

Abstract
The cyclooxygenase (COX) enzymes are responsible for the committed step in prostaglandin biosynthesis, the generation of prostaglandin H2. As a result, these enzymes are pharmacologically important targets for non-steroidal anti-inflammatory drugs, such as aspirin and newer COX-2 selective inhibitors. The cyclooxygenases are functional homodimers, and each subunit contains both a cyclooxygenase and a peroxidase active site. These enzymes are quite interesting mechanistically, as the conversion of arachidonic acid to prostaglandin H2 requires two oxygenation and two cyclization reactions, resulting in the formation of five new chiral centers with nearly absolute regio- and stereochemical fidelity. We have used molecular dynamics (MD) simulations to investigate the equilibrium behavior of both COX-1 and COX-2 enzyme isoforms with bound arachidonate. These simulations were compared with reference simulations of arachidonate in solution to explore the effect of enzyme on substrate conformation and positioning in the active site. The simulations suggest that the substrate has greater conformational freedom in the COX-2 active site, consistent with the larger COX-2 active site volume observed in X-ray crystal structures. The simulations reveal different conformational behavior for arachidonate in each subunit over the course of extended equilibrium MD simulations. The simulations also provide detailed information for several protein channels that might be important for oxygen and water transport to or from active sites, or for intermediate trafficking between the cyclooxygenase and peroxidase active sites. The detailed comparisons for COX-1 versus COX-2 active site structural fluctuations may also provide useful information for design of new isozyme-selective inhibitors.
Cyclooxygenase (COX, or prostaglandin H synthase) is the enzyme responsible for the committed step in prostaglandin biosynthesis, generating the product prostaglandin H2 (PGH2). Various prostaglandin synthases then convert PGH2 into several different prostaglandins and thromboxanes. These prostaglandins and thromboxanes target specific G protein-coupled receptors and play major roles in regulation of renal function, platelet aggregation, protection of the stomach lining, and numerous other homeostatic tasks, as well as mediation of the cellular inflammatory response (1). The former, “housekeeping” functions are attributed to the first of two established COX isoforms, the constitutively expressed COX-1, while the inflammatory response is associated largely with the inducible isoform, COX-2 (2). Inhibition of COX-2 produces the analgesic, antipyretic, and anti-inflammatory effects typical of non-steroidal anti-inflammatory drugs (NSAIDs), while inhibition of COX-1 is responsible for the antithrombotic effects of aspirin and other nonselective NSAIDs, as well as many of their side effects, such as gastric ulcer formation. The many therapeutically useful effects of COX inhibition have made the NSAIDs among the most widely used drugs of the past century. Since selective COX-2 inhibition can provide analgesic and anti-inflammatory effects with reduced undesirable gastric side effects, COX-2 selective inhibitors such as celecoxib and rofecoxib have become some of the most widely used prescription medications in the developed world. However, recent reports that COX-2 selective inhibitors may increase the risk of heart attack in some patients has caused great concern, and stimulated increased interest in these enzymes (3, 4).
Structurally, the two cyclooxygenase enzymes, COX-1 and COX-2, are homodimers of two 70-kDa subunits related by a C2-axis of symmetry (Figure 1). Each monomer consists of a globular, catalytic domain, a membrane-binding domain, and an epidermal growth factor (EGF) type domain. The catalytic domain of each COX monomer has two distinct active sites, a cyclooxygenase site and a heme-containing peroxidase site. The cyclooxygenase site converts arachidonic acid (5,8,11,14-eicosatetraenoic acid or AA) to the hydroperoxy endoperoxide product prostaglandin G2 (PGG2) via two highly regiospecific and stereoselective oxygen additions and two cyclization reactions (Scheme 1). The hydroperoxide intermediate PGG2 then migrates to the peroxidase site, where it is reduced to the final product PGH2. The membrane-binding domain in each monomer consists of four amphipathic helices (helices A-D) arranged in a box-like configuration that anchors the protein to one leaflet of the lipid bilayer. The relatively large space enclosed by these four helices has been referred to as the “lobby” region (5). This membrane-associated domain provides a logical pathway for fatty acid substrates to travel from the membrane into the cyclooxygenase active site through a “gate” constriction, consisting of arginine, tyrosine and glutamate residues (Arg-120, Tyr-355 and Glu-524 using the ovine COX-1 numbering). This gate region engages the carboxylate of fatty acid substrates in a charge-reinforced hydrogen bond network that anchors the substrate in the active site.
