

Abstract
A reductive cyclization strategy was applied to the synthesis of attenol A. This non-traditional approach to the spiroacetal structure illustrated several advantages of the reductive cyclization methodology. The attenol A core was formed in a carbon-carbon bond coupling that gave rise to a previously inaccessible spiroacetal epimer, a new method to synthesize thioketene acetals from a phenyl sulfone was realized, and the configurational stability of a non-anomeric spiroacetal was evaluated. A minor by-product in the reductive cyclization reaction was identified that for the first time allowed direct evaluation of the stereoselectivity in a reductive cyclization of a dialkyloxy alkyllithium reagent.
Keywords: spiroacetal synthesis, spiroketal, alkyllithium, reductive cyclization, orthoester, cyanoacetal
Introduction
Attenol A (1) (Figure 1) was isolated from the extract of the Chinese bivalve Pinna attenuata by Uemura and coworkers.1 The molecular architecture of attenol A features three contiguous stereogenic centers, two homoallylic alcohols, both terminal and Z-disubstituted olefins, and an anomeric stabilized [5.4]-spiroacetal core. Attenol B (2) was isolated from the same extract as attenol A (1), and its structure was determined by Uemura to be an isomeric form of attenol A (1).1 Both attenol A and B show moderate cytotoxic activity against P388 cells.1 Its biological activity and scarcity have made attenol A and interesting synthetic target, and it has been prepared by three independent research groups: D. Uemura’s,2a,b J. Eustache’s,3 and D. Enders’.4 We describe a new synthesis of attenol A using a reductive cyclization strategy.
Figure 1.
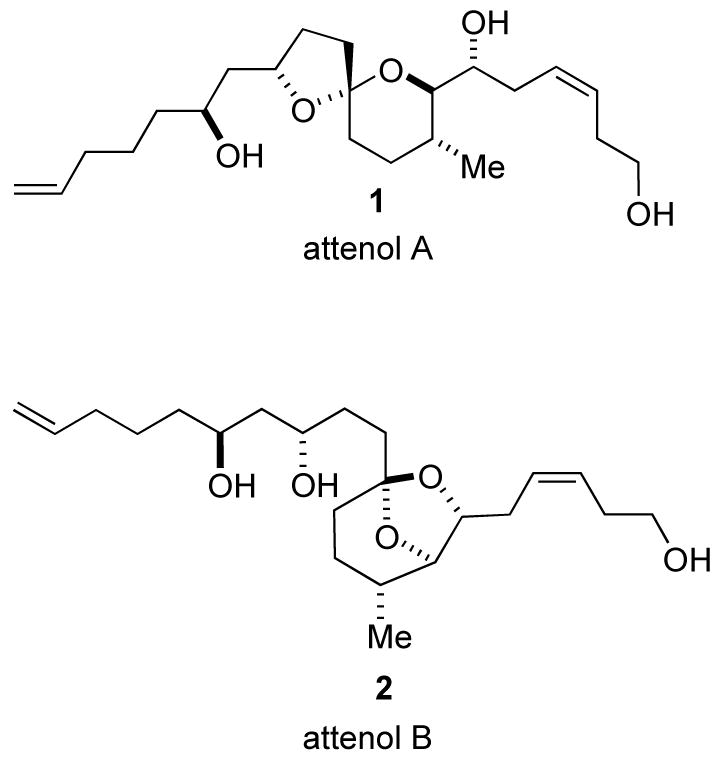
Attenol A and B
The reductive cyclization strategy was developed in our group to facilitate the stereoselective assembly of non-anomeric spiroacetals.5 An example of this cyclization reaction is illustrated in Scheme 1. In contrast to traditional spiroacetal syntheses, this strategy is convergent and gives rise to a non-anomeric stabilized [5.4]-spiroacetal as a single stereoisomer. The resulting non-anomeric spiroacetal equilibrates under acidic conditions to the more stable anomeric epimer.5 Thus both spiroacetal epimers can be accessed from a common intermediate. The synthesis of attenol A illustrates the versatility of a reductive cyclization strategy to efficiently prepare both anomeric and non-anomeric spiroacetals.6
Scheme 1.

Stereoselective preparation of non-anomeric spiroacetal
Many biologically active natural products contain anomeric spiroacetals, and these spiroacetals are presumably more stable than their non-anomeric epimer. On treatment with mild acid, one would expect that the non-anomeric spiroacetal epimer would cleanly isomerize to the natural epimer with a significant change in overall geometry. With such a rearrangement in mind, we considered that non-anomeric spiroacetals might act as pro-drugs for their natural epimers. Solid tumors are known to have unusually low extracellular pH,7 and we considered that spiroacetal epimerization could be triggered by this low pH media and liberate the biologically active anomeric spiroacetal in the presence of the tumor. There are many difficulties with such a scheme, but one prerequisite that could be evaluated was the expected rate of isomerization of a non-anomeric spiroacetal to an anomeric spiroacetal in a low pH aqueous medium. We identified attenol A and its non-anomeric spiroacetal as a convenient test case to evaluate the lability of the spiroacetal linkages, and discuss the results below.
Results
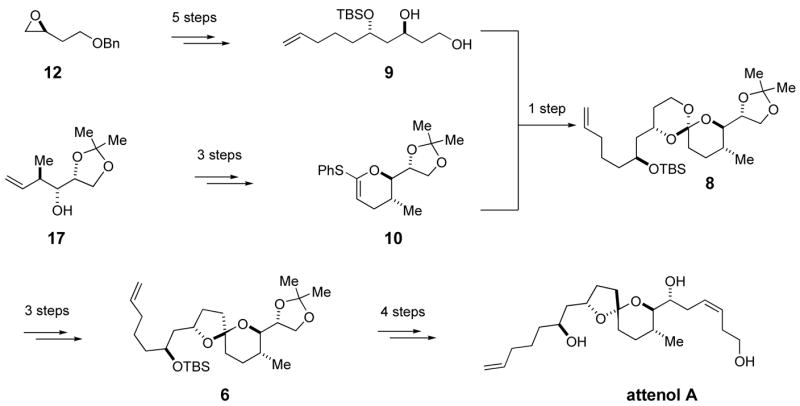
The retrosynthetic analysis of attenol A is illustrated in Figure 2. The epoxide motif in 5, where the planned addition of a vinyl cuprate reagent will install the Z-alkene side chain, lends flexibility to the synthetic route. Acid induced epimerization of the non-anomeric spiro intermediate 6 will give the appropriate configuration of the spiroacetal segment of attenol A (1). Spiroacetal 6 will be synthesized by reductive lithiation of cyanoacetal 7, which itself is derived from spiro orthoester 8. Spiro orthoester 8 will arise from the acid catalyzed coupling of diol 9 and thiophenyl ketene acetal 10, both of which can be derived from readily available chiral materials.
Figure 2.
