

Abstract
Previously, we implicated the opioid receptor (OR), Gβγ subunits, and Ras in the opioid activation of extracellular signal-regulated protein kinase (ERK), a member of the mitogen-activated protein (MAP) kinase family involved in mitogenic signaling. We now report that OR endocytosis also plays a role in the opioid stimulation of ERK activity. COS-7 and HEK-293 cells were cotransfected with the cDNA of δ-, μ-, or κ-OR, dynamin wild-type (DWT), or the dominant suppressor mutant dynamin K44A, which blocks receptor endocytosis. The activation of ERK by opioid agonists in the presence of DWT was detected. In contrast, parallel ectopic coexpression of the K44A mutant with OR, followed by agonist treatment, resulted in a time-dependent attenuation of ERK activation. Immunofluorescence confocal microscopy of δ-OR and DWT-cotransfected COS-7 cells revealed that agonist exposure for 10 min resulted in an ablation of cell surface δ-OR immunoreactivity (IR) and an intensification of cytoplasmic (presumably endosomal) staining as seen in the absence of overexpressed DWT. After 1 hr of δ-agonist exposure the cells displayed substantial internalization of δ-OR IR. If the cells were cotransfected with δ-OR and dynamin mutant K44A, OR IR was retained on the cell surface even after 1 hr of δ-agonist treatment. Parallel immunofluorescence confocal microscopy, using an anti-ERK antibody, showed that agonist-induced time-dependent ERK IR trafficking into perinuclear and nuclear loci was impaired in the internalization-defective cells. Thus, both biochemical and immunofluorescence confocal microscopic evidence supports the hypothesis that the opioid activation of ERK requires receptor internalization in transfected mammalian cells.
Keywords: opioid receptor, opioids, MAP kinase, dynamin, ERK, immunofluorescence confocal microscopy
Growth factors elicit a diversity of cell type-specific responses that regulate cell proliferation. Their signal transduction pathways involve a series of protein phosphorylations that begin with a receptor tyrosine kinase (RTK) on the plasma membrane, continue in the cytoplasm, and culminate in the nucleus. Several receptor–protein interactions, typically via Src homology domains (SH2/SH3), result in Ras/Raf activation. Raf phosphorylates mitogen-activated protein kinase kinase (MEK), which activates the mitogen-activated protein (MAP) kinases. The latter include extracellular signal-regulated protein kinases (ERKs), Jun protein kinases (JNKs), and p38mapk isoforms, all of which phosphorylate different transcription factors in the nucleus (Cobb and Goldsmith, 1995; Segal and Greenberg, 1996; Khokhlatchev et al., 1998). Thus, the MAP kinase phosphorylation cascade plays a central role in growth factor signal transduction.
G-protein-coupled receptors (GPCRs) also mediate MAP kinase activation (Van Biesen et al., 1996; Gutkind, 1998). Diverse GPCR signal transduction pathways converge with RTK phosphorylation cascades. Receptors coupled to pertussis toxin-sensitive G-proteins activate ERKs and JNKs via Gβγ subunits, whereas those coupled to pertussis toxin-insensitive G-proteins typically activate ERK and JNK by Gα.
Opioids stimulate ERK activity in brain (Berhow et al., 1996), in μ-opioid receptor-transfected (OR-transfected) Chinese hamster ovary (CHO) cells (Li and Chang, 1996), and in δ-OR-transfected Rat-1 fibroblasts (Burt et al., 1996). Insight into the molecular mechanism by which opioids activate ERK has been gained. Belcheva et al. (1998)implicated Gβγ subunits of Gi/o proteins and Ras in the opioid agonist modulation of ERK in COS-7 cells transfected with μ-, δ-, or κ-ORs.
Receptors internalize via clathrin-coated vesicles, caveolae, or uncoated vesicles (Haigler et al., 1980; Raposo et al., 1989; Anderson et al., 1992; von Zastrow and Kobilka, 1992). GPCR endocytosis is a dynamic process required for agonist-induced turnover and resensitization. In this sequence the receptors are phosphorylated by GPCR kinases, causing uncoupling from their cognate G-protein, followed by endocytosis. Initial evidence of GPCR internalization via a clathrin-mediated mechanism was obtained by the discovery of OR and β-adrenergic receptor binding in highly purified preparations of clathrin-coated vesicles (Bennett et al., 1985; Chuang et al., 1986). Subsequently, supporting evidence was obtained for these GPCRs (von Zastrow and Kobilka, 1992; Trapaidze et al., 1996; Chu et al., 1997;Segredo et al., 1997). Clathrin-coated vesicle formation entails ATP- and GTP-dependent steps, one of the latter being mediated by dynamin. Dynamin migrates from cytosol to specific sites in clathrin-coated structures in its GDP-bound form. GTP binding causes the redistribution of dynamin and its assembly into a collar at the neck of invaginated coated pits. Subsequent GTP hydrolysis induces a conformational change that leads to a detachment of the coated vesicle from the plasma membrane. Mutation of Lys44 to Ala in the amino acid sequence of dynamin disrupts its GTPase activity and imposes a defect in endocytosis. Vieira et al. (1996) reported that the activation of ERK-1/ERK-2 by EGFR is suppressed in cells transfected with dynamin mutant K44A. Thus, endocytic trafficking of EGFR appears to be important for the full activation of ERKs. Five mechanistically distinct inhibitors of clathrin-mediated endocytosis attenuated ERK activation by GPCR agonists (Luttrell et al., 1997a; Daaka et al., 1998), indicating that GPCR endocytosis is required for ERK activity.
