardiac-specific. Multitissue Northern blotting readily detected mRNA of the MLCK homolog specifically in the heart in both ventricle and atrium at neonatal and adult stages (Figure 1C, top gels).
Figure 1.
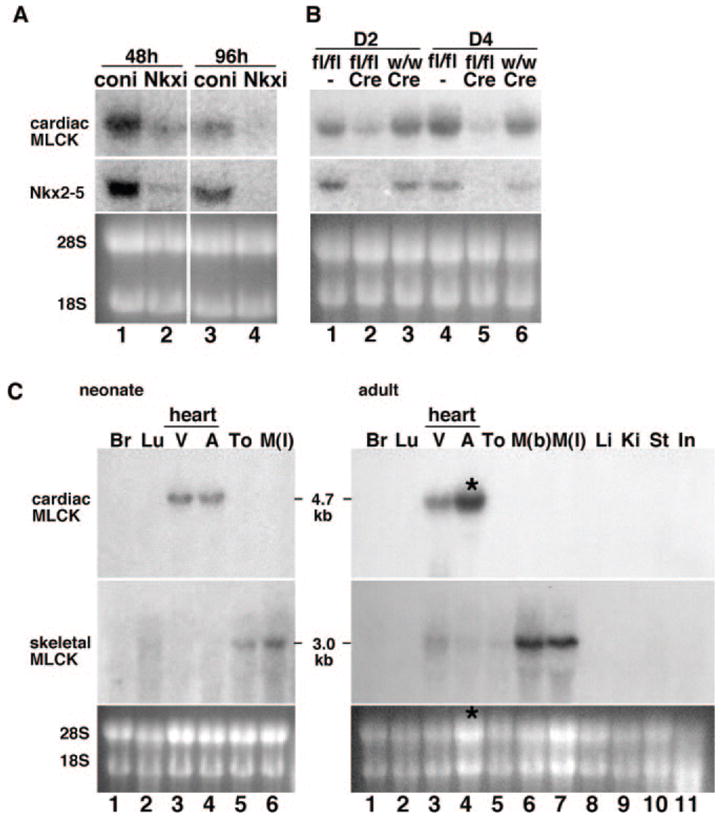
Identification of cardiac-specific MLCK as an Nkx2-5 downstream target gene. A, Northern blotting showed that Nkx2-5 knockdown using adenovirus shRNA-reduced expression of cardiac MLCK 48 hours (lane 1 vs 2) and 96 hours after adenovirus infection (lane 3 vs 4). B, Tamoxifen-induced Nkx2-5 knockout demonstrates reduction of cardiac MLCK expression at postnatal day 2 (D2) (lane 2 vs lanes 1 and 3) and day 4 (D4) (lane 5 vs lanes 4 and 6). Of note, tamoxifen was injected into the pregnant female within 24 hours before delivery. Coni indicates control RNAi; Nkxi, Nkx2-5-RNAi; fl, floxed-Nkx2-5; fl/fl, homozygous for floxed-Nkx2-5; w, wild-type; w/w, homozygous for wild-type; and Cre, Cre-transgene. C, Tissue-specific expression of cardiac MLCK mRNA was examined by Northern blotting (top gels) and compared with that of skeletal MLCK (middle gels) in the neonatal stage (left) and adult stage (right). Increased loading of RNA isolated from adult atrium resulted in higher expression of MLCK in adult atrium vs ventricle (*, lane 4, right). Br indicates brain; Lu, lung; V, ventricle; A, atrium; To, tong; M(l), leg muscle; M(b), back muscle; Li, liver; Ki, kidney; St, stomach; and In, intestine.
Hybridization of the same membrane with the skeletal MLCK-specific probe showed that the expression of skeletal MLCK is below the level of detection in the neonatal heart (Figure 1C, neonate, middle) and only weakly detected in adult ventricle (Figure 1C, adult, middle). The MLCK homolog is hereafter called cardiac MLCK.
The mouse cardiac MLCK gene (chromosome 8) encodes 795 aa with a predicted molecular mass of 86 kDa, excluding posttranslational modifications. The protein consists of a conserved kinase domain at the carboxyl terminus with 58% identity with skeletal MLCK and 44% identity with smooth muscle MLCK; however the amino-terminal domain is quite unique, with no significant homology to other known proteins, including MLCKs. Comparison of protein structure of cardiac, skeletal, and smooth muscle MLCKs, including an alternative gene product, telokin, which lacks a catalytic domain,3 is shown (Figure 2A). The amino acid sequence alignment of carboxyl terminus of cardiac MLCK to skeletal and long-form smooth muscle MLCK is shown in Figure 2B.
Figure 2.
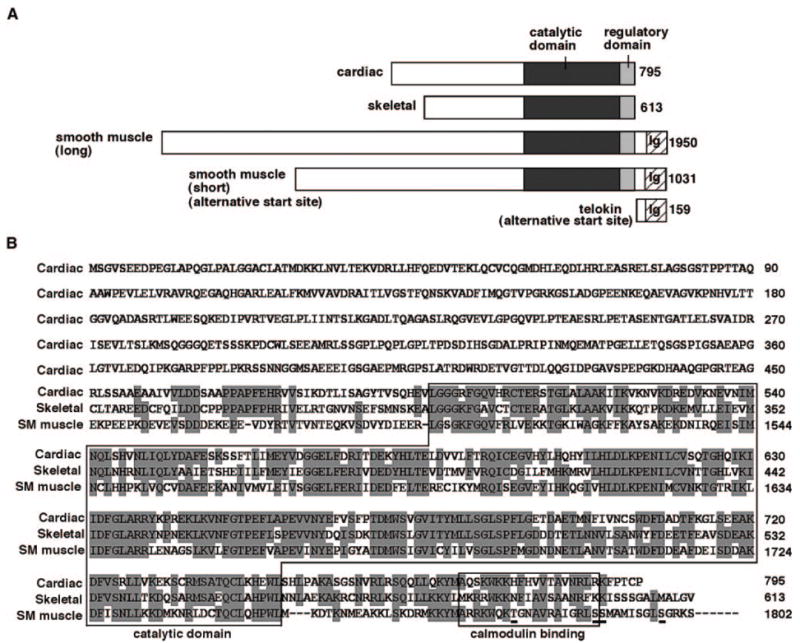
Structure of mouse cardiac MLCK (GenBank accession number EU403565) protein compared with skeletal and smooth muscle MLCKs. A, Schematic of cardiac MLCK protein structure compared with skeletal and long and short forms of smooth muscle MLCK and variant of smooth muscle MLCK, telokin. Protein sequences are retrieved from mouse skeletal MLCK (XP_979674), smooth muscle MLCK (long) (NP647461), smooth muscle MLCK (short),6 and telokin (AAG34169). Ig indicates immunoglobulin C2 like motif. B, Amino acid sequence of cardiac MLCK with alignment among cardiac, skeletal, and smooth muscle (long-form) MLCK at the carboxyl terminus. Identical amino acids between at 2 proteins are shaded. Putative Ca2+/calmodulin binding kinase regulatory domain locating carboxyl terminus to catalytic domain; 2 contiguous serine residues, which are targets of upstream kinases3 and 2 additional autophosphorylation sites29 identified in the smooth muscle MLCK are underlined.