Figure 1.
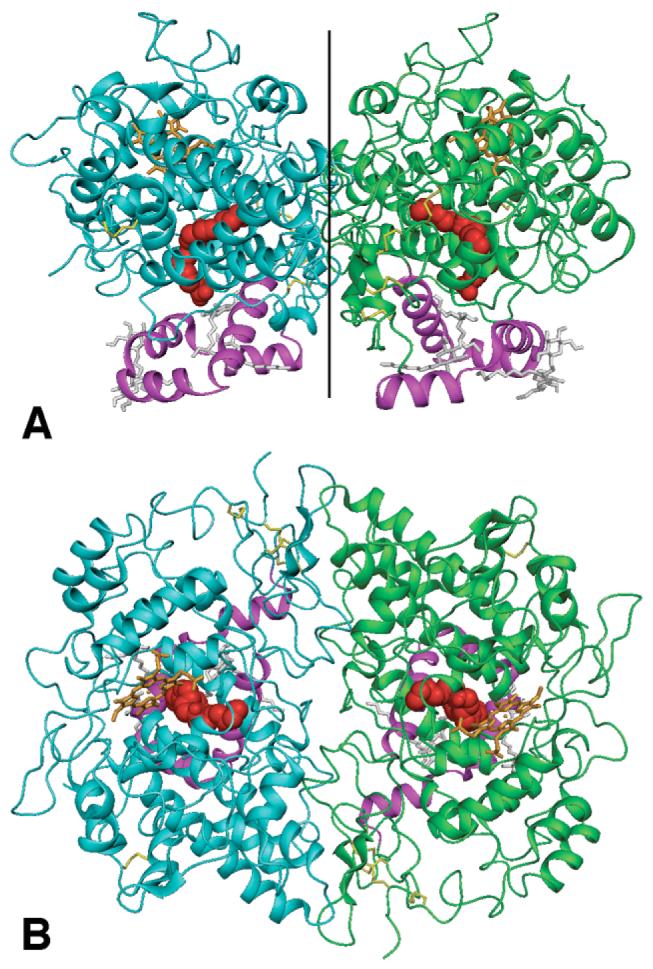
Side (A) and top (B) views of the COX-1 dimer with arachidonate bound. Panel A shows the side view, parallel to the membrane, with the C2-axis relating the monomers indicated by a vertical black line. Panel B is rotated 90° toward the viewer, looking straight down the C2-axis towards the membrane. Monomer A is rendered in cyan and monomer B in green with both membrane-binding domains (helices A-D) highlighted in magenta. Arachidonate molecules are rendered as red CPK structures, heme cofactors as orange tubes, and detergent molecules as gray tubes. Figures 1, 4, and 7-11 were generated using the DINO program (35).
Scheme 1.

Conversion of arachidonic acid to PGH2 by COX enzymes.
From the gate, the largely hydrophobic active site extends upwards away from the membrane, bending in a right angle and continuing towards the dimer interface. This active site geometry enables the enzyme to hold unsaturated fatty acid substrates, like AA, in the bent L-shaped conformation that facilitates conversion to PGG2 (6, 7). The generally accepted cyclooxygenase reaction mechanism (see Scheme 1) involves generation of a tyrosyl radical at Tyr-385 in the cyclooxygenase active site by the oxidized heme cofactor. This tyrosyl radical then stereospecifically abstracts the C13 pro-S hydrogen of AA (6, 8, 9), forming a pentadienyl radical that extends from C11 to C15 (10, 11), and restoring the Tyr-385 hydroxyl group. In the X-ray crystal structure of AA bound to COX-1 (12), the AA C13 pro-S hydrogen atom is well positioned for radical attack by Tyr-385, while the C13 pro-R hydrogen atom is fully shielded from Tyr-385.
A number of MD simulations have been performed for COX with a variety of different bound substrates or inhibitors, in an effort to better understand mechanistic details of COX-mediated prostaglandin biosynthesis, and to provide insight that might facilitate the design of COX-2 selective inhibitors. Structural fluctuation information obtained from MD simulations complements high-resolution X-ray crystal structure data and can provide additional insight for structure-function relationships. Useful simulations for large systems like the COX homodimers are quite expensive and time consuming, so many simulations performed to date have involved approximations or system simplifications that reduced the computational expense. Some simulations have involved only one monomer (13-15), or focused on an even smaller fragment of the protein, such as the membrane-binding domain (16) or the membrane-binding domain and the helices comprising the cyclooxygenase active site (17). Many simulations have been performed in vacuo with a distance-dependent dielectric constant to mimic an aqueous environment (13, 17-19). Still other simulations have used steered or targeted MD to accelerate configurational sampling, so that events occurring over longer timescales (μs to ms) could be simulated reasonably efficiently (14). All of these approaches strike a balance between computational expense and incorporation of enough physical detail to model the relevant biochemical or physiological features.