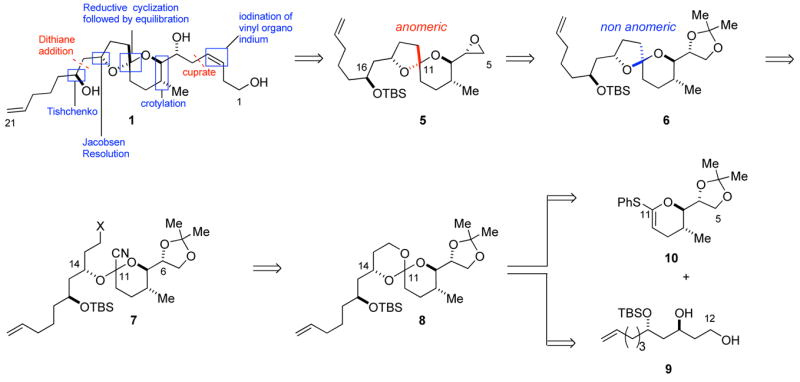
Disconnection plan
The preparation of diol 9 is summarized in Scheme 2. Optically pure epoxide 12, prepared by Jacobsen resolution,8 was treated with two equivalents of the organolithium derived from the known dithiane 11 to give alcohol 13 in 97% yield.9 Hydrolysis of the dithiane with CuCl2/CuO,10 Hg(ClO4)2,11 or (CF3CO2)2IPh12 gave unsatisfactory yields (16–63%) of β-hydroxy ketone 14. Efficient deprotection of 13 finally was achieved with MeI in aqueous acetonitrile, which gave the desired ketone 14 in 94% yield (Scheme 2).13 Ketone 14 was reduced to alcohol 15 (structure not shown) using Schneider’s variation of the Tishchenko reduction catalyzed by Zr(Ot-Bu)4.14 Schneider’s conditions led to the reduction of β-hydroxy ketone 14 with ca. 10:1 selectivity. The reduction of ketone 14 was best carried out at −78 °C in order to obtain the desired anti ester 15 with improved stereoselectivity (98:2 anti:syn)15 and in acceptable yield (83%). Alcohol 15 was converted to tert-butyldimethylsilyl (TBS) ether 16 in 99% yield. The acyl and benzyl protecting groups of silyl ether 16 were simultaneously removed by treatment with Li/NH3 to give diol 9 in 50 to 65% yields. Analysis of the crude reaction mixture revealed a side reaction in which the oxygen atom of the ester had been reductively removed. Similar ester deoxygenations under dissolving metal conditions have been reported previously.16 We hypothesized that a mild reduction or prior acyl hydrolysis would avoid this side reaction. Reduction with sodium and ethanol at −78 °C led to slow cleavage of the benzyl ether; upon warming to 0 °C only trace amounts of the desired diol 9 were detected due to competing migration of the TBS group. Prior acyl cleavage was achieved in situ by adding MeLi to a solution of TBS ether 16 in THF/NH3 to generate LiNH2. Subsequent addition of Li° led to removal of the benzyl ether and gave diol 9 in excellent yield. With the successful synthesis of diol 9 our efforts turned toward the synthesis of its coupling partner.
Scheme 2.
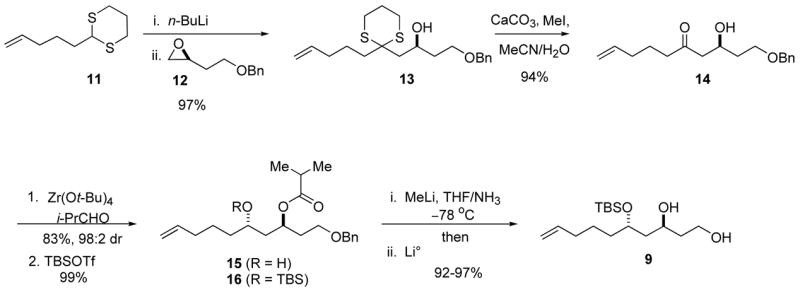
Synthesis of diol
Our initial strategy called for the introduction of the complete Z-alkene side chain prior to thioketene acetal formation. This route is illustrated in Scheme 3. Known homoallylic alcohol 1717 was reacted with allyl bromide to give allyl ether 18. Allyl ether 18 was then subjected to a three-step reaction sequence. First, a RCM reaction using Grubbs’ first generation catalyst gave the dihydropyran 19 (95% yield).18 Hydrolysis of the acetonide protection group followed by cyclization of the resulting diol 20 gave epoxide 21 in good yield.39 Vinyl cuprate 21, prepared from vinyl iodide 25 (Scheme 3),19 reacted with epoxide 26 to give the desired homoallylic alcohol 22 (not shown) in 85% yield.20 The alcohol 22 was converted to triisopropylsilyl (TIPS) ether 23 in quantitative yield under standard reaction conditions. Silyl ether 23 represents half of the attenol A carbon skeleton. Although the route was satisfactory, the sequence placed the ring unsaturation at C3 rather than C2. Isomerization to the C2 enol ether was possible with similar compounds, but the exocyclic alkene complicated the process.21,22 Selective alkene isomerization was not observed with Wilkinson’s catalyst.22 However, both Brimble and Nicolaou have reported base-induced alkene isomerizations of dihydropyrans systems to form cyclic enol ethers.23 When dihydropyran 23 was treated with LDA or t-BuOK under various conditions, only decomposition was observed. Failure of the base-induced alkene isomerization reactions forced us to examine a different approach to the enol ether 29.
Scheme 3.
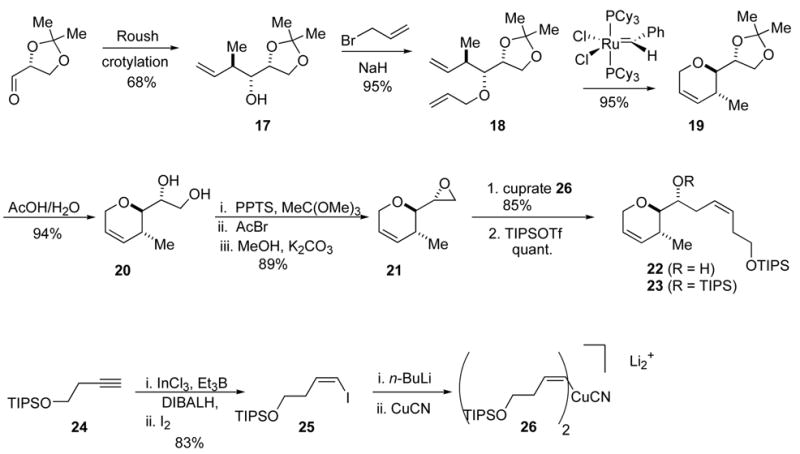
Synthesis of DHP and Organocuprate
A new route to the desired enol ether 29 was investigated, and the outcome is illustrated in Scheme 4. The alkene isomerization of epoxide 21, mediated by Wilkinson’s catalyst,22 gave volatile epoxide 27 in 70% yield. Addition of cuprate 26 to oxirane 27 followed by protection of the resulting homoallylic alcohol 28 completed the synthesis of enol ether 29. All attempts to lithiate enol ether 29 under Boeckman or Myer’s conditions lead to the decomposition of starting enol ether 29.24 Loss of the homoallylic silyl ethers, presumably through Elcb elimination, was evident in the 1H NMR spectrum of the crude reaction mixtures. The unsuccessful kinetic lithiation reaction forced us to develop an alternative method to generate thioketene acetal 30 from enol ether 29.
Scheme 4.