Cardiac MLCK Protein Expression
We generated an affinity-purified antibody against the unique amino terminus of cardiac MLCK (amino acids 28 to 463) (Figure 3A, lanes 1 and 2). The specificity of the antibody was confirmed by its reactivity to hemagglutinin (HA)-tagged full-length cardiac MLCK (Figure 3A, lane 3), but not to HA-tagged full-length skeletal MLCK even at 10-fold abundance (Figure 3A, lanes 4 and 5), to which cardiac MLCK has a higher homology compared with smooth muscle MLCK. In neonatal heart lysates, cardiac MLCK protein with an approximate molecular mass of 90 kDa was readily detected in both ventricle and atrium at similar expression levels (Figure 3B), and its protein expression in heart lysates was ≈0.5 μg/mg (equivalent to 2.3 ng of GST–cardiac MLCK in 5 μg of heart lysates) following densitometric analysis (Figure 3C), which is lower than skeletal MLCK levels in skeletal muscle previously reported (≈5 to 10 ng of skeletal MLCK in 2 μg of skeletal muscle lysates).6 The antibody against the amino terminus of cardiac MLCK does not cross-react with other proteins in skeletal muscle and lung lysates in which skeletal or smooth muscle MLCK is abundantly expressed (Figure 3C, lanes 4 and 5).6 An additional band migrating around 60 kDa was detected using the anti–cardiac MLCK antibody in heart lysates, which may be an alternatively spliced isoform or a degradation product of cardiac MLCK (see Figure 7). We confirmed the expression of 130-kDa short-form smooth muscle MLCK in the heart6 (estimated concentration 0.025 to 0.05 μg/mg), skeletal muscle, and more abundantly in the lung (0.5 to 1 μg/mg) (supplemental Figure I). Thus, cardiac MLCK protein expression is ≈10- to 20-fold more abundant than smooth muscle MLCK in the neonatal heart.
Figure 3.
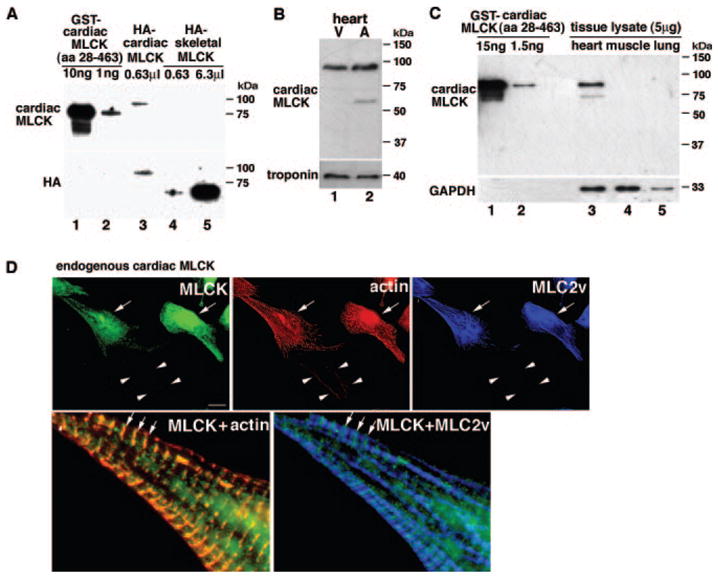
Cardiac MLCK protein expression and intracellular localization. A, Western blotting using anti–cardiac MLCK antibody against GST–cardiac MLCK (amino acids 28 to 463) (lanes 1 and 2) detected HA-tagged full-length cardiac MLCK protein (lane 3) but not HA-tagged full-length skeletal MLCK (lanes 4 and 5). Anti-HA antibody recognizes HA-tagged cardiac and skeletal MLCK (lanes 3 to 5). B, Western blotting using anti–cardiac MLCK antibody detected a similar amount of MLCK protein in protein lysates isolated from mouse neonatal ventricles (lane 1) and atria (lane 2). V indicates ventricle; A, atrium. C, Cardiac MLCK protein expression in heart lysates is equivalent to 2.3 ng of GST–cardiac MLCK in 5 μg of heart lysates. The antibody does not cross-react with other proteins using similar amounts of tissue lysates from skeletal muscle and lung (lanes 4 and 5). D, Coimmunostaining of cardiac MLCK, actin (phalloidin), and MLC2v. Endogenous cardiac MLCK protein is localized diffusely in the cytoplasm with occasional striated patterns (arrows) that overlap with actin but not with striated MLC2v. Arrowheads indicate noncardiomyocyte weakly detected by phalloidin but not by cardiac MLCK nor MLC2 antibodies. Bars=10 μm.
Figure 7.