Here we report biochemical and immunocytochemical evidence for the requirement of OR internalization for ERK activation by agonists.
MATERIALS AND METHODS
Chemicals. Most chemicals were purchased from Sigma (St. Louis, MO) with the exception of [d-Pen2,d-Pen5]enkephalin (DPDPE), [d-ala-d-Leu]enkephalin (DADLE), and [d-ala2,mephe4,gly-ol5] enkephalin (DAMGE), which were obtained from Multiple Peptide Systems (San Diego, CA); anti-ERK antibody (Ab) that cross-reacts with ERK-1 and ERK-2 from Santa Cruz Biotechnology (Santa Cruz, CA); FITC-conjugated donkey anti-rabbit IgG from Jackson ImmunoResearch Laboratories (West Grove, PA); [γ-32P] ATP (3000 Ci/mmol) from Amersham (Arlington Heights, IL); and U69,593 from NIDA Drug Supply (Research Triangle Park, NC).
Cell culture and transfection. COS-7 cells were grown at 37°C in a humidified CO2 (5%) incubator in DMEM and Ham’s nutrient mixture F12 containing 10% heat-inactivated calf serum for six passages after they were transferred from the same medium with 10% fetal calf serum. COS-7 cells were grown in 10 cm Petri dishes and seeded routinely in six-well plates 24 hr before transfection to achieve 50–60% confluency on the day of transfection. Transient transfections were performed with ERK-1 cDNA (in pcDNA-3 vector; kindly provided by Dr. J. Baldassare, St. Louis University, St. Louis, MO) or with δ-OR cDNA (in pCI–neo expression vector) in the presence of DWT or its mutant form K44A (both in pCB1 vector; kindly provided by Dr. Marc Caron, Duke University, Durham, NC), using the DEAE-dextran method (Belcheva et al., 1998). HEK-293 cells were grown in Minimum Essential Medium (MEM) containing 10% fetal bovine serum. Cells were grown in six-well plates, and the transient transfection of rat μ- or κ-OR cDNA (pCMV–neo expression vector) as well as the plasmids described above was performed with Lipofectamine Reagent (Life Technologies, Gaithersburg, MD) according to the manufacturer’s description. Transfection efficiencies were monitored by OR binding assays (Belcheva et al., 1998). Similar levels of δ-OR in DWT and K44A-transfected COS-7 cells were detected by immunoblot analysis.
ERK assay. ERK activity was measured by using the “in-gel” kinase method as described in Belcheva et al. (1998). Briefly, OR and dynamin wild-type (DWT) or K44A-transfected COS-7 or HEK-293 cells that were serum-starved overnight were exposed to type-specific opioids (0.1 μmDADLE or 1 μm DPDPE for δ, 1 μm DAMGE for μ, and 1 μm U69,593 for κ) for the indicated time periods. Then the cells were washed with cold PBS and lysed with buffer containing (in mm) 20 HEPES, 10 EGTA, 40 β-glycerophosphate, 2.5 MgCl2, 2 sodium vanadate, and 1 PMSF plus 1% Nonidet-40, 20 μg/ml aprotinin, and 20 μg/ml leupeptin. Lysates were spun at 14,000 × g for 20 min at 4°C, and the protein concentration of supernatants was determined before the enzyme assay by the micro-Bradford assay (Bio-Rad, Hercules, CA) with bovine serum albumin as a standard. Cell lysates (30–50 μg of protein/lane) were separated by 10% SDS-PAGE. Resolving gels contained 0.5 mg/ml myelin basic protein (MBP). To remove SDS after electrophoresis, we washed the gels twice with 100 ml of 50 mm Tris-HCl, pH 8.0, containing 20% isopropyl alcohol for 30 min. Then a 1 hr wash with 50 mm Tris-HCl, pH 8.0, and 5 mm β-mercaptoethanol was followed by protein denaturation with 100 ml of 6 m guanidine chloride (twice, 30 min each). Finally, proteins in gels were renatured with buffer containing 50 mm Tris-HCl, pH 8.0, 5 mm β-mercaptoethanol, and 0.04% Tween 40 for 16 hr at 4°C with at least four to five changes of buffer. Gels were incubated with 40 mm HEPES buffer, pH 8.0, containing 2 mm DTT and 10 mmMgCl2 at room temperature for 1 hr. The kinase assay was performed by placing gels in 40 mm HEPES buffer, pH 8.0, containing 0.5 mm EGTA, 10 mmMgCl2, 2 μm protein kinase inhibitor (PKI), and 40 μm γ-32P-ATP (50 μCi) for 3 hr at room temperature. Gels then were rinsed with a solution of 5% TCA and 1% sodium pyrophosphate to remove noncovalently bound32P. Kinase activity, as expressed by the degree of phosphorylation of MBP, was quantified with the Storm PhosphoImager (Molecular Dynamics, Sunnyvale, CA) and Image Quant software. Statistical analyses were performed with the Student’s ttest.