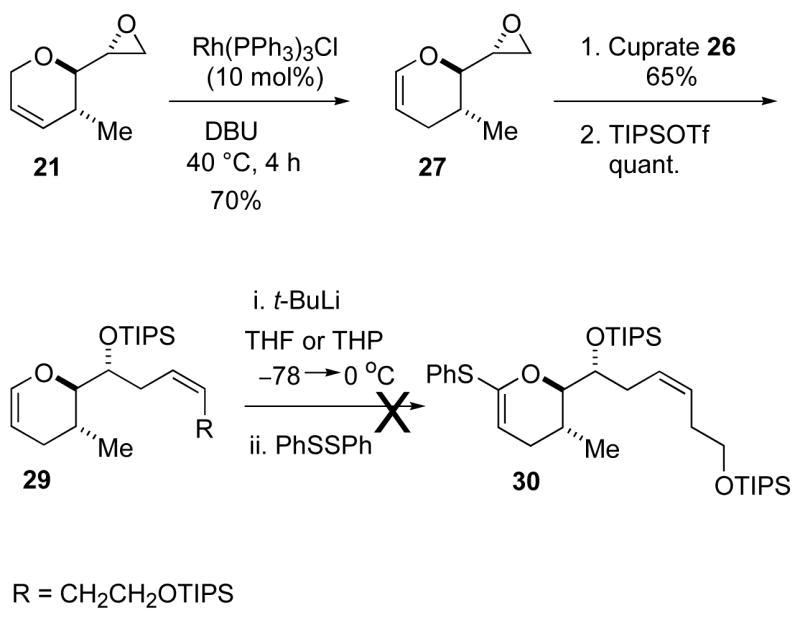
Attempted lithiation
Crich explored generating thioketene acetals directly from arylsulfones, but his endeavor was unsuccessful.25 He showed that lithiation of sulfone 31 and subsequent treatment with diphenyl disulfide gave only dithioorthoester 34, and not the expected thioketene acetal 33 (Scheme 5, part A). We repeated the experiment in our laboratories (Scheme 5, part B) and found that the reaction gave primarily two products. The major constituent was dithioorthoester 34. Surprisingly, the minor component was thioketene acetal 33. Optimization of this reaction led to efficient formation of the desired thioketene acetal. The optimized procedure was applied to the synthesis of attenol A. Enol ether 29 reacted with benzenesulfinic acid to give sulfone 35 in 74–81% yield (Scheme 5, part C). Sulfone 35 was treated with n-BuLi at −100 °C to generate the unstable lithiated sulfone. The lithiated sulfone was quenched with PhSSO2Ph,26 and Et3N was introduced into the reaction vessel to scavenge the benzene sulfinic acid generated in the elimination reaction. These conditions gave the desired thioketene acetal 30 in 65% yield with none of the corresponding dithioacetal detected. With thioketene acetal 30 in hand, the time came to test the spiro orthoester formation.
Scheme 5.
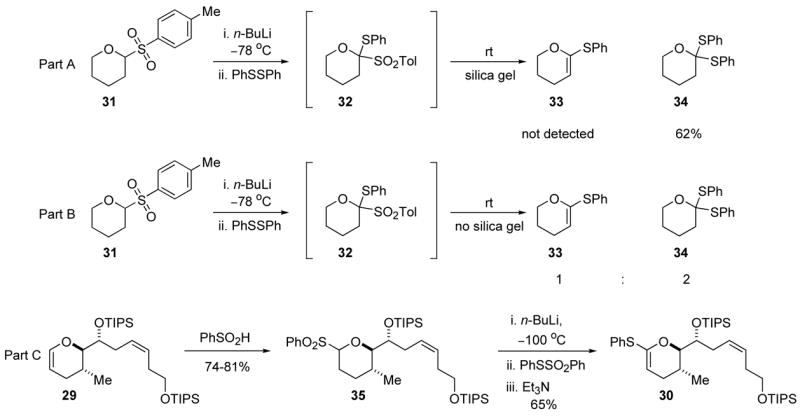
Sulfone mediated thioketene acetal synthesis
Thioketene acetal 30 was reacted with diol 9 under standard reaction conditions (0.1 M in DCM, 1–3 mol % camphorsulfonic acid (CSA) rt, 0.5 h),27 and the desired spiro orthoester 36 was isolated in a disappointing 35% yield (Scheme 6). Several decomposition products were observed by TLC and by spectroscopic analysis of the crude reaction mixture. All attempts to improve the outcome by either using a milder proton source, higher dilution, or rigorously anhydrous solvent were fruitless. The proximity of the bulk secondary TIPS ether apparently disfavors cyclization to the orthoester. With the outcome of these experiments in mind we revaluated the synthetic plan for attenol A.
Scheme 6.
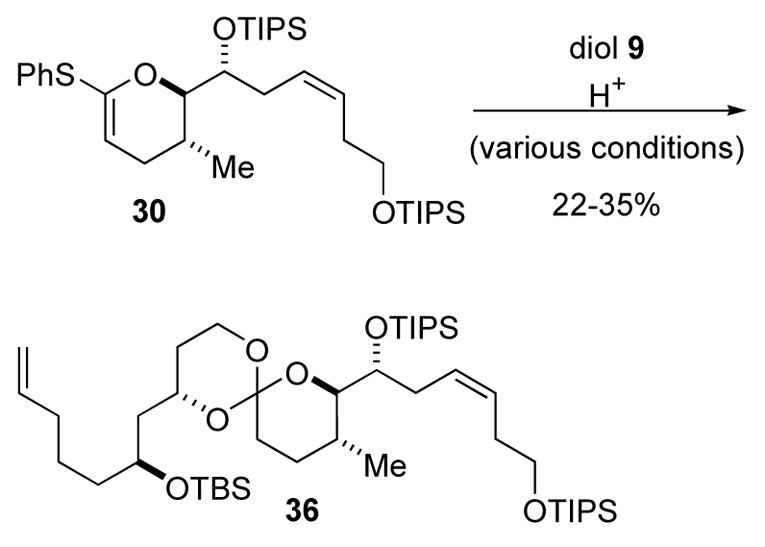
Disappointing orthoester synthesis
A new route was designed that would delay introduction of the side chain until the end of the synthesis. As shown in Scheme 7, enol ether 37 was readily accessed from cyclic allyl ether 19 through an alkene isomerization reaction catalyzed by (PPh3)3RhCl in 90% yield.22 The acetonide protecting group of enol ether 37 was not compatible with t-BuLi treatment, so an alternative thioketene acetal synthesis was required. Applying the phenylsulfone protocol, sulfone 38 was prepared from enol ether 37 and subjected to the same sequence as sulfone 35 (Scheme 5) to produce thiophenylketene acetal 10 in 72% yield. This promising result could be applied to the synthesis of attenol A, but an even more efficient route was identified.
Scheme 7.
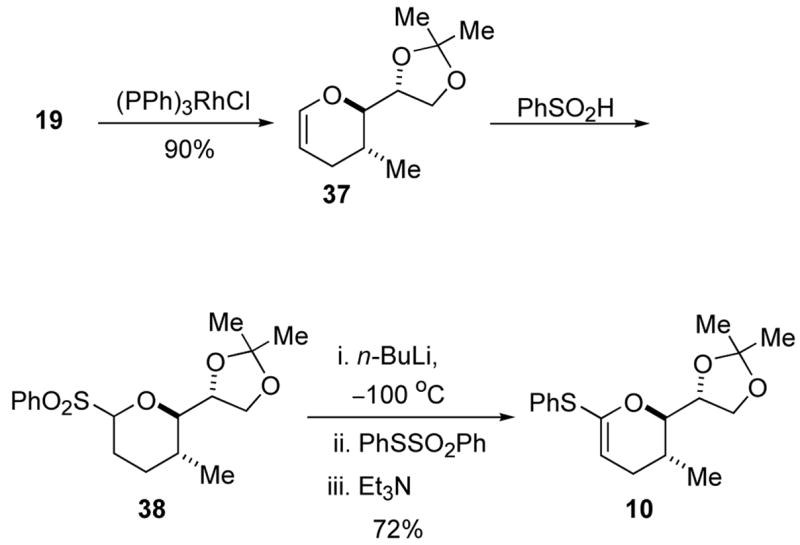
Truncated thioketene acetal
Kocienski recently reported a very effective route to thioketene acetals,28 and application of Kocienski’s strategy might streamline our synthesis of thioketene acetal 10. The new synthetic sequence is shown in Scheme 8. Homoallylic alcohol 17 was converted to vinyl ester 39 in 87% yield.29 Lactone 41 was accessed by a sequential RCM/hydrogen reaction with both steps mediated by Grubbs’ second generation catalyst.30 Kocienski’s Ni0 catalyzed protocol was applied to lactone 41, and gave the desired thioketene acetal 10 in 75% yield. Clearly, this three-pot sequence to thioketene acetal 10 was superior to the previously explored routes. With thioketene acetal 10 in hand, the time came to test the orthoester formation.
Scheme 8.