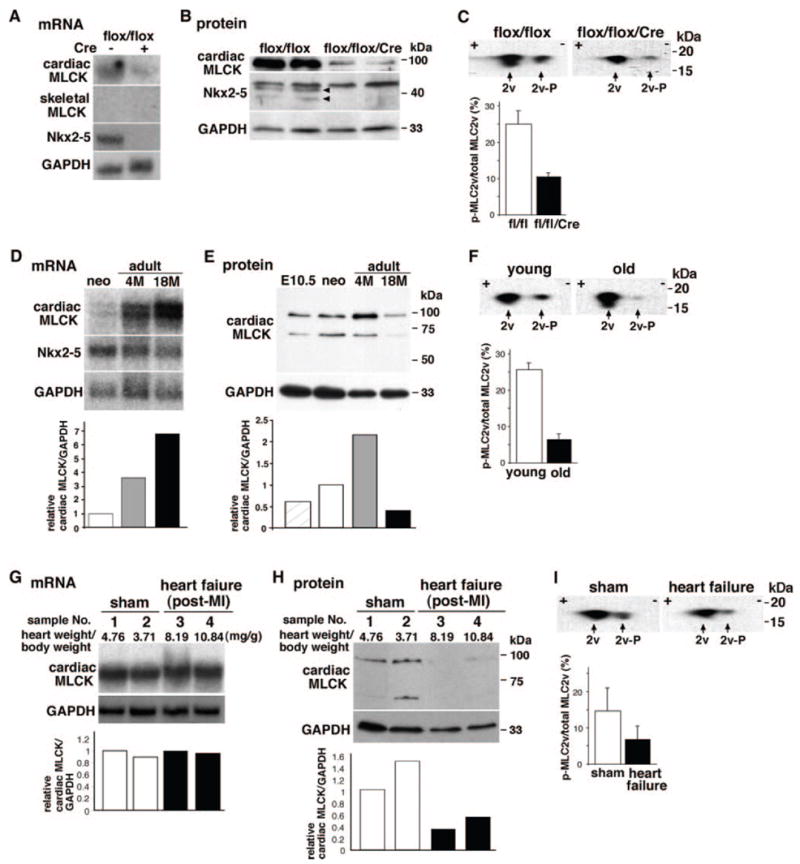
Cardiac MLCK expression and MLC2v phosphorylation in mice with Nkx2-5 knockout, aging, and post–myocardial infarction (MI) heart failure. A and B, Decreased expression of cardiac MLCK mRNA and protein in Nkx2-5 knockout hearts at postnatal day 12. Skeletal MLCK mRNA was not detected by Northern blotting. C, Unphosphorylated (left, with higher pI) and phosphorylated (right, with lower pI) MLC2v examined in 2D electrophoresis, followed by Western blotting with anti-MLC antibody. Relative amounts of phosphorylated to total MLC2v are shown (mean±SE, n=2). D, Expression of cardiac MLCK and Nkx2-5 mRNA in neonatal, adult (4 months) and aged hearts (18 months). Relative expression of cardiac MLCK normalized to GAPDH is shown with the value in neonatal heart defined as 1. E, Expression of cardiac MLCK protein in embryonic day 10.5, neonatal, adult, and aged hearts. Relative expression of cardiac MLCK normalized to GAPDH is shown with the value in neonatal heart defined as 1. F, Level of MLC2v phosphorylation in young (PD 12) and aged hearts. Relative amounts of phosphorylated to total MLC2v are shown (mean±SE; young, n=3; old, n=6 from 2 mice at 18 and 21 months). G, Noninfarcted upper septal tissue dissected from mice 3 weeks after coronary ligation (3 month old) was analyzed for cardiac MLCK mRNA expression: 2 sham-operated (lanes 1, 2) and 2 heart failure (lanes 3 and 4) mice. Values of heart weight/body weight are indicated. Additional experimental conditions and parameters of cardiac function have been described previously35 and in Materials and Methods. Relative expression of cardiac MLCK normalized to GAPDH is shown with the value in sample 1 defined as 1. H, Cardiac MLCK protein expression in tissue lysates from the same mice used in G is shown with the value in sample 1 defined as 1. I, Level of MLC2v phosphorylation examined in mice with sham-operated and heart failure. Relative amounts of phosphorylated to total MLC2v are shown (mean±SE; sham, n=4 from 2 mice; heart failure, n=4 from 2 mice).
Endogenous cardiac MLCK is diffusely localized in the cytoplasm of cardiomyocytes (Figure 3D, green, arrows; arrowheads, noncardiomyocyte); however, in some areas, striated staining of cardiac MLCK was observed. Interestingly, enlarged image of cardiomyocytes coimmunostained to detect actin (Figure 3D, red, localizing at I bands) and MLC2v (Figure 3D, blue, localizing at A bands) demonstrated that striated MLCK staining was colocalized with actin (Figure 3D, green and red) but not with MLC2v (Figure 3D, green and blue). Specificity of immunostaining and additional endogenous cardiac MLCK stainings are shown in supplemental Figure II.
Cardiac MLCK Phosphorylates MLC2 In Vitro and in Cardiomyocytes as Well as Potentially MLCK Itself
In vitro kinase assay demonstrated incorporation of 32P to GST-MLC2v and GST-MLC2a fusion proteins using HA-tagged cardiac MLCK expressed in 293 cells in the absence of Ca2+/calmodulin (supplemental Figure III), indicating that ectopically expressed cardiac MLCK is sufficient for phosphorylation of MLC2v and MLC2a without Ca2+/calmodulin. Because catalytic activity of skeletal and smooth muscle MLCK is Ca2+/calmodulin-dependent, this finding was tested in additional kinase assays in a quantitative manner and confirmed that cardiac MLCK phosphorylated MLC2v in the absence of Ca2+/calmodulin (Figure 4A, top, cardiac MLCK, control), and that addition of EGTA (Figure 4A, top, +EGTA), or Ca2+/calmodulin (Figure 4A, top, +Ca2+/calmodulin) had little effect on the catalytic activity of cardiac MLCK. Under the same condition, control experiments using HA-tagged skeletal MLCK performed side by side demonstrated strong Ca2+/calmodulin-dependent kinase activities to MLC2v consistent with previous studies (Figure 4A, bottom, skeletal MLCK). In addition, HA-tagged cardiac as well as skeletal MLCKs purified with proteinase inhibitors appeared as single bands in Western blotting (Figure 3A, lanes 3 to 5). These results indicate that Ca2+/calmodulin-independent cardiac MLCK kinase activity is not likely attributable to our experimental conditions or to selective proteolytic cleavages of the Ca2+/calmodulin binding domain in cardiac MLCK.
Figure 4.