Phosphatidyl inositide (PI) turnover. COS-7 cells were transfected with δ-OR and either DWT or dynamin mutant K44A. After 48 hr of starvation in serum-free MEM, cells were labeled overnight in the same medium with 1.5 μCi myo-[2-3H(N)]-inositol (20 Ci/mmol; New England Nuclear, Boston, MA) per milliliter. The medium was replaced with fresh medium containing 5 mm LiCl for a 30 min incubation before DADLE (1 μm, 1 hr) treatment. The cells were washed twice in cold PBS and collected in PBS–EDTA with scraping. Inositol phosphates were extracted in a 1:1 ratio of methanol/chloroform and eluted from Bio-Rad AG1 × 8 columns with 1 m ammonium formate in 0.1 m formic acid, as described (Barg et al., 1994).
Immunohistochemistry. Polyclonal Abs were generated against a mouse δ-OR CTe peptide with the sequence TRERVTACTPSDGPG (amino acids 353–367). Cells transfected with δ-OR and ERK or DWT or K44A mutant were grown on coverslips and were treated with different ligands for the indicated time periods. Cells then were fixed in 4% paraformaldehyde for 15 min. After three 5 min washes with TBS (20 mm Tris, pH 7.4, 150 mmNaCl, and 1 mm CaCl2), the cells were permeabilized with 0.1% Triton X-100 in PBS for 15 min. The cells were washed again three times with TBS and blocked for 30 min in 3% milk dissolved in 50 mm Tris, pH 7.4, containing 0.1% Triton X-100. After washing, a primary Ab (Anti-CTe δ-OR or anti-ERK) was applied and incubated at 4°C for 18 hr with constant shaking. Then the cells were washed five times and incubated with secondary Ab (FITC-conjugated donkey anti-rabbit IgG) for 1 hr at room temperature. Coverslips were washed five times with TBS and twice in H2O and mounted on slides that used n-propyl gallate as fluorescence protectant. Slides were examined with a Zeiss LSM 410 scanning laser confocal microscope system (Oberkochen, Germany). The system uses a Zeiss 135 Axiovert inverted microscope fit with an Omnicron argon/krypton laser for confocal excitation. The cells, mounted on coverslips, were viewed with either a 63× magnification, 1.25 numerical aperture oil or water immersion objective. For the FITC fluorophore, fluorescence was excited with the 488 nm laser line, and emission was collected in the 510–540 nm region. In experiments on δ-OR and DWT or K44A-expressing COS-7 cells, 50 untreated and 100 agonist-treated cells were quantified for staining to identify representative micrographs. Of these cells ∼30% displayed the δ-OR staining patterns shown.
RESULTS
Biochemical evidence for the requirement of OR internalization in the activation of ERK
To determine the effect of overexpression of DWTand mutant K44A on classical opioid second messenger signaling, we assayed COS-7 cells, cotransfected with cDNA of δ-OR and the dynamins for DADLE-stimulated PI turnover. As seen in Figure1, DADLE significantly increased PI turnover in cells transfected with DWT or K44A. The same PI stimulation was observed in cells expressing DWT or K44A. The data are in agreement with a report that overexpression of the dynamin mutant K44E did not affect opioid inhibition of cAMP accumulation in HEK-293 cells (Chu et al., 1997). They are also consistent with our previous findings that overexpressed δ-OR (naltrindole binding parameters, KD = 0.7 ± 0.4 nm; Bmax = 1.3–2.3 pmol/mg protein) in COS-7 cells is functional (Belcheva et al., 1998).
Fig. 1.

Dynamin mutant K44A overexpression does not affect DADLE-induced PI turnover in COS-7 cells. Cells transfected with δ-OR and DWT or mutant K44A were treated for 1 hr with 1 μm DADLE and assayed for inositol phosphate accumulation.Open bars, Control cells; hatched bars, DADLE-treated cells. The data are expressed as a percentage of the control of PI turnover in untreated cells (average counts for controls in DWT, 6918 dpm/well; in K44A, 9373 dpm/well) and are represented as the mean of four experiments performed in duplicate ± SEM; *p < 0.05.
Because it is established that different agonists have different efficacies in inducing receptor internalization and that mechanisms of GPCR-induced ERK activation are cell type-dependent, it was important to show the effects of the dynamin mutant on at least two cell lines tested with several opioid agonists. COS-7 cells, cotransfected with the cDNA of δ-OR and DWT or mutant K44A, were treated with DADLE or DPDPE for different time intervals, and the ERK activity was measured. As seen in Figure 2, a 2.5- to 3.5-fold activation of ERK by 10 min of agonist treatment was detected in the presence of DWT. Although ERK activation by 1 μm DPDPE continued for 60 min, 0.1 μmDADLE stimulation of the enzyme did not persist as long. In contrast, parallel ectopic coexpression of the K44A mutant with δ-OR, followed by treatment with either agonist, resulted in the inhibition of ERK stimulation by 33–44% at 10 min.
Fig. 2.

Evidence for the requirement of OR internalization in the opioid activation of ERK in COS-7 cells. Cells cotransfected with δ-OR and DWT or mutant K44A were treated with 1 μm DPDPE (A) or 0.1 μm DADLE (B) for different time intervals. Top, Representative autoradiograms showing the phosphorylated MBP bands. Bottom, Quantification of ERK activity. n = 2–6; *p < 0.05.