We have performed detailed molecular dynamics (MD) simulations for both COX-1 and COX-2 homodimers with bound AA, including water, crystallographically resolved detergent molecules, and counterions explicitly, in order to investigate the equilibrium baseline behavior of the enzyme-substrate complexes. Comparison of these simulations with reference simulations of AA free in solution demonstrates the effect of the enzymes on substrate conformation and positioning in the active site over time. The simulations also provide detailed information for several protein channels that might be important for oxygen and water transport, or possibly even for trafficking of the intermediate, PGG2, between the cyclooxygenase and peroxidase active sites. These detailed simulations also allow us to compare dynamical features (i.e., structural fluctuations) for COX-1 versus COX-2 isoforms, and may provide useful information for future design of inhibitors with increased isoform selectivity. This possibility is especially significant, given the recent problems and concerns regarding the risk of heart attack associated with many COX-2 selective inhibitors used as anti-inflammatory drugs (3, 4).
Materials and Methods
Parameter Development
AA (Figure 2) contains four homoconjugated double bonds, and this system is not well represented by parameters available in the standard AMBER ff99 force field (20), so new parameters were derived. A new atom type, CE, was created for the homoconjugated carbons in AA. A gas phase minimum energy structure for AA was generated by Hartree-Fock calculations with a 6-31G* basis set using Gaussian98 (21). Equilibrium bond lengths and angles were taken directly from this minimum energy structure, and force constants were interpolated using reference values in the ff99 force field (20). The torsional parameters for the standard AMBER double bond were adopted for the new CE-CE bond. New torsional parameters were needed to properly model the dihedral angles for AA fragments CE-CE-CT-CE and CE-CE-CT-CT (where CT is the standard AMBER sp3 carbon atom type). Torsion energy profiles over the range 0-360° for (Z,Z)-2,5-heptadiene and (Z)-2-pentene, which mimic the AA fragments CE-CE-CT-CE and CE-CE-CT-CT, respectively, were calculated using density functional theory at the B3LYP/6-31G* level of theory (22). Potential function parameters for the CE-CE-CT-CE and CE-CE-CT-CT torsion angles were then adjusted to reproduce the quantum mechanical torsion profiles. A molecular electrostatic potential for AA was calculated over a grid of points using the Hartree-Fock optimized geometry and a 6-31G* basis set. Partial charges for AA were derived by fitting the electrostatic potential to an atom-centered point charge model according to the procedure of Cornell et al (23).
Figure 2.
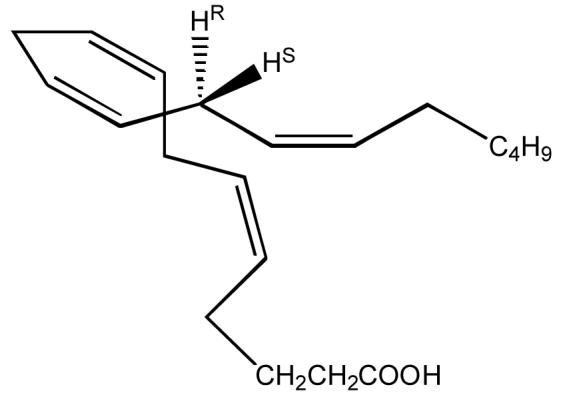
Arachidonic acid (5Z,8Z,11Z,14Z-eicosatetraenoic acid). The C13 pro-R and pro-S hydrogen atoms are highlighted.
Homodimer Model Setup and Simulation
The 3.0 Å resolution X-ray crystal structure of the ovine COX-1/AA complex (12), pdb entry 1DIY, was used to generate the initial model. The cobalt atom present in the 1DIY crystal structure porphyrin was replaced with iron, and the heme groups were tethered to the axial histidine ligand (His-388) with a weak harmonic restraint (20 kcal/mol-Å) to prevent dissociation from the homodimer complex during the long simulations. The crystal structure also includes four β-octyl glucoside (BOG) detergent molecules per monomer surrounding the membrane binding domains. While detergent molecules do not appear to be essential to stabilize the membrane binding domain during simulations, we included the BOG molecules to better mimic the aqueous detergent solution conditions used for many experimental studies (5). All crystallographic waters were retained in the homodimer model. Hydrogen atoms were added using the LEaP module in AMBER 7.0 (24).