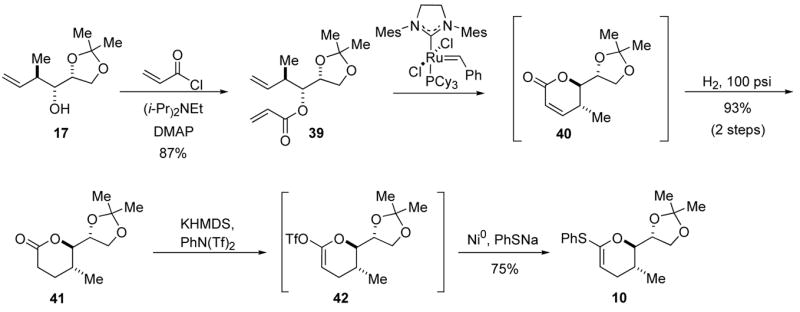
Revised strategy to thioketene acetal
Thioketene acetal 10 was coupled to diol 9 using catalytic CSA.27 The desired spiro orthoester 8 was isolated in 86% yield as a single diastereomer (Scheme 9).31 The [5.5]-spiro orthoester 8 was opened with BF3·OEt2 and TMSCN with excellent regio- and chemoselectivity to give alcohol 44 (Scheme 9) in 71% yield as a single diastereomer.5 The reaction proceeded to completion faster than the analogous reaction of a [5.4]-spiro orthoester. In our previous synthetic endeavors the electrophile, a primary alkyl chloride, was installed prior to cyanoacetal synthesis.5 Unfortunately, the attenol A architecture does not lend itself to this approach, and the electrophile had to be installed after the cyanoacetal. Previous studies have shown that the cyanoacetal motif is incompatible with Ph3P/CCl4.32 An alternative alcohol to alkyl chloride conversion, mesylate formation followed by treatment with Lid in DMF, has been used successfully.5 However, LiCl/DMF is known to cleave TBS ethers.33 Phosphate esters can also serve as leaving groups in reductive cyclization reactions,34 and primary alcohol can be converted to phosphate ester under mild reaction conditions. Thus, alcohol 44 was treated with (EtO)2P(O)Cl and N-methylimidazole to give the cyclization precursor, phosphate ester 45, in 97% yield as a single stereoisomer (Scheme 9).35
Scheme 9.
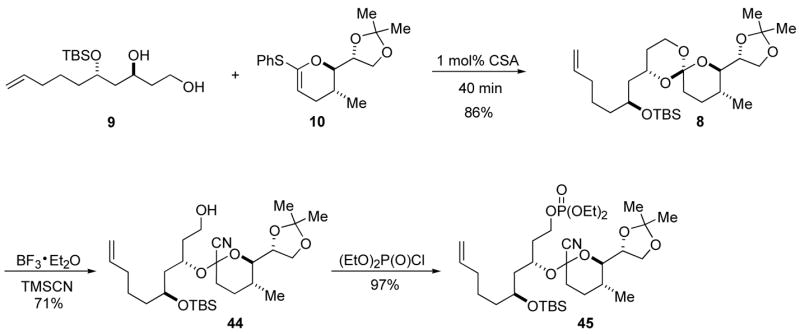
Spiro orthoester and cyclization precursor synthesis
The reductive cyclization of phosphate 45 produced the non-anomeric spiroacetal 6 as the major product in a remarkable 94% yield (Scheme 10). Both cyanoacetal 48, a product of the competitive reduction of the phosphate motif by lithium di-tert-butylbiphenylide (LiDBB)36 and anomeric spiroacetal 47 were identified as minor products. Analysis of the crude reaction mixture by GC returned a 78:1 ratio of non-anomeric spiroacetal 6 and anomeric spiroacetal 47. NOESY correlation confirmed the non-anomeric configuration of spiroacetal 6 (Scheme 10). The key reductive cyclization was a very efficient process, and spiroacetal 6 was amendable to preparative scale synthesis.
Scheme 10.
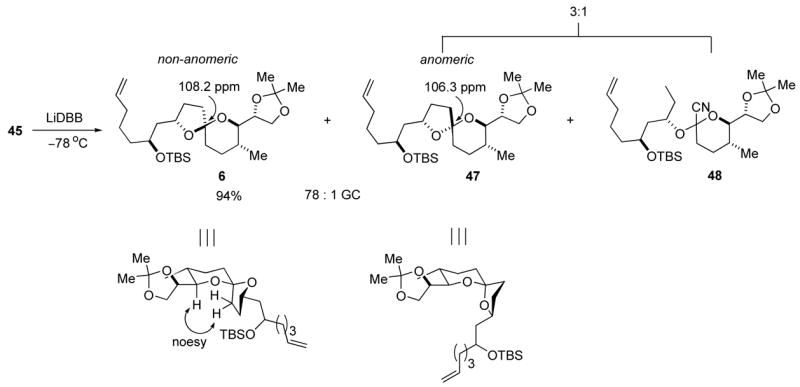
Reductive cyclization
Non-anomeric spiroacetal 6 was subjected to equilibrating conditions (PPTS/MeOH). The reaction was complicated by cleavage of the TBS ether and equilibration of spiroacetals 49 and 50, the attenol B core.37 Attempts to suppress the equilibration were unsuccessful, and therefore the mixture was used in the next reaction. The traditional technique of selective sulfonation and subsequent cyclization gave poor yields of epoxide 5. Poor regioselectivity in the sulfonation of triol 49 by both MsCl, and 2,4,6-triisopropylsulfonylchloride was responsible for the inefficient epoxidation reaction.38 An alternative epoxidation technique was required, and the Sharpless-Moffatt cyclization was investigated.39 The desired epoxide 5 was generated in a satisfactory 67% yield over two steps (Scheme 11). The C16 alcohol did not interfere with the cyclization. Epoxide 5 reacted with an excess of vinyl cuprate 26 to yield alcohol 51 in 85% yield. Finally, removal of the TIPS silyl ether completed the total synthesis of attenol A.
Scheme 11.

Completion of the synthetic sequence
Equilibration
Solid tumors have low extracellular pH (ca. 6.8), and exploiting this property to selectively target tumor cells has been proposed.7 Acidic media could trigger the isomerization of non-anomeric spiroacetals to their anomeric isomers, in which case the non-anomeric spiroacetal would act as a pro-drug for its epimer. We selected non-anomeric spiroacetal 53 (Table 1) as a test case. The effect of pH on the rate of isomerization of spiroacetal 53 with its more stable epimer in aqueous buffer solutions was evalauted.37 Spiroacetal 53 was prepared by removal of the TBS group from spiroacetal 6 to improve the solubility properties. The epimerization of the spiroacetal is summarized in Table 1. Spiroacetal 53 was dissolved in a NaOAc/HOAc buffer system using THF as a cosolvent. After three days at pH 5.8, epimerization of spiroacetal 53 was not evident by TLC or GC analysis of the reaction mixture. Epimerization was not observed in a pH 5.0 environment over a 48 h time span. Partial epimerization of spiroacetal 53 took place in a pH 4.0 buffer, leading to a 10:1 ratio of non-anomeric to anomeric isomers. The half-life for the epimerization event could not be accurately deduced from this data due to concurrent acetonide cleavage, but it would be at least several days.40 Under normal physiological conditions the non-anomeric spiroacetal 53 would be stable indefinitely. Thus the use of this class of non-anomeric spiroacetals as pro-drugs for solid tumors is impractical because the epimerization would be too slow at an extracellular pH of ca. 6.8.7
Table 1.