Similar results were gained in time course experiments that used δ-, μ-, or κ-OR-transfected HEK-293 cells (Fig.3). Agonist-induced ERK activation was reduced in endocytosis-defective cells for all receptor types and for the κ-OR at all time points. DPDPE stimulation of ERK also was reduced at the three earliest time points that were studied, whereas DADLE effects were attenuated only at 10 min in the presence of K44A. These experiments suggest that the inhibition of receptor internalization attenuated the activation of ERK at early time points, which coincided with the highest levels of ERK activation by opioid agonists.
Fig. 3.

Evidence for the requirement of OR internalization in the opioid activation of ERK in HEK-293 cells. Cells cotransfected with δ-, μ-, or κ-OR, DWT, or mutant K44A were treated with 1 μm DPDPE (A), 0.1 μm DADLE (B), 1 μmDAMGE (C), or 1 μm U69,593 (D) for different time intervals.Top, Representative autoradiograms showing the phosphorylated MBP bands. Bottom, Quantification of ERK activity. n = 2–6; *p < 0.05.
Agonist-induced δ-OR immunoreactivity (IR) trafficking in COS-7 cells
To monitor the intracellular migration of δ-OR IR and ERK IR under the same conditions as described above, we plated COS-7 cells on microscope coverslips, treated them with agonists, and subjected them to immunofluorescence confocal microscopy. The specificity of anti-δ-OR Ab was examined by using NG108–15 and parental COS-7 cells. NG108–15 cells express relatively large amounts of δ-OR (0.6 pmol/mg protein) and have been used extensively as a model system for the study of δ-opioid action. NG108–15 cells incubated with anti-δ-OR Ab that had been preadsorbed with the antigenic peptide showed little or no staining again as seen for preimmune serum treatment (Fig. 4). Parental COS-7 cells displayed no staining with anti-δ-OR Ab, and they resembled δ-OR-transfected controls treated with preimmune serum (Fig. 4).
Fig. 4.
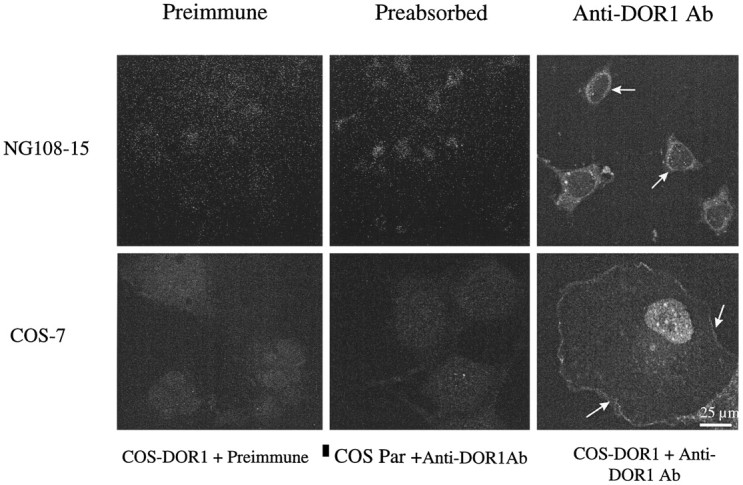
Specificity of anti-δ-OR Ab. NG108–15 cells were fixed, permeabilized, and stained with preimmune serum (1:1000), anti-δ-OR (Anti-DOR1 Ab; 1:1000) preadsorbed with the antigenic peptide (0.3 mg/ml), and FITC-conjugated secondary Ab or anti-δ-OR Ab (1:1000 dilution). COS-7 cells overexpressing δ-OR were processed as above and stained with preimmune serum or anti-δ-OR Ab (1.5 μg/ml). Parental COS-7 cells were stained with anti-δ-OR Ab (1.5 μg/ml). Shown are representative micrographs from two to six independent experiments.
COS-7 cells cotransfected with δ-OR and DWT or mutant K44A were treated with DADLE for 10 min or 1 hr and stained with anti-δ-OR Ab. COS-7 cell surface staining of δ-OR seen in the untreated DWT control cells dissipated after 10 min of exposure to agonist; instead, more granular cytoplasmic staining suggestive of endosomal compartmentation was observed (Fig.5). Similarly, in dual immunofluorescence confocal microscopy experiments, 10 min of agonist exposure elicited colocalization of δ-OR IR and transferrin–Texas Red in NG108–15 cells, indicative of endosomal compartmentation (data not shown). After 1 hr of agonist treatment the plasma membrane staining was dissipated and fluorescence was localized predominantly in perinuclear/nuclear regions of the cell (Fig. 5). Immunofluorescence confocal microscopy experiments on δ-OR-transfected HEK-293 cells displayed similar receptor endocytosis except that less perinuclear/nuclear staining was observed (data not shown). However, in cells transfected with the K44A mutant, δ-OR IR was retained on the cell surface at all time intervals in both COS-7 and HEK-293 cells.
Fig. 5.

Confocal microscopy of δ-OR IR in agonist-treated COS-7 cells. Cells were cotransfected with δ-OR and DWT or mutant K44A. Control and agonist-treated (0.1 μm DADLE for 10 min or 1 hr) cells were fixed, permeabilized, and immunostained with anti-δ-OR Ab (2 μg/ml) and FITC-conjugated secondary Ab. Shown are representative micrographs from four independent experiments.