For the COX-2 simulations, the 2.4 Å resolution X-ray crystal structure for the mouse COX-2/AA complex (25), pdb entry 1CVU, was used to generate the initial model. However, in this crystal structure, AA is bound in a catalytically unproductive, inverted orientation, with the carboxylate interacting with Tyr-385 and Ser-530 in the middle of the cyclooxygenase active site, and the hydrocarbon chain positioned just above the gate region. A previous theoretical model for COX-2 with a properly oriented AA molecule does exist (pdb entry 1DCX), but the docked conformation of AA in this complex appears to be inconsistent with the proposed reaction mechanism. The first step in the cyclooxygenase reaction involves stereospecific abstraction of the C13 pro-S hydrogen atom by the catalytic tyrosyl radical Tyr-385 (6, 8, 9). Both the C13 pro-R and pro-S hydrogen atoms are directly accessible to Tyr-385 in the theoretical model 1DCX. By contrast, only the C13 pro-S hydrogen atom is accessible to Tyr-385 in the COX-1/AA crystal structure 1DIY, because the substrate adopts a different conformation in this structure. We therefore decided to use the COX-1/AA crystal structure to guide our construction of a catalytically viable COX-2/AA complex.
The COX-2 construct used to obtain the 1CVU crystal structure has a point mutation, His-207-Ala, which was incorporated to prevent heme binding and thus inactivate the enzyme. A BOG detergent molecule occupies the peroxidase active site in this crystal structure in the absence of the heme cofactor. To create a “catalytically competent” COX-2 homodimer model, the detergent molecule was removed from the peroxidase active site, the His-207 residue and heme groups were restored, and AA was repositioned in the cyclooxygenase active site in a catalytically viable orientation. To facilitate positioning of the heme group and substrate molecule in the COX-2 complex, the COX-1 and COX-2 active site regions were superimposed using the graphics program PSSHOW (26). The RMSD for COX-1 versus COX-2 active site region backbone atoms is 0.44 Å, and 0.65 Å for backbone plus side chain atoms, so the two active sites are structurally nearly identical. Both heme and AA in the COX-2 model were overlaid on their corresponding positions in the COX-1 complex to generate the final COX-2 substrate complex. The reorientation of AA in the COX-2 active site and reinsertion of the heme group were quite simple and straightforward procedures. Neither generated any unfavorable steric interactions with protein residues, and the only structural modification necessary during the entire model-building procedure was adjustment of the reintroduced His-207 side chain so as to ligate the heme iron atom.
One of the four BOG detergents surrounding the membrane binding domain in the COX-1 crystal structure is also present in the COX-2 crystal structure; the other three detergent molecules were added to the COX-2 model to insure that differing numbers of detergent molecules in the two complexes did not induce different structural fluctuations during the simulations. Two crystallographic water molecules occupying the upper channel of the cyclooxygenase active site in the COX-2 1CVU crystal structure overlapped with the new AA position, and these waters were deleted along with three additional water molecules present in the “empty” peroxidase site in the 1CVU crystal structure; reinsertion of the heme group in the 1CVU structure precludes the presence of these three water molecules in the peroxidase site. All remaining crystallographic waters were retained in the final COX-2 homodimer model. As with the COX-1 model, hydrogen atoms were added using the LEaP program, and the heme groups were tethered to the axial histidine (His-388) with a weak harmonic restraint (20 kcal/mol-Å).
MD simulations were run using AMBER 7.0 (24) with the AMBER ff99 force field (20) and the SPC/E water model (27). Both COX-1 and COX-2 complexes were solvated in a truncated octahedron periodic box, with a minimum 11 Å distance from any protein complex atom to the closest box edge. Sodium counterions were added to each solvated complex to establish charge neutrality for the systems. The resulting COX-1 system consisted of the homodimer, two heme groups, two AA molecules, eight detergent molecules, 28,401 water molecules and 6 sodium counterions, for a total of 104,234 atoms. The COX-2 system consisted of the homodimer, two hemes, two AA molecules, eight detergent molecules, 32,776 waters and 12 sodium counterions, for a total of 118,858 atoms. Each homodimer complex was then energy minimized using a steepest decent gradient method to relieve any residual unfavorable steric interactions introduced during the model building and solvation procedures. First, the protein, substrates, heme cofactors and detergent molecules were minimized for 300 steps while water molecules and sodium ions were held fixed. Then, the water and ion positions were minimized for 1000 steps while the protein-substrate complex was restrained. Finally, the full system was minimized for an additional 300 steps.