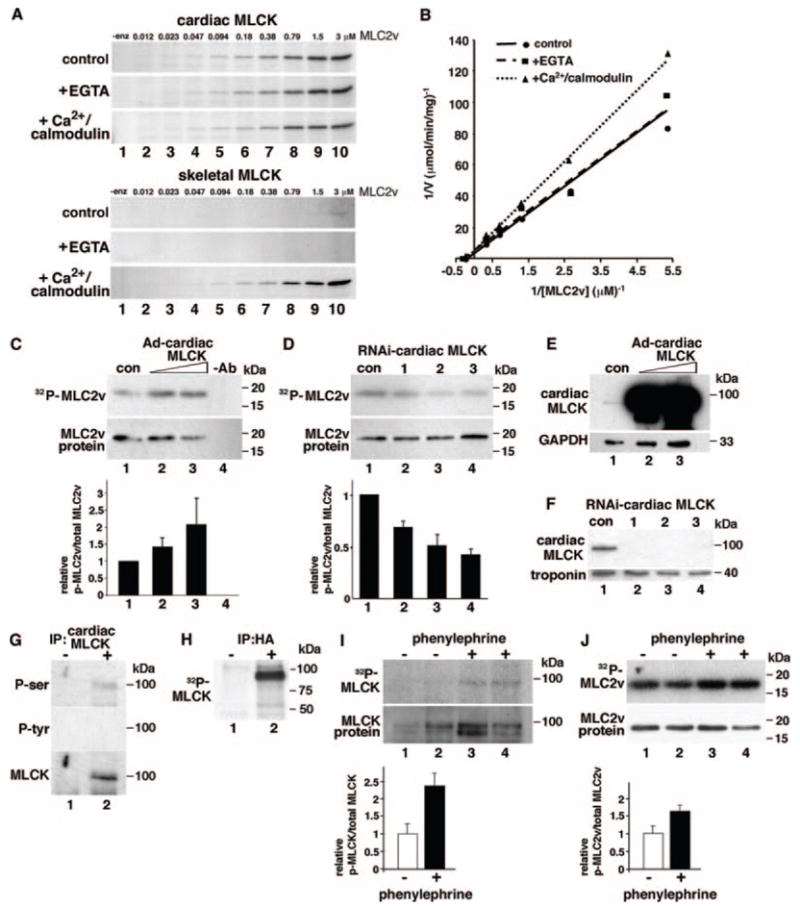
Cardiac MLCK phosphorylates MLC2 and potentially MLCK itself. A, Autoradiogram of GST-MLC2v phosphorylation at various concentrations (0.012 to 3 μmol/L) catalyzed by cardiac MLCK (upper gels) and skeletal MLCK (lower gels). Control experiments without EGTA or Ca2+/calmodulin (control), with EGTA (+EGTA, 5 mmol/L), or with Ca2+/calmodulin (+Ca2+/calmodulin, 1 mmol/L Ca2+, and 1 μmol/L calmodulin) are shown. B, Kinetic analysis of cardiac MLCK using double reciprocal Lineweaver–Burk plot (0.18 to 3 μmol/L). Calculated kinetic values of cardiac MLCK were as follows: Km (μmol/L) 4.3±1.5 (without EGTA or Ca2+/calmodulin), 2.9±0.8 (with EGTA), and 3.9±1.2 (with Ca2+/calmodulin); Vmax (μmol/min per milligram): 0.26±0.06, (without EGTA or Ca2+/calmodulin), 0.18±0.03 (with EGTA), and 0.18±0.03 (with Ca2+/calmodulin). Values are expressed as means±SE (n=2). C, MLC2v phosphorylation was examined in neonatal cardiomyocytes with increased expression of cardiac MLCK by infection of Ad-cardiac MLCK (lanes 2, 3 vs lane 1). The relative amounts of phosphorylated to total MLC2v protein are shown below in comparison with control cardiomyocytes. Lane 1: Ad-βgal, 10 multiplicities of infection (mois); lane 2: Ad-cardiac MLCK, 2 mois; lane 3: Ad-cardiac MLCK, 10 mois. The relative values are expressed as means±SE (n=2). D, Decreased expression of cardiac MLCK using 3 different shRNAs (lanes 2 to 4) decreased MLC2v phosphorylation compared with scrambled adenovirus-shRNA (lane 1). The relative values of phosphorylated to total MLC2v protein in comparison with control cardiomyocytes are expressed as means±SE (n=4). E, Increased expression of cardiac MLCK by infection of Ad-cardiac MLCK adenovirus detected by Western blotting with anti–cardiac MLCK antibody. Lane 1: Ad-βgal, 10 mois; lane 2: Ad-cardiac MLCK, 2 mois; lane 3: Ad-cardiac MLCK, 10 mois. F, Reduced expression of cardiac MLCK by 3 different shRNA is detected by Western blotting with anti–cardiac MLCK antibody. Lane 1, shRNA with scrambled sequence; lanes 2 to 4, shRNA targeting to 3 different sequences. G, Cardiac MLCK immunoprecipitated from neonatal ventricular cardiomyocytes was blotted with anti–phospho-serine or –phospho-tyrosine antibodies. Phospho-serine antibody reacted to cardiac MLCK protein. H, In vitro kinase assay showed 32P-incorporated HA-tagged MLCK expressed in 293 cells. I, Phenylephrine (30 μmol/L for 30 minutes) increased cardiac MLCK phosphorylation in cardiomyocytes infected with Ad-HA cardiac MLCK (1 moi). The relative amounts of phosphorylated cardiac MLCK to total cardiac MLCK protein are shown below in comparison with control cardiomyocytes without phenylephrine treatments (mean±SE, n=2). J, Phenylephrine (30 μmol/L for 30 minutes) increased cardiac MLC2v phosphorylation in rat neonatal cardiomyocytes. The relative values of phosphorylated to total MLC2v protein in comparison with control cardiomyocytes without phenylephrine are shown (mean±SE, n=2).
The estimated kinetic constants determined by Lineweaver–Burk plot were as follows: Km, 4.3±1.5 μmol/L; Vmax, 0.26±0.06 μmol/min per milligram; Vmax/Km ratio, 0.06 (without EGTA and Ca2+/calmodulin) (Figure 4B). The low Km value of cardiac MLCK indicating high affinity to the substrate is equivalent to skeletal MLCK to skeletal MLC (3.5 μmol/L) and smooth muscle MLCK to smooth muscle MLC (6 to 11 μmol/L).15,16 However, an indicator of efficiency of catalysis, Vmax/Km ratio of cardiac MLCK, is lower than this ratio for skeletal and smooth muscle MLCK toward their MLC substrates isolated from the same tissue (9.3 and 3.5, respectively).17 Therefore cardiac MLCK appears to have a high affinity and relatively low catalytic efficiency to MLC2v.
In cardiomyocytes, overexpression of cardiac MLCK increases MLC2v phosphorylation nearly 2.1-fold in a dose-dependent manner (Figure 4C). Conversely, decreased MLCK expression achieved by infection of 3 MLCK–short hairpin (sh)RNA adenoviruses targeting different MLCK sequences (Figure 4D, RNA interference [RNAi] cardiac MLCK-1, -2, -3) decreased MLC2v phosphorylation levels by 30% to 55%. The levels of cardiac MLCK protein expression in overexpressed (Figure 4E) and knockdown cardiomyocytes (Figure 4F) are shown.