Agonist-induced ERK IR trafficking in COS-7 cells
ERK IR also was displaced after DADLE or DPDPE exposure of COS-7 cells cotransfected with δ-OR and ERK. Untreated controls and acute (10 min) DADLE- or DPDPE-treated cells showed diffuse cytoplasmic staining (Fig. 6). After 1 hr of agonist exposure the staining became less diffuse and more localized in the perinuclear/nuclear regions of the cell (Fig. 6). This distribution of ERK IR is consistent with subcellular fractionation data and is typical of what is seen in other immunohistochemical studies of nuclear import of ERK [Khokhlatchev et al. (1998) and references cited therein]. Antagonist (1 μm naltrexone) pretreatment 1 hr before DADLE prevented ERK nuclear import. Naltrexone alone had no effect on ERK trafficking (data not shown).
Fig. 6.
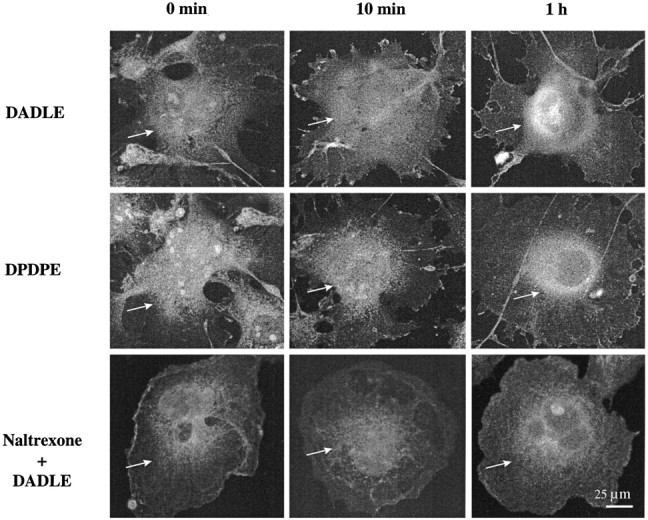
Confocal microscopy of ERK IR in agonist-treated COS-7 cells. Cells were cotransfected with ERK-1 and δ-OR. DADLE (0.1 μm) or DPDPE (1 μm) was added to the medium for 0 or 10 min or 1 hr. The bottom panel displays the effect of a 1 hr pretreatment of cells with 1 μmnaltrexone before DADLE exposure. After fixation and permeabilization the cells were stained with anti-ERK Ab (0.8 μg/ml) and FITC-conjugated secondary Ab. Shown are representative micrographs from three to four independent experiments.
When COS-7 cells were cotransfected with δ-OR and DWT or mutant K44A, DADLE initiated a time-dependent ERK IR nuclear import only in DWT-expressing cells (Fig.7). Again, diffuse cytoplasmic ERK IR staining was observed after 10 min of agonist exposure. By 30 min of agonist treatment the ERK IR accumulated in perinuclear/nuclear regions, which persisted for at least 1 hr. Endocytosis-defective cells displayed only cytoplasmic staining of ERK at all time intervals, resembling that seen for 0 and 10 min in DWT-overexpressing cells. These results suggest that ERK activation and nuclear import require receptor internalization.
Fig. 7.

Confocal microscopy of ERK IR in agonist-treated dynamin-transfected COS-7 cells. Cells were cotransfected with DWT or mutant K44A and δ-OR. DADLE (0.1 μm) was added to the medium for 0, 10, or 30 min or 1 hr. After fixation and permeabilization the cells were stained with anti-ERK Ab (0.8 μg/ml) and FITC-conjugated secondary Ab. Shown are representative micrographs from four to six independent experiments.
DISCUSSION
By using receptor endocytosis-defective cells, we provide both biochemical and immunofluorescence confocal microscopic evidence to demonstrate that receptor internalization is required for ERK activation. The biochemical findings reveal that μ-, δ-, and κ-opioid activation of ERK is attenuated in cells overexpressing dynamin mutant K44A. In a previous communication on the essential role of GPCR endocytosis for ERK activation, defective β-adrenergic sequestration was monitored by cell sorting (Daaka et al., 1998). The immunohistochemical data shown here reveal that δ-OR internalization and intracellular migration as well as ERK trafficking to the nucleus are impeded under the same conditions in receptor endocytosis-defective cells.
The existence of intracellular ORs has been recognized for some time (Roth et al., 1981); initially, it was thought that internalization was required only for resensitization and turnover. Although it is clear that OR endocytosis is not necessary for classical GPCR signaling to generate cytosolic second messengers, recent evidence suggests that the internalization of ORs and other GPCRs may play a role in gene transcription (Belcheva et al., 1993, 1996; Tencheva et al., 1997; Lu et al., 1998; Ventura et al., 1998). The data gained here support this hypothesis.