Next, each enzyme-substrate complex was restrained during a 20 ps, constant volume MD simulation, while water and sodium ions were allowed to move freely to relax the protein hydration shell. A thermalization procedure was then used to prepare each full system for equilibrium MD simulation. Initial velocities for each atom were assigned from a Boltzmann distribution at 298 K, followed by a 0.2 ps, constant pressure dynamics simulation. This procedure was repeated ten times with a different Boltzmann velocity distribution in each cycle. This thermalization procedure serves to distribute atomic velocities more uniformly, eliminating potential “hot” and “cold” zones in the system. Isothermal-isobaric ensemble MD simulations were then run for 10 ns for each complex.
An additional 10 ns MD simulation was run for AA in a periodic water box with a sodium counterion, to establish baseline equilibrium behavior for the substrate molecule free in solution. All covalent bonds containing hydrogen were fixed at equilibrium lengths using the SHAKE algorithm (28). A 1 fs integration time step was used for all simulations, and configurations were collected every 1 ps for subsequent analysis. Constant temperature and pressure were maintained via weak coupling to a thermal reservoir (tautp = 1.0 ps) and Berendsen piston (taup = 1.0 ps) (29). A 9 Å real-space nonbonded cutoff was used in all simulations, and a particle-mesh Ewald summation method was used to compute long-range electrostatic energy and force corrections (30). All MD trajectories were analyzed visually using MDDISPLAY (31). The ptraj module of AMBER was used to perform most numerical analyses of the MD trajectories.
Channel Analysis
A new protocol for channel analysis was developed for this work. In previous simulation work with acetylcholinesterase, Tai et al. demonstrated that iterative calculations of solvent-accessible surfaces with varying probe radii could be used to estimate the radius of a channel in a protein (32). The basic concept underlying the “channel_finder” algorithm developed in this work is quite similar, and differs primarily in the criteria used to differentiate open and closed states for a channel. If the probe sphere used for a molecular surface calculation is forced to initially touch an atom inside an interior cavity, it will not escape the cavity as long as the probe radius is larger than the radius of the channel leading to the exterior of the protein. If, however, the probe radius is smaller than the radius of the channel, it will escape and generate a molecular surface for the entire protein, yielding a large total surface area and volume. The largest probe sphere radius that can enable successful “exit” from the protein interior corresponds quite closely with the associated channel radius. In order to use this method to measure a specific channel radius, alternate possible escape routes from the internal cavity must be blocked. This can be accomplished through the placement of “blocker” spheres in all but the channel of interest. For probe spheres smaller than 0.7 Å, it becomes increasingly difficult to block all of the potential escape routes in a robust manner, and the channel mapping results may be ambiguous. However, any channel whose maximum radius observed over the course of a long dynamics trajectory is less than 0.7 Å is too small to permit passage of water and most ions, much less typical enzyme substrates, products, or cofactors, and we classify these as closed channels. Molecular surfaces needed for the channel analysis were calculated with the MSMS program (33). Channel analysis was performed over the final ∼8.5 ns of each trajectory, and the procedure was repeated several times for each channel using different initial probe sphere placements to insure that results were not dependent upon probe starting position in the channel.
Electrostatic Potential Calculations
Electrostatic potential maps were calculated using GRASP (34). All cofactors, ligands, ions, water and detergent molecules were removed from the structures, so that the electrostatic potential maps reflect the apoprotein properties. Atomic charges for the GRASP calculations were taken from the AMBER “all_amino94.lib” library file. Molecular surfaces were calculated using MSMS (33) with a 1.5 Å probe radius. The electrostatic potential was displayed on the molecular surface using DINO (35).
Results
Stability of Complexes
Root mean square deviations (RMSD) for backbone atoms of each complex relative to the initial crystal structure models are shown in Figure 3. The two monomers of each homodimer are referred to as monomers “A” and “B.” The plots illustrate that the structural fluctuations for each complex are well behaved and consistent over the entire trajectory. The structural fluctuations for the membrane binding domains (Figure 3B, E) are somewhat more pronounced and irregular than the rest of the protein, but relatively modest nonetheless, with an average RMSD slightly above 1.5 Å. The active site regions deviate only minimally from the reference crystal structures, with average RMSD values around 0.5 Å.