Acetal Equilibration Studies
 | |||
|---|---|---|---|
| pH | T (°C) | Time (h) | ratio (nonanomeric:anomeric) |
| 5.8 | 23 | 72 | no epimerization |
| 5 | 23 | 48 | no epimerization |
| 4 | 23 | 22 | 10:1 (nonanomeric:anomeric) |
Discussion
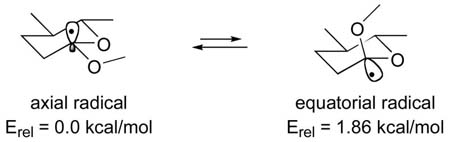
The key reductive cyclization reaction proved to be a very efficient process, but we were interested in the origin of the minor anomeric spiroacetal 47. The experiments summarized in Table 1 demonstrate that the non-anomeric spiroacetal 53, and presumably 6, is not susceptible to epimerization in a neutral aqueous solution. Detailed analysis of the radical lithiation reaction revealed that more than one pathway could be responsible for the genesis of anomeric spiroacetal 47. These competing pathways are depicted in Scheme 12. Initial SET and subsequent C-CN bond fission reveals equilibrating radical species 55 and 59.41 Preference for the radical to occupy the axial position has been calculated to be ~1.86 kcal/mol in a closely related model system (R1, R2 = Me).42 If we take this enthalpy as an estimate for the ΔG between the two radical epimers, it would predict a 122:1 ratio favoring the axial radical at −78 °C. This value is in good agreement with the 78:1 ratio observed for between the spiroacetals 6 and 47.
Scheme 12.

Mechanism of Cyclization
The observed selectivity for non-anomeric spiroacetal 6 is a bit lower than predicted by the DFT modeling, but it falls within the uncertainty of the calculations. The difference may indicate a modest erosion of selectivity from the thermodynamic ratio between radicals 55 and 59. If simplified Winstein-Holness kinetics apply,43 the ratio of alkyllithium reagents 56:60 would reflect that of the corresponding radicals 55 and 59. Any change in selectivity would result from differential efficiency in the reactions of 56 and 60, or from cyclization with partial inversion of configuration.44 Examples have been reported were stereogenic, sp3-hybridized, α-hetero alkyllithium species undergo invertive electrophilic substitution reactions,45 but the process has not been documented with α,α-dialkyloxy alkyllithium reagents. A second possibility is that the alkyllithium product ratio in the second SET does not precisely reflect the ratio of 55:59. This outcome would be expected in a more general Curtin-Hammett situation where the rates of ET differ for the two radicals.43 The minor isomer 47 then would arise from a larger proportion of alkyllithium 60 formed in the reduction. We know from previous studies that the calculated ratio of anomeric radicals is a good predictor of the product ratios,41a so the change in ratio that might be attributed to differential reduction rates is modest. The minor anomeric spiroacetal 47 may reflect the true equilibrium ratio between radicals 55 and 59, or there may be a minor loss of selectivity attributable to either of the pathways discussed above. The formation of spiroacetal 47 is the first characterized anomeric spiroacetal formed directly in a reductive cyclization reaction.
Conclusion
The synthesis of attenol A is summarized in Scheme 13. The target molecule 1 was synthesized in 13 steps (longest linear sequence), starting from enantiomerically pure epoxide 12, in 21.4% overall yield.46 The key intermediates, thioketene acetal 10, spiro orthoester 8, and spiroacetal 6 were efficiently accessed on preparative scales. The attenol A core was formed by a carbon-carbon bond reductive cyclization event that gave rise to a previously inaccessible spiroacetal epimer.
Scheme 13.
Summary of the Synthesis
This non-traditional approach to an anomeric spiroacetal target illustrates several features of the reductive cyclization methodology. The synthesis is more efficient than previous approaches to attenol A, and demonstrates potential of using reductive cyclization reactions to afford anomeric spiroacetals. A thioketene acetal and [5.5]-spiro orthoester, two uncommon chemical entities, were efficiently synthesized en route to the synthesis of attenol A. A new method to synthesize thioketene acetals from a phenyl sulfone was developed. The key reductive cyclization reaction once again proved to be an efficient process. The configurational stability of the non-anomeric spiroacetal moiety was also evaluated, and it was found to be unsuitable as a pH trigger in a pro-drug. For the first time an anomeric spiroacetal was isolated from the reductive cyclization reaction, providing insight into the inherent selectivity of the cyclization.
Experimentals
Thioketene acetal 10
The following procedure was adopted from the protocol developed by Kocienski.27
Flask 1
A solution of KHMDS (0.5 M in toluene, 24.8 mL, 12.4 mmol) and THF (37 mL) was cooled to −78 °C. A mixture consisting of PhN(Tf)2 (3.80 g, 10.6 mmol), lactone 41 (1.90 g, 8.87 mmol), and THF (44 mL) was prepared and added drop wise over a 1 h 15 min period. After the addition was completed, the mixture was stirred at −78 °C for 15 min.
Flask 2
To a suspension of NaH (60% dispersion in mineral, 0.710 g, 17.7 mmol), in THF (8.8 mL), at room temperature, was added PhSH (1.09 mL, 10.6 mmol) dropwise. After the evolution of gas had ceased, the mixture was stirred at room temperature for 15 min and then cooled to −78 °C.
Flask 3
To a mixture of Ni(PPh3)2Br2 (0.659 g, 0.887 mmol), PPh3 (0.465 g, 1.77 mmol), and zinc powder (0.348 g, 5.32 mmol) was added THF (21 mL). The green suspension was stirred at room temperature for 20 min, after which the color of the reaction mixture became deep red.
The contents of flask 1 were transferred to flask 2 (via cannula), and the contents of flask 3 were transferred to flask 2 by syringe. The deep red mixture was removed from the cooling bath, warmed to room temperature, and stirred for 3.5 h. The reaction mixture was diluted with 5% NaOH(aq) (50 mL), and extract with Et2O(3 × 100 mL). The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure. The crude product mixture was subsequently purified by silica gel chromatography (deactivated silica gel, 5–10% Et2O/hexanes) to afford 2.05 g (75% yield) of the title compound as a colorless oil: Rf = 0.43 (15% Et2O/hexanes); [α]D −40 (c 0.36, CH2Cl2); 1H NMR (500 MHz, C6D6) δ 7.47 (dd, 2H, J = 1.5, 8.7 Hz), 7.05 (t, 2H, J = 7.5 Hz), 6.96 (m, 1H), 5.23 (dd, 1H, J = 2.7, 4.9 Hz), 3.95 (dt, 1H, J = 2.4, 7.3 Hz), 3.79 (t, 1H, J = 7.7 Hz), 3.61 (app t, 1H, J =7.1 Hz), 2.93 (dd, 1H, J = 2.4, 9.1 Hz), 2.01 (m, 1H), 1.79 (dt, 1H, J = 5.3, 17.5 Hz), 1.46 (ddd, 1H, J = 2.7, 10.3, 17.5 Hz), 1.39 (s, 3H), 1.33 (s, 3H), 0.72 (d, 3H, J = 6.8 Hz) ppm; 13C NMR (125 MHz, C6D6) δ 147.6, 135.4, 130.8, 129.4, 127.2, 109.8, 107.7, 81.1, 75.0, 65.3, 31.5, 28.7, 26.7 (2C), 17.2 ppm; IR (neat) 2914, 1634, 1584, 1480, 1379, 1070 cm−1; HRMS (ESI) calcd for C17H23O3S [M + H]+ 307.1368; found 307.1367.