Catalytic activities of smooth muscle and skeletal MLCKs are regulated by phosphorylation by upstream kinases as well as MLCKs themselves.18 Phosphorylation of cardiac MLCK was detected with anti–phospho-serine antibody (Figure 4G, P-ser) but not with anti–phospho-tyrosine antibody (Figure 4G, P-tyr; supplemental Figure IV). We also detected incorporation of 32P to cardiac MLCK itself in an in vitro kinase assay in which immunoprecipitated exogenous cardiac MLCK proteins expressed in 293 cells were used (Figure 4H). This result suggests that cardiac MLCK autophosphorylates MLCK or that MLCK upstream kinases physically interact with cardiac MLCK in 293 cells. The level of cardiac MLCK phosphorylation was increased with phenylephrine stimulation (Figure 4I, lanes 1 and 2 versus lanes 3 and 4), accompanied by an increased MLC2v phosphorylation (Figure 4J, lanes 1 and 2 versus lanes 3 and 4). Potential phosphorylation sites and kinases predicted by amino acid sequence conserved between mouse and rat cardiac MLCK, including protein kinase A, are listed in supplemental Table I.
Cardiac MLCK Promotes Sarcomere Organization and Increases Cardiomyocyte Contractility
We observed that cardiomyocytes in which cardiac MLCK was overexpressed displayed organized sarcomere structures characterized by straight, thick, striated actin bundles, as had been seen with overexpression of skeletal MLCK (Figure 5A, Ad-β-galactosidase [βgal] versus Ad-cardiac MLCK).19 Phalloidin intensity in individual cardiomyocyte was increased in the MLCK-overexpressing cardiomyocytes compared with control βgal-infected cardiomyocytes (Figure 5A and 5B). In MLCK knockdown cardiomyocytes using 3 RNAis showed slight changes in peripheral structure up to 96 hours after adenoviral infection (Figure 5C, RNAi control versus RNAi cardiac MLCK) accompanied by reduced phalloidin intensity in individual cardiomyocyte compared with cardiomyocytes infected with control adenoviral RNAi (Figure 5D). These results suggest that cardiac MLCK is involved in sarcomere organization.
Figure 5.
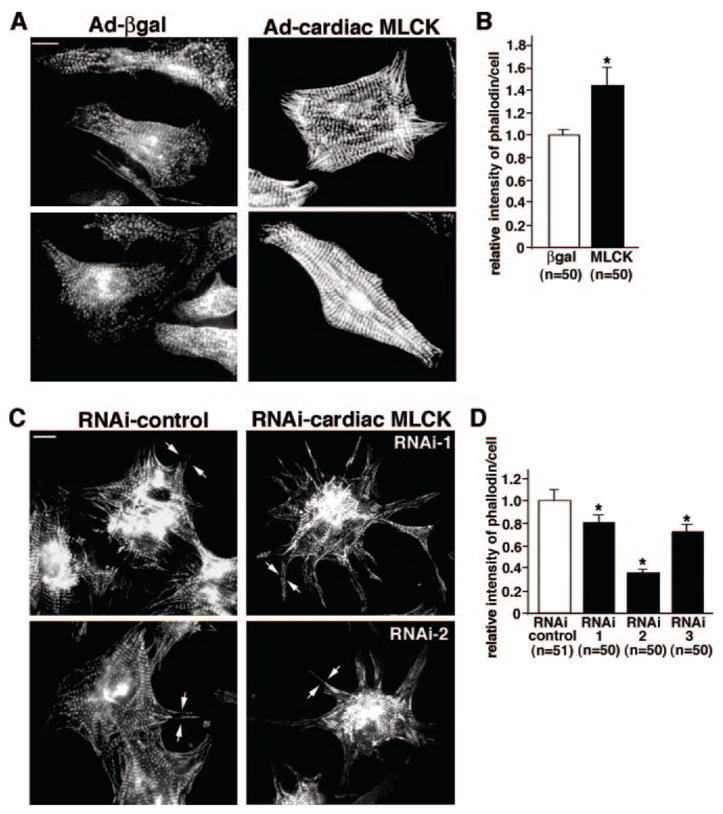
Effects of cardiac MLCK in sarcomere organization. A, Overexpression of cardiac MLCK by Ad-cardiac MLCK adenovirus (10 mois) promotes sarcomere organization detected by phalloidin compared with cells infected with Ad-βgal (10 mois). Bars=10 μm. B, Relative intensity of phalloidin staining in individual cardiomyocytes overexpressing cardiac MLCK is shown with the value in control cardiomyocytes defined as 1 (mean±SE). C, Reduced expression of cardiac MLCK using 2 different shRNAs, RNAi-1 and RNAi-2, does not disturb sarcomere organization centrally with slight changes in peripheral structure (arrows). Bars=10 μm. D, Relative intensity of phalloidin staining in individual cardiomyocyte treated with 3 different shRNAs is shown with the value in control cardiomyocytes defined as 1 (mean±SE).
Consistent with morphological changes, overexpression of MLCK resulted in significantly increased cardiomyocyte contraction amplitude and kinetics of contraction and relaxation without a significant change in intracellular Ca2+ transients (Figure 6A through 6C, Ad-βgal versus Ad-cardiac MLCK). On the other hand, MLCK knockdown, targeting 3 specific sequences was without effect on cardiomyocyte contraction at the standard Ca2+ superfusate concentration (1.2 mmol/L) but with significant effect at the higher Ca2+ superfusate concentration (2.5 mmol/L), with the exception of −dL/dT in RNAi-1 (data not shown). These results suggest that alteration in sarcomere organization in MLCK knockdown cardiomyocytes may have an effect on function under increased demand.
Figure 6.
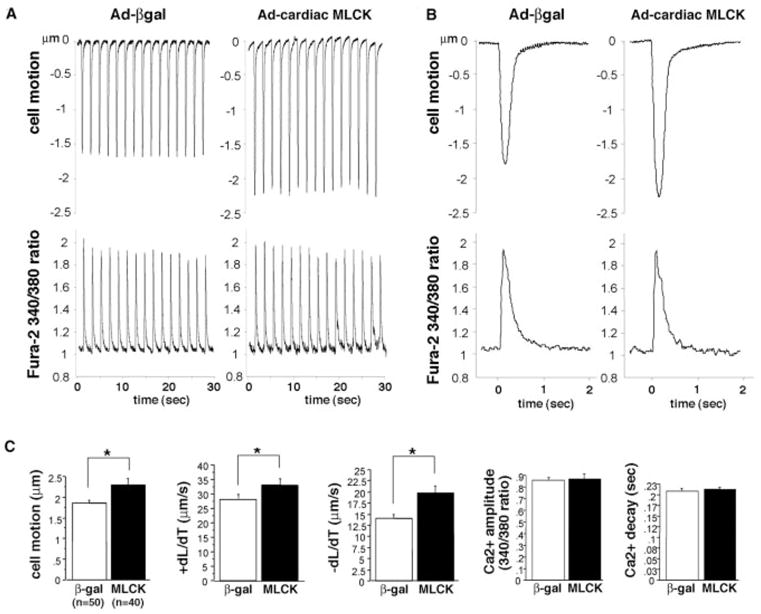
Cardiac MLCK potentiates cardiomyocyte contraction. A, Representative tracings of cell motion (top) and simultaneous Ca2+ transients (bottom) in an isolated rat neonatal cardiomyocytes obtained continuously for 30 seconds. Ten multiplicities of infection of adenovirus encoding βgal or cardiac MLCK were infected. B, Representative tracings of a single contraction are shown. C, Summarized data obtained from multiple cardiomyocytes demonstrate that increased cardiac MLCK expression significantly increases cell motion, +dL/dT (contraction) and −dL/dT (relaxation), without a significant change in Ca2+ amplitude and decay. Values are expressed as means±SE.