Figure 3.

Root-mean-square deviations for configurations taken at 1 ps intervals from the COX-1 (A, B, C) and COX-2 (D, E, F) simulations, relative to their respective crystal structures, 1DIY and 1CVU. Panels A and D show superposition of backbone atoms (C, CA, N) of monomer A (black) and monomer B (red). Panels B and E show superposition of the backbone atoms of the membrane binding domains alone, residues 73-123. Panels C and F show superposition of the backbone atoms of active site residues, Arg-120, Phe-205, Phe-209, Val-344, Tyr-348, Val-349, Leu-352, Tyr-355, Ile-377, Phe-381, Tyr-385, Trp-387, Ile-523, Gly-526, Ala-527, Ser-530, Gly-533 and Leu-534.
The COX-1 complex starting model possesses C2 symmetry, but this symmetry is broken during the simulation. The structural deviations for monomer A versus monomer B are more noticeable for the COX-1 homodimer compared to COX-2, although the differences are not dramatic: COX-1 monomer A exhibits an average backbone RMSD relative to the starting crystal structure of ∼1.5 Å, while the monomer B average backbone RMSD value is nearly 1.7 Å. The COX-2 homodimer complex displays more modest RMSD values relative to the crystal structure reference (average backbone RMSD is ∼ 1.25 Å). Monomer A in the COX-2 complex appears to undergo a small conformational change after approximately 7 ns (Figure 3D), but this change has no impact on the active site structure (Figure 3F). Overall, the COX-1 and COX-2 complexes display only modest deviations from the reference crystal structures during the entire 10 ns simulations, suggesting that the models, which include both detergent molecules and counterions, are quite stable.
Average structures for both COX-1 and COX-2 complexes, computed over the latter half of the trajectories, are shown in Figure 4, together with the reference crystal structures. AA displays considerably more conformational flexibility in the COX-2 complex, evident in the different bound conformations in monomer A vs. B. Two COX-2 average structures were computed, reflecting subtly different major (Figure 4B) and minor (Figure 4C) conformations along C11-C15 of AA observed over the final 6 ns of the trajectory. The AA molecule adopts a conformation and orientation in the COX-1 crystal structure 1DIY that is completely consistent with the proposed reaction mechanism described above, and this AA position is duplicated in our COX-2/AA model. The AA carboxyl group forms a hydrogen bond network with gate residues Tyr-355, Arg-120 and Glu-524, and this hydrogen bond network remained intact throughout both the COX-1 and COX-2 trajectories. In both monomers of the COX-1 simulation the AA carboxyl group maintained a bifurcated, charge-reinforced hydrogen bond with Arg-120 (Figure 4A), while in the COX-2 simulation, this interaction was often reduced to a single charge-reinforced hydrogen bond (Figure 4B, C). In COX-2 monomer A, the AA carboxyl group simultaneously interacted with Arg-120 and Arg-513 (His-513 in COX-1) on the opposite side of the gate region.
Figure 4.

Stereo views of identical orientations of the COX-1 (A) and COX-2 (B, C) active sites. In each panel, the X-ray crystal structure is shown in gray, the average structure of monomer A in blue and monomer B in red. For COX-2, major (B) and minor (C) average structures are shown, representing two slightly different arachidonate conformations along C11-C15, compared to crystal structure 1CVU, with the productive conformation of arachidonate from the COX-1 crystal structure replacing the inverted arachidonate from the original 1CVU structure.
While AA shows greater positional and conformational variability in COX-2, the COX-2 enzyme structure remains very consistent, evident in the RMSD plots as well as the average structures. For COX-1, on the other hand, a small, local helix backbone distortion around residues 529-533 of monomer B results in a ∼2 Å backbone shift around Ser-530, and a ∼2 Å shift of lobby helix D (residues 108-121, includes Arg-120) towards the dimer interface. The shift of helix D expands the lobby and widens the gate without disrupting the gate hydrogen bond network. These two helix shifts may be coupled, as the lobby helix D and the active site helix containing Ser-530 make significant van der Waals contact in this region. The local expansion of the upper active site channel in monomer B appears to allow an AA conformational change that reorients the C13 pro-S hydrogen atom away from Tyr-385 (Figure 4A), resulting in what appears to be a catalytically unfavorable conformation that is similar to that observed in the X-ray crystal structure for a COX-1/eicosapentaenoic acid complex (36). Malkowski et al. suggested that this misaligned conformation could account for the inefficient oxygenation of eicosapentaenoic acid by COX-1.