Orthoester 8
To a solution of thiophenyl ketene acetal 10 (1.07 g, 3.49 mmol), in CH2Cl2 (17 mL), was added a solution of diol 9 (1.16 g, 3.83 mmol), in CH2Cl2 (19 mL), and 4 Å molecular sieves (700 mg). The mixture was stirred at room temperature for 2 h, and filtered through a 45 μm Whatman syringe filter into another flask that had been equipped with a magnetic stir bar. The solution was treated with CSA (8 mg, 3.49 μmol), stirred at room temperature for 40 min. The reaction was quenched with Et3N (0.10 mL, 0.717 mmol), and the solution was concentrated under reduced pressure. The resulting oil was purified by silica gel chromatography (15% Et2O/hexanes) to afford 1.49 g (86% yield) of the desired orthoester 8 as a colorless oil: Rf = 0.61 (20% Et2O/hexanes); [α]D +2.7 (c 0.75, CH2Cl2); 1H NMR (500 MHz, C6D6) δ 5.79 (ddt, 1H, J = 6.7, 10.2, 16.9 Hz), 5.04 (dd, 1H, J = 1.8, 17.1 Hz), 4.99 (dd, 1H, J = 1.1, 10.2 Hz), 4.51 (ddt, 1H, J = 3.5, 7.3, 11.4 Hz), 4.15 (dt, 1H, J = 3.5, 7.1 Hz), 4.04–3.96 (m, 2H), 3.84 (t, 1H, J = 6.9 Hz), 3.75 (ddd, 1H, J = 2.6, 11.0, 13.2 Hz), 3.49 (dd, 1H, J = 4.3, 10.9 Hz), 3.18 (dd, 1H, J = 3.5, 10.1 Hz), 2.04–1.98 (m, 3H), 1.79–1.71 (m, 3H), 1.63–1.52 (m, 8H), 1.51–1.45 (m, 2H), 1.41–1.35 (m, 4H), 1.17–1.11 (m, 1H), 1.05 (s, 9H), 0.73 (d, 3H, J = 6.6 Hz), 0.25 (s, 3H), 0.16 (s, 3H) ppm; 13C NMR (125 MHz, C6D6) δ 139.4, 115.1, 110.7, 109.3, 77.2, 76.7, 69.8, 66.8, 65.8, 58.8, 45.1, 38.3, 35.3, 34.7, 32.1, 31.8, 30.2, 27.1, 26.7, 26.3, 24.8, 18.7, 17.4, −3.6, −3.7 ppm; IR (neat) 2930, 1462, 1379, 1256, 1063, 985 cm−1; HRMS (ESI) calcd for C27H51O6Si [M + H]+ 499.3455; found 499.3471.
Spiroacetal 6
A solution of phosphate 45 (0.730 g, 1.10 mmol), 1,10-phenanthroline (0.1 mg), and THF (11 mL), was cooled to −78 °C. To the vigorously stirring solution was added LiDBB (2 mL, over a 0.5 h period) in a dropwise fashion, directly to the center of the reaction mixture, whereby the green color of LiDBB was allowed to dissipate before the addition of the following drop. Another 3 mL of LiDBB were added over a 40 min period in an identical manner. The solution was stirred for 5 min at −78 °C, and then another 2.5 mL of LiDBB were added over a 35 min period in the same manner as described above. Then 0.8 mL of LiDBB was added over a 10 min period (8.3 mL total volume). After addition of the final drop of LiDBB its dark color persisted for 15 seconds, and then dissipated to give a red colored solution. The solution was allowed to stir at −78 °C for 2 h, and excess LiDBB was quenched by the addition of MeOH (1 mL). The mixture was warmed to room temperature, diluted with 40 mL of water, and extracted with Et2O (3 × 100 mL). The combined organic layers were washed with brine (1 × 40 mL), dried over anhydrous Na2SO4 filtered and concentrated under reduced pressure. The crude product was determined to be a 78:1 ratio of Spiroacetal 6 and Spiroacetal 47 by GC analysis (Rt−6 = 21.75 min, Rt−47 = 19.55 min).47 The resulting residue was purified by silica gel chromatography, eluting first with 100% hexanes (to remove DBB), then 15% Et2O/hexanes. The desired Spiroacetal 6 (0.502 g, 94% yield) was isolated as a colorless oil: Rf = 0.44 (15% Et2O/hexanes); [α]D +20 (c 0.23, CH2Cl2); 1H NMR (500 MHz, C6D6) δ 5.79 (ddt, 1H, J = 6.7, 10.2, 16.9 Hz), 5.04 (dq, 1H, J = 1.6, 17.1 Hz), 4.99 (ddt, 1H, J = 1.2, 2.2, 10.2 Hz), 4.50 (m, 1H), 4.22 (dq, 1H, J = 2.8, 6.9 Hz), 4.06–3.99 (m, 2H), 3.82 (t, 1H, J = 6.8 Hz), 2.74 (dd, 1H, J = 2.8, 9.3 Hz), 2.00 (app q, 2H, J = 7.4 Hz), 1.93–1.85 (m, 2H), 1.83–1.75 (m, 2H), 1.67–1.62 (m, 2H), 1.60 (s, 3H), 1.56–1.51 (m, 2H), 1.50–1.45 (m, 4H), 1.44 (s, 3H), 1.39–1.29 (m, 2H), 1.04 (s, 9H), 0.96 (m, 1H), 0.79 (d, 3H, J = 6.6 Hz), 0.28 (s, 3H), 0.15 (s, 3H) ppm; 13C NMR (125 MHz, C6D6) δ 139.4, 115.1, 109.6, 108.2, 78.1, 76.4, 75.9, 70.4, 65.7, 44.7, 38.5, 35.1, 34.7, 32.9, 31.6 (2C), 31.5, 27.1, 26.7, 26.6, 24.9, 18.7, 17.6, −3.6, −3.8 ppm; IR (neat) 2933, 1624, 1460, 1251, 1064 cm−1; HRMS (ESI) calcd for C27H50O5SiNa [M + Na]+ 505.3325; found 505.3335.
Epoxide 5
Spiroacetal 6 (0.150 g, 0.311 mmol), PPTS (0.312 g, 1.24 mmol) and MeOH (3.1 mL) were combined and stirred at room temperature for 28 h. Then 4 mL of saturated NaHCO3(aq) was added, and the mixture was vigorously stirred for 5 min and then extracted with Et2O (2 × 60 mL). The organic layers were combined, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to give 0.112 g of an oil that was dried by azeotropic removal of moisture with benzene.
The oil (0.112 g, 0.34 mmol) was dissolved in CH2Cl2 (1.7 mL), treated with trimethyl orthoacetate (0.10 mL, 0.82 mmol), and PPTS (9 mg, 0.034 mmol). The mixture was stirred at room temperature for 45 min, concentrated under reduced pressure, and placed under a vacuum (0.2 mmHg) for 2 min. The resulting residue was dissolved in CH2Cl2 (1.7 mL), treated with Et3N (0.01 mL, 0.072 mmol), and the reaction vessel was then placed in a room temperature water bath. The mixture was treated with acetyl bromide (0.061 mL, 0.82 mmol), stirred for 40 min, and then concentrated under reduced pressure. The resulting residue was dissolved in MeOH (3.2 mL), and the solution was then treated K2CO3(S) (0.471 g, 3.41 mmol). The mixture was stirred vigorously at room temperature for 15 h, diluted with 4 mL of brine, and extracted with Et2O (2 × 20 mL). The combined organic layers were dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The resulting oil was purified by silica gel chromatography (40% EtOAc/hexanes) to afford 0.0645 g (67% yield) of the title compound as a colorless oil: Rf = 0.38 (40% EtOAc/hexanes); [α]D−32 (c 1.3, CHCl3); 1H NMR (500 MHz, C6D6) δ 5.80 (ddt, 1H, J = 6.7, 10.2, 17.0 Hz), 5.05 (dq, 1H, J = 1.7, 17.1 Hz), 4.99 (ddt, 1H, J = 1.1, 2.3, 10.2 Hz), 4.32 (m, 1H), 3.86 (m, 1H), 3.25 (dd, 1H, J = 6.9, 10.0 Hz), 2.78 (ddd, 1H, J = 2.6, 4.0, 6.8 Hz), 2.45 (dd, 1H, J = 2.6, 5.5 Hz), 2.39 (dd, 1H, J = 4.0, 5.3 Hz), 2.05–1.97 (m, 3H), 1.96–1.90 (m, 1H), 1.89–1.82 (m, 2H), 1.74–1.68 (m, 1H), 1.65–1.55 (m, 4H), 1.51–1.41 (m, 7H), 0.67 (d, 3H, J = 6.3 Hz) ppm; 13C NMR (125 MHz, C6D6) δ 139.6, 115.0, 106.1, 78.4, 78.2, 69.8, 53.4, 45.5, 45.0, 39.1, 38.0, 34.6, 34.4, 33.2, 31.0, 29.9, 25.8, 17.9 ppm; IR (neat) 3439, 2927, 1640, 1459, 1373, 1015 cm−1; HRMS (ESI) calcd for C18H30O4Na [M + Na]+ 333.2042; found 333.2048.