ERK IR appears to be in both cytosol and nuclei of untreated COS-7 cells (Figs. 6, 7). This cannot be attributable to overexpression of ERK in the COS-7 cells, because ERK-containing plasmids were included only in the initial experiment (Fig. 6), but not the second (Fig. 7). The data from both experiments indicate that upon agonist treatment a time lag exists between ERK activation and its nuclear import. ERK is phosphorylated maximally within 10 min, and some nuclear ERK is found at 0 and 10 min time points. Nevertheless, ERK IR that is localized in perinuclear/nuclear regions is observed only after 30 min. The classical paradigm predicts that ERK is activated in the cytoplasm and then is translocated into the nucleus [Khokhlatchev et al. (1998) and references cited therein]. This is consistent with the marked redistribution of ERK IR that occurs 30 min after opioid agonist treatment. The smaller amounts of nuclear ERK IR at earlier time points could be residual phosphorylated and unphosphorylated molecules persisting there from previous activation [Khokhlatchev et al. (1998)and references cited therein]. Moreover, phospho-ERK IR has been found in the cytoplasm and nuclei in HEK-293T and HEK-293 cells; within 90 min of agonist exposure, nuclear labeling is displayed (Joneson et al., 1998) (E. Ignatova, unpublished observations). Another possibility recently raised is that ERK migration into the nucleus precedes its activation by MEK. MEK has a nuclear export signal, and there is evidence to suggest that it enters the nucleus where it could phosphorylate ERK (Jaaro et al., 1997; Kim and Kahn, 1997). However, ERK-3, a widely distributed isoform that is restricted to the nucleus, may be the target of nuclear MEK (Cheng et al., 1996). The results obtained here are more compatible with the notion that the MEK activation of ERK precedes trafficking into the nucleus, but carefully controlled kinetic studies that use ERK subtype specific Abs for both phosphorylated and unphosphorylated forms must be performed to resolve this issue.
The requirement of EGF receptor internalization for ERK stimulation has been reported despite the fact that membrane-delimited “upstream” effectors in the EGF pathway such as PLCγ, Shc, and Raf are activated in endocytosis-defective cells (Vieira et al., 1996; Daaka et al., 1998). Here we show that opioid activation of PLC, presumably the β-isoform, is also insensitive to endocytosis blockade. The data in the literature suggest that a multi-adaptor receptor complex containing components of both GPCR and RTK pathways may come together within the cell to trigger the activation of ERK via the cytoplasmic kinase, MEK. Many membrane-delimited intermediates of this pathway are believed to be arranged on a protein scaffold that serves to assemble different components of phosphorylation cascades (Levin and Errede, 1995; Wang et al., 1996; Luttrell et al., 1997b). The scaffold may be the RTK itself in some cells (Luttrell et al., 1997b), whereas in others caveolin is a candidate (Mineo et al., 1996; Couet et al., 1997). In fact, EGFR may undergo endocytosis via clathrin- or caveolin-mediated pathways, both of which can be blocked by the dynamin mutant K44A (Haigler et al., 1980; Mineo et al., 1996; Couet et al., 1997; Henley et al., 1998; Oh et al., 1998). In addition, it has been proposed that growth factors stimulate the release of dynamin from a complex with the adaptor protein Grb2, thereby promoting receptor endocytosis (Vidal et al., 1998). Taken together with the above-mentioned evidence for cytoplasmic phosphorylation of ERK, an attractive hypothesis is that the internalized receptor or receptors play a direct role in activating ERK and possibly even in transferring it to the nucleus. The MAP kinase phosphorylation cascade has been shown to turn the cell cycle in oocytes on and off in a “switch-like” manner (Ferrell and Machleder, 1998; Koshland, 1998). Regulation of ERK by receptor endocytosis may be a part of the “switch” mechanism.
Footnotes
This work was supported by National Institutes of Health Grant DA05412. We thank Dr. John Freeman for performing the confocal microscopy, Matt Mabery for assistance in densitometric analysis, and Myra Kim for maintaining cell culture and for transfections.
Correspondence should be addressed to Dr. Carmine J. Coscia, Department of Biochemistry and Molecular Biology, St. Louis University School of Medicine, St. Louis, MO 63104.