Visual analysis of the COX-1 trajectory revealed a factor that appears to explain the local helix distortion in monomer B. After ∼500 ps, a water molecule penetrated past the gate region in monomer B and entered the active site. This water molecule remained in the active site for several nanoseconds, forming a bridging hydrogen bond between the Ser-530 hydroxyl side chain and the AA carboxyl group, before finally exiting back to bulk solvent. No water molecules ever penetrated into the monomer A active site, and no comparable local backbone distortion around residue Ser-530 was observed for monomer A. Instead, the Ser-530 side chain formed a hydrogen bond with the Gly-526 carbonyl oxygen, comparable to an interaction observed in a COX-1/dihomo-γ-linolenic acid crystal structure (37). The tighter upper channel diameter in monomer A appears to help maintain AA in a “productive” orientation vis-à-vis Tyr-385.
The position of the Co3+-protoporphyrin IX cofactor in the COX-1/AA X-ray crystal structure (1DIY) is displaced ∼0.8 Å relative to the heme position observed in other COX-1 structures, including 1Q4G, a high-resolution structure of COX-1 with an inhibitor bound (38). Apart from the cofactor, this structure is quite similar to the 1DIY structure, with an overall backbone RMSD of only 0.51 Å. In 1Q4G, the more buried of the two heme propionate arms forms two hydrogen bonds with the enzyme; one oxygen atom interacts with residue Asn-382 and the other with Thr-212. In the 1DIY structure, this propionate arm adopts a different conformation in which a single cofactor oxygen interacts with both residues. In both monomers of the COX-1 and COX-2 simulations, the propionate arm moved to a conformation similar to that in the 1Q4G crystal structure, establishing the same hydrogen bond pattern. This change was accompanied by a shift and slight rotation of the heme group that resulted in an overall position more like that observed in 1Q4G.
Arachidonate Conformational Dynamics
Optimal positioning of the C13 pro-S hydrogen atom of AA for abstraction by Tyr-385· is strongly dependent on the bound substrate conformation, especially the C11-C12-C13-C14 torsion angle. To obtain a more complete picture of AA conformations within the cyclooxygenase active site, we monitored torsion angles for all six rotatable bonds of the homoconjugated double bond fragment during each trajectory, and the results are shown in Figure 5. The torsion angles monitored include: C5-C6-C7-C8 (hereafter labeled C6-C7 for convenience), C6-C7-C8-C9 (C7-C8), C8-C9-C10-C11 (C9-C10), C9-C10-C11-C12 (C10-C11), C11-C12-C13-C14 (C12-C13), and C12-C13-C14-C15 (C13-C14). We also monitored these six torsion angles for simulations of AA free in solution. We used density functional theory calculations to compute a reference torsion angle energy profile for 2Z,5Z-heptadiene to identify local minima and torsion barrier heights. The torsion angles for homoconjugated double bonds display degenerate local minima at 120° and 240°. The computed energy barriers are ∼1.4 kcal/mol at 180° and ∼3.8 kcal/mol at 0°, suggesting facile inter-conversion between 120° and 240° local minima is likely. As seen in Figure 5, all six torsion angles do sample both local minima extensively in the simulations of AA free in solution, while AA is conformationally constrained to varying extents in the different enzyme complexes.
Figure 5.

Color-coded histograms showing torsion angle sampling for arachidonate homoconjugated methylene torsion angles simulated free solution (Soln) as well as bound to COX-1 and COX-2. Colors indicate percentage of the trajectory spent in each torsion angle range. The numbers on the left indicate the torsion described, e.g. 6-7 refers to the C5-C6-C7-C8 torsion angle. Vertical black lines indicate the torsion angle value observed in the COX-1 crystal structure complex (1DIY), and the crystallographic structure of arachidonate is shown on the left for reference.