Spiroacetal 51
A solution of vinyl iodide 25 (0.58 g, 1.6 mmol), in Et2O (3.3 mL), was cooled to −78 °C, and treated with n-BuLi (2.5 M in hexanes, 0.65 mL, 1.6 mmol). The colorless solution was stirred at −78 °C for 1 h, solid CuCN (0.076 g, 0.85 mmol) was then added in one portion. The reaction flask was placed in a −30 °C bath, stirred for 1 h, and recooled to −78 °C. A solution of epoxide 5 (0.063 g, 0.20 mmol), in Et2O (1 mL), was then added dropwise. The mixture was again placed in a −30 °C bath, stirred for 1.5 h, and then 5 mL of concentrated NH4OH(aq) was added in one portion. The mixture was warmed to room temperature, diluted with saturated NH4Cl(aq) (1 ml), and stirred until the green copper salts were digested (~20 min). The blue colored solution was extracted with Et2O (3 × 70 mL), the organic layers were combined, washed with saturated NaHCO3(aq) (1 × 20 mL), and then brine (1 × 20 mL). The organic extracts were dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure to give an oil that was purified by silica gel chromatography (30% EtOAc/hexanes) to afford 0.093 g (85% yield) of the title compound as a colorless oil: Rf = 0.33 (30% EtOAc/hexanes); [α]D−4.8 (c 0.60, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 5.80 (ddt, 1H, J = 6.7, 10.2, 16.9 Hz), 5.59–5.52 (m, 2H), 5.00 (dq, 1H, J = 1.6, 17.1 Hz), 4.94 (ddt, 1H, J = 1.2, 2.3, 10.1 Hz), 4.32 (m, 1H), 3.84 (m, 1H), 3.71–3.65 (m, 3H), 3.31 (dd, 1H, J = 1.0, 10.3 Hz), 2.50–2.43 (m, 1H), 2.38–2.30 (m, 2H), 2.21 (br s, 1H), 2.14–2.05 (m, 3H), 2.04–1.96 (m, 2H), 1.86–1.61 (m, 9H), 1.57–1.41 (m, 5H), 1.08–1.04 (m, 21H), 0.86 (d, 3H, J = 6.5 Hz) ppm; 13C NMR (125 MHz, CDCl3) δ 138.6, 128.3, 127.7, 114.5, 106.3, 77.7, 77.6, 70.1, 69.9, 63.0, 43.8, 38.5, 36.7, 33.9, 33.7, 32.7, 31.4, 30.9, 30.2, 29.0, 25.1, 18.0, 17.2, 12.0 ppm; IR (neat) 3430, 2942, 1641, 1461, 1380 cm−1; HRMS (ESI) calcd for C31H58O5SiNa [M + Na]+ 561.3951; found 561.3946.
Supplementary Material
SUPPORTING INFORMATION AVAILABLE: Experimental details for preparation of the cyclization substrates and the characterization and correlation of the products. Proton and 13C NMR spectra are included. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The National Institutes of Health (GM65388) provided support for this project. Novartis Pharmaceuticals is gratefully acknowledged for fellowship support to TEL.
Endnotes
- 1.Takada N, Suenaga K, Yamada K, Zheng SZ, Chen HS, Uemura D. Chem Lett. 1999:1025–1026. [Google Scholar]
- 2.(a) Suenaga K, Araki K, Sengoku T, Uemura D. Org Lett. 2001;3:527–529. doi: 10.1021/ol006905z. [DOI] [PubMed] [Google Scholar]; (b) Araki K, Suenaga K, Sengoku T, Uemura D. Tetrahedron. 2002;58:1983–1995. [Google Scholar]
- 3.Aoun D, Boiteau JG, Eustache J, Van de Weghe P. Org Lett. 2002;4:4105–108. doi: 10.1021/ol0268438. [DOI] [PubMed] [Google Scholar]
- 4.Enders D, Lenzen A. Synlett. 2003:2185–2187. [Google Scholar]
- 5.Takaoka LR, Buckmelter AJ, La Cruz TE, Rychnovsky SD. J Am Chem Soc. 2005;127:528–529. doi: 10.1021/ja044642o. [DOI] [PubMed] [Google Scholar]
- 6.Recent reviews on spiroacetal synthesis: Mead KT, Brewer BN. Curr Org Chem. 2003;7:227–256.Aho JE, Pihko PM, Rissa TK. Chem Rev. 2005;105:4406–4440. doi: 10.1021/cr050559n. See also Jacobs MF, Kitching W. Curr Org Chem. 1998;2:395–36.Perron F, Albizati KF. Chem Rev. 1989;89:1617–1661.Boivin TLB. Tetrahedron. 1987;43:3309–3362.
- 7.(a) Montcourrier P, Silver I, Farnoud R, Bird I, Rochefort H. Clin Exp Metastasis. 1997;15:382–392. doi: 10.1023/a:1018446104071. [DOI] [PubMed] [Google Scholar]; (b) Stubbs M, McSheehy PMJ, Griffiths JR, Bashford CL. Mol Med Today. 2000;6:15–19. doi: 10.1016/s1357-4310(99)01615-9. [DOI] [PubMed] [Google Scholar]; (c) Han SK, Na K, Bae YH. Colloids Surf A: Physicochem Eng Aspects. 2003;214:49–59. [Google Scholar]; (d) Reddy LH. J Pharm Pharmacol. 2005;57:1231–1242. doi: 10.1211/jpp.57.10.0001. [DOI] [PubMed] [Google Scholar]
- 8.Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J Am Chem Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- 9.For an account of natural product synthesis based on a dithiane-strategy see: Smith AB, III, Adams CM. Acc Chem Res. 2004;37:365–377. doi: 10.1021/ar030245r. and references therein.
- 10.Chikashita H, Kittaka E, Kimura Y, Itoh K. Bull Chem Soc Jpn. 1989;62:833–837. (b) Also see Uemura, reference 2b.
- 11.Furuta H, Hase M, Noyori R, Mori Y. Org Lett. 2005;7:4061–4064. doi: 10.1021/ol051679k. [DOI] [PubMed] [Google Scholar]
- 12.Stork G, Zhao K. Tetrahedron Lett. 1989;30:287–290. [Google Scholar]
- 13.Petri AF, Kühnert SM, Scheufler F, Maier ME. Synthesis. 2003:940–955. [Google Scholar]
- 14.(a) Schneider C, Klapa K, Hansch M. Synlett. 2005:91–94. [Google Scholar]; (b) Schneider C, Hansch M, Weide T. Chem Eur J. 2005;11:3010–3021. doi: 10.1002/chem.200400951. [DOI] [PubMed] [Google Scholar]
- 15.The anti relationship of the two stereocenters of product 14 was confirmed by 13C analysis of acetonide 14a, where the appropriate 13C resonance signals of both the acetal and methyl carbon atoms were observed, refer to the supporting information section, Rychnovsky SD, Skalitzky DJ. Tetrahedron Lett. 1990;31:945–948.Rychnovsky SD, Rogers B, Yang G. J Org Chem. 1993;58:3511–3515.