REFERENCES
- 1.Anderson RG, Kamen BA, Rothberg KG, Lacey SW. Potocytosis: sequestration and transport of small molecules by caveolae. Science. 1992;255:410–411. doi: 10.1126/science.1310359. [DOI] [PubMed] [Google Scholar]
- 2.Barg J, Belcheva MM, Levy R, Saya D, McHale RJ, Johnson FE, Coscia CJ, Vogel Z. Opioids inhibit endothelin-mediated DNA synthesis, phosphatidylinositol turnover, and Ca2+ mobilization in rat C6 glioma cells. J Neurosci. 1994;14:5858–5864. doi: 10.1523/JNEUROSCI.14-10-05858.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belcheva M, Barg J, Rowinski J, Gloeckner CA, Ho A, Gao X-M, Chuang D-M, Coscia CJ. Novel intracellular opioid binding sites associated with the nuclei of neurohybrid cells. J Neurosci. 1993;13:104–114. doi: 10.1523/JNEUROSCI.13-01-00104.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belcheva MM, Ignatova EG, Young EC, Coscia CJ. Agonist-induced desensitization and down-regulation of δ-opioid receptors alter the levels of their 125I-β-endorphin cross-linked products in subcellular fractions from NG108–15 cells. Biochemistry. 1996;35:14818–14824. doi: 10.1021/bi961579+. [DOI] [PubMed] [Google Scholar]
- 5.Belcheva MM, Vogel Z, Ignatova E, Avidor-Reiss T, Zippel R, Levy R, Young EC, Barg J, Coscia CJ. Opioid modulation of extracellular signal-regulated protein kinase activity is Ras-dependent and involves Gβγ subunits. J Neurochem. 1998;70:635–645. doi: 10.1046/j.1471-4159.1998.70020635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett DB, Laskowski MB, Roth BL, Spain JW, Coscia CJ. Stereo-specific opiate binding sites occur in coated vesicles. J Neurosci. 1985;5:3010–3015. doi: 10.1523/JNEUROSCI.05-11-03010.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berhow MT, Hiroi N, Nestler EJ. Regulation of ERK (extracellular signal-regulated kinase), part of the neurotrophin signal transduction cascade, in the rat mesolimbic dopamine system by chronic exposure to morphine or cocaine. J Neurosci. 1996;16:4707–4715. doi: 10.1523/JNEUROSCI.16-15-04707.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burt AR, Carr IC, Mullaney I, Anderson NG, Milligan G. Agonist activation of p42 and p44 mitogen-activated protein kinases following expression of the mouse δ-opioid receptor in Rat-1 fibroblasts: effects of receptor expression levels and comparisons with G-protein activation. Biochem J. 1996;320:227–235. doi: 10.1042/bj3200227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng M, Boulton TG, Cobb MH. ERK-3 is a constitutively nuclear protein kinase. J Biol Chem. 1996;271:8951–8958. doi: 10.1074/jbc.271.15.8951. [DOI] [PubMed] [Google Scholar]
- 10.Chu P, Murray S, Lissin D, von Zastrow M. Delta and kappa opioid receptors are differentially regulated by dynamin-dependent endocytosis when activated by the same alkaloid agonist. J Biol Chem. 1997;272:27124–27130. doi: 10.1074/jbc.272.43.27124. [DOI] [PubMed] [Google Scholar]
- 11.Chuang D-M, Carter D, Spain JW, Bennett DB, Roth BL, Laskowski MB, Coscia CJ. Detection and characterization of β-adrenergic receptors in coated vesicles of bovine brain. J Neurosci. 1986;6:2578–2584. doi: 10.1523/JNEUROSCI.06-09-02578.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cobb MH, Goldsmith EJ. How MAP kinases are regulated. J Biol Chem. 1995;270:14843–14846. doi: 10.1074/jbc.270.25.14843. [DOI] [PubMed] [Google Scholar]
- 13.Couet J, Sargiacomo M, Lisanti MP. Interaction of a receptor tyrosine kinase, EGF-R, with caveolins. Caveolin binding negatively regulates tyrosine and serine/threonine kinase activities. J Biol Chem. 1997;272:30429–30438. doi: 10.1074/jbc.272.48.30429. [DOI] [PubMed] [Google Scholar]
- 14.Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SG, Caron MG, Lefkowitz RJ. Essential role for G-protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- 15.Ferrell JE, Machleder EM. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science. 1998;280:895–898. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- 16.Gutkind JS. The pathways connecting G-protein-coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J Biol Chem. 1998;273:1839–1842. doi: 10.1074/jbc.273.4.1839. [DOI] [PubMed] [Google Scholar]
- 17.Haigler HT, Maxfield FR, Willingham MC, Pastan I. Dansylcadaverine inhibits internalization of 125I-epidermal growth factor in BALB 3T3 cells. J Biol Chem. 1980;255:1239–1241. [PubMed] [Google Scholar]
- 18.Henley JR, Krueger EWA, McNiven MA. Dynamin-mediated internalization of caveolae. J Cell Biol. 1998;141:85–99. doi: 10.1083/jcb.141.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaaro H, Rubinfeld H, Hanoch T, Seger R. Nuclear translocation of mitogen-activated protein kinase kinase (MEK-1) in response to mitogenic stimulation. Proc Natl Acad Sci USA. 1997;94:3742–3747. doi: 10.1073/pnas.94.8.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joneson T, Fulton JA, Volle DJ, Chaika OV, Barsagi D, Lewis RE. Kinase suppressor of Ras inhibits the activation of extracellular ligand-regulated (ERK) mitogen-activated protein (MAP) kinase by growth factors, activated Ras, and Ras effectors. J Biol Chem. 1998;273:7743–7748. doi: 10.1074/jbc.273.13.7743. [DOI] [PubMed] [Google Scholar]