Cardiac MLCK Expression and MLC2v Phosphorylation in Mice With Nkx2-5 Knockout, Aging, and Post–Myocardial Infarction Heart Failure
To examine catalytic activities of cardiac MLCK in vivo, we first examined cardiac MLCK expression and MLC2v phosphorylation levels in Nkx2-5 knockout hearts at postnatal day 12 when expression of cardiac MLCK mRNA (Figure 7A) and protein (Figure 7B) were markedly reduced. Skeletal MLCK mRNA expression in Nkx2-5 knockout hearts was below the level of detection by Northern blotting (Figure 7A, skeletal MLCK); however, using quantitative real-time PCR, skeletal (2.55±0.04 fold, n=2) and smooth muscle MLCK (1.48±0.02 fold, n=2) expression were increased. Despite this apparent compensatory increase, the level of MLC2v phosphorylation was decreased in hearts from Nkx2-5 knockout mice by nearly 60% compared with age-matched control mice (Figure 7C).
Cardiac MLCK mRNA increased during development from neonatal to adult stages and persisted in the aged hearts (Figure 7D). Of note, greater separation of RNA by agarose gel electrophoresis revealed 2 hybridized bands near 4.7 kb with similar intensities. Cardiac MLCK protein increased in hearts from embryonic day 10.5, neonates and adult, but was decreased in aged hearts (Figure 7E). Consistent with decreased cardiac MLCK protein in aged hearts (18 and 21 months old), MLC2v phosphorylation was decreased in aged hearts compared with postnatal day 12 hearts (Figure 7F).
We next examined cardiac MLCK expression in a post–myocardial infarction mouse model of heart failure 3 weeks after coronary artery ligation. At the mRNA level, cardiac MLCK expression in noninfarcted upper ventricular septal tissue was similar to tissue from sham-operated age-matched mice (Figure 7G, lanes 1 and 2 versus lanes 3 and 4). In contrast, cardiac MLCK protein was decreased in heart failure tissue compared with control tissue (Figure 7H, lanes 1 and 2 versus lanes 3 and 4). Furthermore, levels of MLC2v phosphorylation were decreased in heart failure compared with controls (Figure 7I). The lack of concordance between mRNA versus protein levels in neonatal and aged hearts and in failed hearts suggests altered posttranscriptional regulations of cardiac MLCK in aging and heart failure.
Discussion
In this study, we isolated a homolog of skeletal and smooth muscle MLCK that is preferentially expressed in the heart, herein named cardiac MLCK. Expression of cardiac MLCK mRNA was markedly downregulated shortly after reduction of Nkx2-5 expression by Nkx2-5 knockdown and inducible Nkx2-5 knockout. Nkx2-5 expression is nearly restricted to the heart in the postnatal stage,12–14 and expression of cardiac MLCK, its downstream target (either direct or indirect), was detected only in the heart using multitissue Northern blotting.
Cardiac MLCK has a similar overall structure to known skeletal and smooth muscle MLCKs and has a high affinity to MLC2v similar to skeletal MLCK to skeletal muscle MLC2 and smooth muscle MLCK to smooth muscle MLC2.17 However, its catalytic efficiency is lower, and it was not regulated by Ca2+/calmodulin or EGTA in vitro. Notably, for smooth muscle MLCK, which is also expressed in the heart, the amino acid sequence of substrates appears to be critical for affinity and catalytic activity, particularly an arginine residue in the third-position amino terminus to the phosphorylated serine residue (smooth muscle MLC [Arg-Ala-Thr-Ser]).15,16,20 The catalytic activity of smooth muscle MLCK toward skeletal MLC2, in which the critical Arg residue is replaced with Gly similar to MLC2v (skeletal MLC [Gly-Gly-Ser-Ser], MLC2v [Gly-Gly-Thr-Ser]), was reported as a Km value of 94 μmol/L and a Vmax/Km ration of 0.03.17 If similar values are applicable to MLC2v, these data imply that cardiac MLC2v may be as good a substrate for cardiac MLCK (Vmax/Km 0.06) as it is for smooth muscle MLCK but with distinct expression levels in neonatal hearts.
Under physiological conditions, the level of MLC2v phosphorylation is maintained relatively constant by well-balanced phosphorylation and phosphatase-induced dephosphorylation.21 Elevation of cytoplasmic [Ca2+] induced by infusion of Ca2+ did not increase MLC2v phosphorylation consistently.22–24 If Ca2+/calmodulin-independent catalytic activity, as well as high-affinity and relatively low catalytic efficiencies of cardiac MLCK toward MLC2v demonstrated in vitro, are applicable to in vivo, these previous studies may reflect functions of cardiac MLCK in the heart.
Increased expression of cardiac MLCK induced sarcomere organization in neonatal cardiomyocytes, as has been observed by overexpression of skeletal MLCK.19 Ser19 phosphorylation of MLC2 leading to potentiation of the force and speed of contraction has been well studied in smooth and skeletal muscle.3,18 Our findings demonstrate that overexpression of cardiac MLCK enhances cardiomyocyte contraction, likely because of a combination of increased MLC2 phosphorylation and organized sarcomere structure stably formed within 36 to 48 hours after MLCK adenoviral infection. On the other hand, decreased expression of cardiac MLCK by RNAi resulted, at most, in a 55% reduction of MLC2v phosphorylation and contributed to a reduction in overall cell motion only under increased demand at a 2.5 mmol/L Ca2+ superfusate concentration. Less dramatic functional effects observed with loss of cardiac MLCK expression may be attributable to remaining MLC2v phosphorylation by other MLC2 kinases, such as smooth muscle MLCK and protein kinase C,6,25 and by counterbalancing MLC2v phosphatase activities.