- 16.Barrett AGM, Godfrey CRA, Hollinshead DM, Prokopiou PA, Barton DHR, Boar RB, Joukhadar L, McGhie JF, Misra SC. J Chem Soc Perkin Trans 1. 1981:1501–509. [Google Scholar]
- 17.(a) Ronald RL, Roush WR. J Am Chem Soc. 1986;108:294–296. [Google Scholar]; (b) Coe JW, Roush WR. J Org Chem. 1989;54:915–930. [Google Scholar]; (c) Roush WR, Ando K, Powers DB, Palkowitz AD, Halterman RL. J Am Chem Soc. 1990;112:6339–6348. [Google Scholar]
- 18.Schmidt B. J Org Chem. 2004;69:7672–7687. doi: 10.1021/jo048937w. [DOI] [PubMed] [Google Scholar]
- 19.(a) Takami K, Yorimitsu H, Oshima K. Org Lett. 2002;4:2993–2995. doi: 10.1021/ol026401w. [DOI] [PubMed] [Google Scholar]; (b) Takami K, Mikami S, Yorimitsu H, Shinokubo H, Oshima K. J Org Chem. 2003;68:6627–6631. doi: 10.1021/jo0344790. [DOI] [PubMed] [Google Scholar]
- 20.(a) Lipshutz BH. Synthesis. 1987:325–341. [Google Scholar]; (b) Solladié G, Hamdouchi C, Ziani-Chérif C. Tetrahedron Asym. 1991;2:457–469. [Google Scholar]
- 21.(a) Leeuwenburgh MA, Overkleeft HS, van der Marel GA, van Boom JH. Synlett. 1997:1263–1264. [Google Scholar]; (b) Clark JS, Kettle JG. Tetrahedron Lett. 1997;38:127–130. [Google Scholar]
- 22.Corey EJ, Suggs JW. J Org Chem. 1973;38:3224. [Google Scholar]
- 23.(a) Nicolaou KC, Veale CA, Hwang CK, Hutchinson J, Prasad CVC, Ogilvie WW. Angew Chem, Int Ed Engl. 1991;30:299–303. [Google Scholar]; (b) Furkert DP, Brimble MA. Org Lett. 2002;4:3655–3658. doi: 10.1021/ol026605c. [DOI] [PubMed] [Google Scholar]
- 24.For a review on the metalation of vinyl ethers see: Friesen RW. J Chem Soc Perkin Trans 1. 2001:1969–2001.
- 25.Crich D, Ritchie TJ. Tetrahedron. 1988;44:2319–2328. [Google Scholar]
- 26.The sulfinating reagent PhSSO2Ph was crucial to the success of the reaction: Billard T, Langlois BR, Large S, Anker D, Roidot N, Roure P. J Org Chem. 1996;61:7545–7550. doi: 10.1021/jo960619c.
- 27.La Cruz TE, Rychnovsky SD. Synlett. 2004:2013–2015. [Google Scholar]
- 28.Milne JE, Kocienski PJ. Synthesis. 2003:584–592. [Google Scholar]
- 29.Statsuk AV, Liu D, Kozmin SA. J Am Chem Soc. 2004;126:9546–9547. doi: 10.1021/ja046588h. [DOI] [PubMed] [Google Scholar]
- 30.(a) Louie J, Bielawski CW, Grubbs RH. J Am Chem Soc. 2001;123:11312–11313. doi: 10.1021/ja016431e. [DOI] [PubMed] [Google Scholar]; (b) Cossy J, Bauer D, Bellosta V. Tetrahedron. 2002;58:5909–5922. [Google Scholar]
- 31.The configuration of orthoester 8 was determined by 2D NMR analysis. The minor orthoester arising from the reaction of thioketene acetal 10 and syn stereoisomer of diol 9 was removed at this stage by column chromatography.
- 32.Buckmelter AJ. PhD thesis. University of California; Irvine: 2000. [Google Scholar]
- 33.Farras J, Serra C, Vilarrasa J. Tetrahedron Lett. 1998;39:327–330. [Google Scholar]
- 34.(a) Zeller E, Sajus H, Grierson DS. Synlett. 1991:44–46. [Google Scholar]; (b) Zeller E, Grierson DS. Synlett. 1991:878–880. [Google Scholar]; (c) Wolckenhauer SA, Rychnovsky SD. Tetrahedron. 2005;61:3371–3381. [Google Scholar]
- 35.Calaza MI, Hupe E, Knochel P. Org Lett. 2003;5:1059–1061. doi: 10.1021/ol0340742. [DOI] [PubMed] [Google Scholar]
- 36.Guijarro D, Mancheño B, Yus M. Tetrahedron. 1994;50:8551–8558. [Google Scholar]
- 37.(a) Pihko PM, Aho JE. Org Lett. 2004;6:3849–3852. doi: 10.1021/ol048321t. [DOI] [PubMed] [Google Scholar]; (b) Bender T, Schuhmann T, Magull J, Grond S, von Zezschwitz P. J Org Chem. 2006;71:7125–7132. doi: 10.1021/jo060149e. [DOI] [PubMed] [Google Scholar]
- 38.(a) Hicks DR, Fraser-Reid B. Synthesis. 1974:203. [Google Scholar]; (b) Cink RD, Forsyth CJ. J Org Chem. 1995;60:8122–8123. [Google Scholar]
- 39.Kolb HC, Sharpless KB. Tetrahedron. 1992;48:10515–10530. [Google Scholar]
- 40.Acetonide cleavage is accompanied by formation of the attenol B core, structure 50.
- 41.(a) Rychnovsky SD, Powers JP, Lepage TJ. J Am Chem Soc. 1992;114:8375–8384. [Google Scholar]; (b) Rychnovsky SD, Hata T, Kim AI, Buckmelter AJ. Org Lett. 2001;3:807–810. doi: 10.1021/ol006866r. [DOI] [PubMed] [Google Scholar]
-
42.The relative energy difference between equatorial and axial radical isomers of 2-methoxyl-5,6-dimethyltetrahydropyran-2-yl were calculated (6–31G(d)/UB3LYP//6–31G(d)/UB3LYP) and found to favor the axial radical by 1.86 kcal/mol. (With zero point correction; without ZPE correction the difference was 2.03 kcal/mol.) Geometries and energies of the structures are included in the supporting information.
- 43.Seeman JI. Chem Rev. 1983;83:83. [Google Scholar]
- 44.Reviews on chiral organolithiums: Basu A, Thayumanavan S. Angew Chem Int Ed. 2002;41:716–738. doi: 10.1002/1521-3773(20020301)41:5<716::aid-anie716>3.0.co;2-z.Hoppe D, Marr F, Brueggemann M. Enantioselective synthesis by lithiation adjacent to oxygen and electrophile incorporation. Top Organomet Chem. 2003;5:61–137.
- 45.Inversion of configuration at the lithium bearing carbon of α-oxy alkyllithiums have been observed in [1,2]- and [2,3]-Wittig rearrangements: Verner EJ, Cohen T. J Am Chem Soc. 1992;114:375–377.Hoffmann R, Brückner R. Angew Chem Int Ed Engl. 1992;31:647–649.Hoffmann R, Rückert T, Brückner R. Tetrahedron Lett. 1993;34:297–300. For a computational study of this phenomenon see: Capó M, Saá JM. J Am Chem Soc. 2004;126:16738–16739. doi: 10.1021/ja045203s.
- 46.The longest linear sequence proceeds from commercial 3-butene-l-ol, a precursor of epoxide 12, in 16 steps and 7.3% overall yield, which includes a 44% yield on the resolution step.
- 47.GC conditions are reported in Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION AVAILABLE: Experimental details for preparation of the cyclization substrates and the characterization and correlation of the products. Proton and 13C NMR spectra are included. This material is available free of charge via the Internet at http://pubs.acs.org.