- 21.Khokhlatchev AV, Canagarajah B, Wilsbacher J, Robinson M, Atkinson M, Goldsmith E, Cobb MH. Phosphorylation of the MAP kinase ERK-2 promotes its homodimerization and nuclear translocation. Cell. 1998;93:605–615. doi: 10.1016/s0092-8674(00)81189-7. [DOI] [PubMed] [Google Scholar]
- 22.Kim SJ, Kahn CR. Insulin regulation of mitogen-activated protein kinase kinase (MEK), mitogen-activated protein kinase, and casein kinase in the cell nucleus—a possible role in the regulation of gene expression. Biochem J. 1997;323:621–627. doi: 10.1042/bj3230621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koshland DE. The era of pathway quantification. Science. 1998;280:852–853. doi: 10.1126/science.280.5365.852. [DOI] [PubMed] [Google Scholar]
- 24.Levin DE, Errede B. The proliferation of MAP kinase signaling pathways in yeast. Curr Opin Cell Biol. 1995;7:197–202. doi: 10.1016/0955-0674(95)80028-x. [DOI] [PubMed] [Google Scholar]
- 25.Li L-Y, Chang K-J. The stimulatory effect of opioids on mitogen-activated protein kinase in Chinese hamster ovary cells transfected to express μ-opioid receptors. Mol Pharmacol. 1996;50:599–602. [PubMed] [Google Scholar]
- 26.Lu D, Yang H, Shaw G, Raizada MK. Angiotensin II-induced nuclear targeting of the angiotensin type 1 (AT1) receptor in brain neurons. Endocrinology. 1998;139:365–375. doi: 10.1210/endo.139.1.5679. [DOI] [PubMed] [Google Scholar]
- 27.Luttrell LM, Daaka Y, Dellarocca GJ, Lefkowitz RJ. G-protein-coupled receptors mediate two functionally distinct pathways of tyrosine phosphorylation in rat-1a fibroblasts. Shc phosphorylation and receptor endocytosis correlate with activation of ERK kinases. J Biol Chem. 1997a;272:31648–31656. doi: 10.1074/jbc.272.50.31648. [DOI] [PubMed] [Google Scholar]
- 28.Luttrell LM, Dellarocca GJ, Van Biesen T, Luttrell DK, Lefkowitz RJ. Gβγ subunits Src-dependent phosphorylation of the epidermal growth factor receptor. A scaffold for G-protein-coupled receptor-mediated Ras activation. J Biol Chem. 1997b;272:4637–4644. doi: 10.1074/jbc.272.7.4637. [DOI] [PubMed] [Google Scholar]
- 29.Mineo C, James GL, Smart EJ, Anderson RG. Localization of epidermal growth factor-stimulated Ras/Raf-1 interaction to caveolae membrane. J Biol Chem. 1996;271:11930–11935. doi: 10.1074/jbc.271.20.11930. [DOI] [PubMed] [Google Scholar]
- 30.Oh P, McIntosh DP, Schnitzer JE. Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium. J Cell Biol. 1998;141:101–114. doi: 10.1083/jcb.141.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raposo G, Dunia I, Delavier-Klutchko C, Kaveri S, Strosberg AD, Benedetti EL. Internalization of β-adrenergic receptor in A431 cells involves noncoated vesicles. Eur J Cell Biol. 1989;50:340–352. [PubMed] [Google Scholar]
- 32.Roth BL, Laskowski MB, Coscia CJ. Evidence for distinct subcellular sites of opiate receptors: demonstration of opiate receptors in smooth microsomal fractions isolated from rat brain. J Biol Chem. 1981;256:10117–10121. [PubMed] [Google Scholar]
- 33.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 34.Segredo V, Burford NT, Lameh J, Sadee W. A constitutively internalizing and recycling mutant of the μ-opioid receptor. J Neurochem. 1997;68:2395–2404. doi: 10.1046/j.1471-4159.1997.68062395.x. [DOI] [PubMed] [Google Scholar]
- 35.Tencheva Z, Belcheva MM, Velichkova AA, Lissichkova EG, Coscia CJ. Opioid regulation of AP-1 DNA binding activity in NG108–15 cells under conditions of opioid receptor adaptation. Mol Brain Res. 1997;48:156–158. doi: 10.1016/s0169-328x(97)00128-9. [DOI] [PubMed] [Google Scholar]
- 36.Trapaidze N, Keith DE, Cvejic S, Evans CJ, Devi LA. Sequestration of the δ-opioid receptor role of the C terminus in agonist-mediated internalization. J Biol Chem. 1996;271:29279–29285. doi: 10.1074/jbc.271.46.29279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Biesen T, Luttrell LM, Hawes BE, Lefkowitz RJ. Mitogenic signaling via G-protein-coupled receptors. Endocr Rev. 1996;17:698–714. doi: 10.1210/edrv-17-6-698. [DOI] [PubMed] [Google Scholar]
- 38.Ventura C, Maioli M, Pintus G, Posadino A-M, Tadolini B. Nuclear opioid receptors activate opioid peptide gene transcription in isolated myocardial nuclei. J Biol Chem. 1998;273:13383–13386. doi: 10.1074/jbc.273.22.13383. [DOI] [PubMed] [Google Scholar]
- 39.Vidal M, Montiel JL, Cussac D, Cornille F, Duchesne M, Parker F, Tocque B, Roques BP, Garbay C. Differential interactions of the growth factor receptor-bound protein 2 N-SH3 domain with son of sevenless and dynamin. Potential role in the Ras-dependent signaling pathway. J Biol Chem. 1998;273:5343–5348. doi: 10.1074/jbc.273.9.5343. [DOI] [PubMed] [Google Scholar]
- 40.Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]
- 41.von Zastrow M, Kobilka BK. Ligand-regulated internalization and recycling of human β2-adrenergic receptors between the plasma membrane and endosomes containing transferrin receptors. J Biol Chem. 1992;267:3530–3538. [PubMed] [Google Scholar]
- 42.Wang Z, Tung PS, Moran MF. Association of p120 Ras GAP with endocytic components and colocalization with epidermal growth factor (EGF) receptor in response to EGF stimulation. Cell Growth Differ. 1996;7:123–133. [PubMed] [Google Scholar]