The amino terminus of cardiac MLCK, lacking homologies to known proteins, may have functions specific for cardiac MLCK. For instance, cardiac MLCK occasionally showed a striated expression pattern not overlapping with MLC2v in A bands but overlapping with actin in I bands. This finding may be interpreted that, locally, the interaction between MLCK and its substrate, MLC2v, may be transient or that cardiac MLCK may have additional functions including phosphorylation of other proteins. Of note, long-form smooth muscle MLCK also colocalizes with actin depending on the actin binding sequence consisting of repeat motifs (DFRXXL) located in the amino terminus,18,26 which was not found in cardiac MLCK.
Cardiac MLCK appeared to be phosphorylated; however, the phosphorylation sites of other MLCKs important for regulating their activities are not conserved in cardiac MLCK. These include 2 contiguous serine residues in the carboxyl terminus of the Ca2+/calmodulin binding sequence of smooth muscle MLCK by protein kinase A, protein kinase C, CaMKII (Ca2+/calmodulin-dependent protein kinase II) and PAK3 (789R, 790K; in Figure 2B); the autophosphorylation site of skeletal MLCK (amino terminus to the catalytic domain), smooth muscle MLCK (in the calmodulin binding domain and carboxyl terminus to this domain), and dictyostelium MLCK (between the catalytic and calmodulin binding domain).27–30 Phenylephrine stimulation resulted in increased phosphorylation of both cardiac MLCK and MLC2v. Whether increased cardiac MLCK phosphorylation directly increases MLC2v phosphorylation remains to be studied; however, this observation demonstrates 1 pathway for phosphorylation of cardiac MLCK, which indeed has several potential protein kinase A phosphorylation sites.
Cardiac MLCK protein expression appeared to be decreased in aged hearts and in heart failure in mice accompanied by decreased MLC2v phosphorylation. Because previous studies have demonstrated that MLC2v phosphorylation is decreased in patients with heart failure,31,32 and expression of a mutant MLC2v in transgenic mouse hearts that cannot be phosphorylated (Ser14, -15, and 19 to Ala mutations) leads to heart failure,33 it is possible that decreased cardiac MLCK protein expression may contribute to compromised contractile function in aging and in heart failure. Of note, a recent study reported upregulated cardiac MLCK mRNA expression in heart failure.34 In the present study, we found lack of concordance between mRNA and protein levels of cardiac MLCK that was likely attributable to altered posttranscriptional regulation of cardiac MLCK in aging and heart failure in mice.
In summary, we report the initial characterization of cardiac MLCK, which is likely a new regulatory factor for cardiac contraction and sarcomere organization.
Supplementary Material
Acknowledgments
We thank E. Chan, T. Seki, Y. Ikeda, K. Ito, M. Nakayama, and N. Terada for valuable suggestions and technical support. During revision of the present manuscript, cardiac MLCK (mylk3; GenBank accession number EU403565) was identified and reported independently by Kitakaze and colleagues.34
Sources of Funding
This work was supported by an American Heart Association National Scientist Development Grant and NIH grant HL081577 (to H.K.).
Footnotes
Disclosures
None.
References
- 1.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. 4. New York: Garland Publishing Inc; 2002. [Google Scholar]
- 2.Murthy KS. Signaling for contraction and relaxation in smooth muscle of the gut. Annu Rev Physiol. 2006;68:345–374. doi: 10.1146/annurev.physiol.68.040504.094707. [DOI] [PubMed] [Google Scholar]
- 3.Kamm KE, Stull JT. Dedicated myosin light chain kinases with diverse cellular functions. J Biol Chem. 2001;276:4527–4530. doi: 10.1074/jbc.R000028200. [DOI] [PubMed] [Google Scholar]
- 4.Stelzer JE, Patel JR, Moss RL. Acceleration of stretch activation in murine myocardium due to phosphorylation of myosin regulatory light chain. J Gen Physiol. 2006;128:261–272. doi: 10.1085/jgp.200609547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moss RL, Fitzsimons DP. Myosin light chain 2 into the mainstream of cardiac development and contractility. Circ Res. 2006;99:225–227. doi: 10.1161/01.RES.0000236793.88131.dc. [DOI] [PubMed] [Google Scholar]
- 6.Herring BP, Dixon S, Gallagher PJ. Smooth muscle myosin light chain kinase expression in cardiac and skeletal muscle. Am J Physiol Cell Physiol. 2000;279:C1656–C1664. doi: 10.1152/ajpcell.2000.279.5.C1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blue EK, Goeckeler ZM, Jin Y, Hou L, Dixon SA, Herring BP, Wysolmerski RB, Gallagher PJ. 220- and 130-kDa MLCKs have distinct tissue distributions and intracellular localization patterns. Am J Physiol Cell Physiol. 2002;282:C451–C460. doi: 10.1152/ajpcell.00333.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis JS, Hassanzadeh S, Winitsky S, Lin H, Satorius C, Vemuri R, Aletras AH, Wen H, Epstein ND. The overall pattern of cardiac contraction depends on a spatial gradient of myosin regulatory light chain phosphorylation. Cell. 2001;107:631–641. doi: 10.1016/s0092-8674(01)00586-4. [DOI] [PubMed] [Google Scholar]
- 9.Herring BP, Stull JT, Gallagher PJ. Domain characterization of rabbit skeletal muscle myosin light chain kinase. J Biol Chem. 1990;265:1724–1730. [PMC free article] [PubMed] [Google Scholar]
- 10.Zhi G, Ryder JW, Huang J, Ding P, Chen Y, Zhao Y, Kamm KE, Stull JT. Myosin light chain kinase and myosin phosphorylation effect frequency-dependent potentiation of skeletal muscle contraction. Proc Natl Acad Sci U S A. 2005;102:17519–17524. doi: 10.1073/pnas.0506846102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohlmann P, Tesse A, Loichot C, Ralay Ranaivo H, Roul G, Philippe C, Watterson DM, Haiech J, Andriantsitohaina R. Deletion of MLCK210 induces subtle changes in vascular reactivity but does not affect cardiac function. Am J Physiol Heart Circ Physiol. 2005;289:H2342–H2349. doi: 10.1152/ajpheart.00511.2004. [DOI] [PubMed] [Google Scholar]
- 12.Lints TJ, Parsons LM, Hartley L, Lyons I, Harvey RP. Nkx-2.5: a novel murine homeobox gene expressed in early heart progenitor cells and their myogenic descendants. Development. 1993;119:419–431. doi: 10.1242/dev.119.2.419. [DOI] [PubMed] [Google Scholar]
- 13.Komuro I, Izumo S. Csx: a murine homeobox-containing gene specifically expressed in the developing heart. Proc Natl Acad Sci U S A. 1993;90:8145–8149. doi: 10.1073/pnas.90.17.8145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasahara H, Bartunkova S, Schinke M, Tanaka M, Izumo S. Cardiac and extracardiac expression of Csx/Nkx2.5 homeodomain protein. Circ Res. 1998;82:936–946. doi: 10.1161/01.res.82.9.936. [DOI] [PubMed] [Google Scholar]
- 15.Herring BP, Gallagher PJ, Stull JT. Substrate specificity of myosin light chain kinases. J Biol Chem. 1992;267:25945–25950. [PubMed] [Google Scholar]
- 16.Ikebe M, Reardon S, Schwonek JP, Sanders CR, 2nd, Ikebe R. Structural requirement of the regulatory light chain of smooth muscle myosin as a substrate for myosin light chain kinase. J Biol Chem. 1994;269:28165–28172. [PubMed] [Google Scholar]
- 17.Zhi G, Herring BP, Stull JT. Structural requirements for phosphorylation of myosin regulatory light chain from smooth muscle. J Biol Chem. 1994;269:24723–24727. [PubMed] [Google Scholar]
- 18.Soderling TR, Stull JT. Structure and regulation of calcium/calmodulin-dependent protein kinases. Chem Rev. 2001;101:2341–2352. doi: 10.1021/cr0002386. [DOI] [PubMed] [Google Scholar]
- 19.Aoki H, Sadoshima J, Izumo S. Myosin light chain kinase mediates sarcomere organization during cardiac hypertrophy in vitro. Nat Med. 2000;6:183–188. doi: 10.1038/72287. [DOI] [PubMed] [Google Scholar]
- 20.Kemp BE, Pearson RB, House C. Role of basic residues in the phosphorylation of synthetic peptides by myosin light chain kinase. Proc Natl Acad Sci U S A. 1983;80:7471–7475. doi: 10.1073/pnas.80.24.7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olsson MC, Patel JR, Fitzsimons DP, Walker JW, Moss RL. Basal myosin light chain phosphorylation is a determinant of Ca2+ sensitivity of force and activation dependence of the kinetics of myocardial force development. Am J Physiol Heart Circ Physiol. 2004;287:H2712–H2718. doi: 10.1152/ajpheart.01067.2003. [DOI] [PubMed] [Google Scholar]
- 22.Jeacocke SA, England PJ. Phosphorylation of myosin light chains in perfused rat heart. Effect of adrenaline and increased cytoplasmic calcium ions. Biochem J. 1980;188:763–768. doi: 10.1042/bj1880763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.High CW, Stull JT. Phosphorylation of myosin in perfused rabbit and rat hearts. Am J Physiol. 1980;239:H756–H764. doi: 10.1152/ajpheart.1980.239.6.H756. [DOI] [PubMed] [Google Scholar]
- 24.Herring BP, England PJ. The turnover of phosphate bound to myosin light chain-2 in perfused rat heart. Biochem J. 1986;240:205–214. doi: 10.1042/bj2400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venema RC, Raynor RL, Noland TA, Jr, Kuo JF. Role of protein kinase C in the phosphorylation of cardiac myosin light chain 2. Biochem J. 1993;294(pt 2):401–406. doi: 10.1042/bj2940401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poperechnaya A, Varlamova O, Lin PJ, Stull JT, Bresnick AR. Localization and activity of myosin light chain kinase isoforms during the cell cycle. J Cell Biol. 2000;151:697–708. doi: 10.1083/jcb.151.3.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao ZH, Moomaw CR, Hsu J, Slaughter CA, Stull JT. Autophosphorylation of skeletal muscle myosin light chain kinase. Biochemistry. 1992;31:6126–6133. doi: 10.1021/bi00141a024. [DOI] [PubMed] [Google Scholar]
- 28.Sobieszek A. Calmodulin-dependent autophosphorylation of smooth muscle myosin light chain kinase: intermolecular reaction mechanism via dimerization of the kinase and potentiation of the catalytic activity following activation. Biochemistry. 1995;34:11855–11863. doi: 10.1021/bi00037a025. [DOI] [PubMed] [Google Scholar]
- 29.Tokui T, Ando S, Ikebe M. Autophosphorylation of smooth muscle myosin light chain kinase at its regulatory domain. Biochemistry. 1995;34:5173–5179. doi: 10.1021/bi00015a031. [DOI] [PubMed] [Google Scholar]
- 30.Tokumitsu H, Hatano N, Inuzuka H, Ishikawa Y, Uyeda TQ, Smith JL, Kobayashi R. Regulatory mechanism of Dictyostelium myosin light chain kinase A. J Biol Chem. 2004;279:42–50. doi: 10.1074/jbc.M309621200. [DOI] [PubMed] [Google Scholar]
- 31.van Der Velden J, Klein LJ, Zaremba R, Boontje NM, Huybregts MA, Stooker W, Eijsman L, de Jong JW, Visser CA, Visser FC, Stienen GJ. Effects of calcium, inorganic phosphate, and pH on isometric force in single skinned cardiomyocytes from donor and failing human hearts. Circulation. 2001;104:1140–1146. doi: 10.1161/hc3501.095485. [DOI] [PubMed] [Google Scholar]
- 32.Palmer BM. Thick filament proteins and performance in human heart failure. Heart Fail Rev. 2005;10:187–197. doi: 10.1007/s10741-005-5249-1. [DOI] [PubMed] [Google Scholar]
- 33.Sanbe A, Fewell JG, Gulick J, Osinska H, Lorenz J, Hall DG, Murray LA, Kimball TR, Witt SA, Robbins J. Abnormal cardiac structure and function in mice expressing nonphosphorylatable cardiac regulatory myosin light chain 2. J Biol Chem. 1999;274:21085–21094. doi: 10.1074/jbc.274.30.21085. [DOI] [PubMed] [Google Scholar]
- 34.Seguchi O, Takashima S, Yamazaki S, Asakura M, Asano Y, Shintani Y, Wakeno M, Minamino T, Kondo H, Furukawa H, Nakamaru K, Naito A, Takahashi T, Ohtsuka T, Kawakami K, Isomura T, Kitamura S, Tomoike H, Mochizuki N, Kitakaze M. A cardiac myosin light chain kinase regulates sarcomere assembly in the vertebrate heart. J Clin Invest. 2007;117:2812–2824. doi: 10.1172/JCI30804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kasahara H, Wakimoto H, Liu M, Maguire CT, Converso KL, Shioi T, Huang WY, Manning WJ, Paul D, Lawitts J, Berul CI, Izumo S. Progressive atrioventricular conduction defects and heart failure in mice expressing a mutant Csx/Nkx2.5 homeoprotein. J Clin Invest. 2001;108:189–201. doi: 10.1172/JCI12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.