Most of the AA torsion angles sampled similar ranges in both COX-1 monomers, on average remaining near crystallographic values. The primary exception was the C12-C13 torsion, which was restricted to different local minima in monomer A vs. B, resulting in the distinct bound AA conformations in the average structure (Figure 4A). In monomer A, the AA C12-C13 torsion angle fluctuates around 240° (near the crystallographic value of 215°), which orients the C13 pro-S hydrogen atom directly at Tyr-385. In monomer B, however, the C12-C13 torsion angle fluctuates around 120°, which orients both C13 hydrogen atoms away from Tyr-385. Interestingly, in both COX-1 monomers, the C13-C14 torsion angle adopts an equilibrium value near 180° despite the small energy barrier, highlighting the conformational constraint the COX-1 active site imposes on the substrate. X-ray crystal structures for COX-1 complexed with several fatty acids of varying chain lengths and degree of saturation, arachidonic, linoleic, and dihomo-γ-linolenic acid, all display similar conformational trends for the C12-C13 angle (215°, 225° and 237°, respectively) and the C13-C14 torsion angle (169°, 195° and 180°) (12, 36, 37).
In the COX-2 simulation, the AA molecules in both monomers exhibit much larger conformational fluctuations than observed in the COX-1 simulation (Figure 5). The C12-C13 torsion angle sampled both 120° and 240° local minima extensively, and as a result, many AA conformations observed in the COX-2 trajectory appear to shield the C13 hydrogen atoms from Tyr-385, although detailed analysis reveals that the C13 pro-R hydrogen is shielded to a much greater extent (see Supporting Information for plots of the distances between Tyr-385 and the two C13 hydrogen atoms as a function of time in both COX-1 and COX-2 simulations). Unlike COX-1, the COX-2 active site does not constrain the C13-C14 torsion angle near 180°.
Channels
Four distinct channels connect the cyclooxygenase active site to the exterior of the protein. We profiled all four channels in the average structures from the COX-1 and COX-2 simulations, and used the channel_finder utility to monitor structural fluctuations in these channels during the final 8.5 ns of each trajectory (Figure 6). The channel_finder protocol determines the largest spherical particle that can transit the entire channel in each trajectory configuration, and reveals channel constriction or opening as a function of time.
Figure 6.

Channel radii histograms for the COX-1 and COX-2 trajectories. Panel A shows measurements of the main channel from the lobby region into the active site; panel B displays the primary dimer interface channel (D1), panel C shows second dimer interface channel (D2), and panel D displays the side channel or I/V523 side pocket.
The main channel (M1) leads from the lobby through the gate constriction into the cyclooxygenase active site, and is the proposed entrance/exit route for the substrate. The average structure of COX-1 monomer A closely resembles the COX-1/AA crystal structure, with a constricted (i.e., closed) gate region and an empty pocket below Ser-530 (Figure 7A). The presence of this pocket may be mechanistically important, as it is the proposed staging site for molecular oxygen before addition at C11 of the AA pentadienyl radical (12). The COX-1 monomer A gate region remained closed throughout the trajectory, with radius of 0.9 ± 0.1 Å, on average. The carboxylate and first few methylene groups of AA were retained in the active site for all M1 channel measurements, so the value reflects the size of probe spheres that can transit the M1 channel while AA is bound. If molecular oxygen is considered to be an ellipsoid molecule with radii of approximately 1.6 and 2.2 Å along its two principal axes, no channel openings large enough to allow molecular oxygen passage were observed for COX-1 monomer A.
Figure 7.


Solid surface side views showing three of the four active site channels for COX-1 (A, B) and COX-2 (C, D) average structures calculated from the MD trajectories. In each panel, arachidonate is shown in red with the two C13 hydrogens highlighted in white and heme cofactors in orange. Each structure has been sliced with a vertical clipping plane (cut surface is white) to show the interior cavity surface (gray) generated with a 1.0 Å probe sphere. The portion of the surface corresponding to the sidechain atoms of Tyr-385 is highlighted in cyan since the residue itself is not visible through the solid surface rendering. The D1 channel in COX-2 monomer B is closed to this probe, as is the side channel in COX-1 monomer A. A separate clipping plane was used for arachidonate in order to show the entire molecule. Panel E shows a ribbon rendering of the view used in panels A-D along with the global orientation of the dimer, with coloring as described in Figure 1. Surfaces for figures 7-11 were generated using MSMS (33).
Early in the COX-1 simulation, a water molecule entered the active site of monomer B through the M1 channel, apparently causing a structural perturbation (Figures 4A and 7B). Residue Ser-530 shifted ∼2 Å towards the membrane-binding domain, reducing the volume of the hydrophobic pocket beneath Ser-530 (i.e., the putative molecular oxygen staging site), and helix D shifted ∼2 Å towards the dimer interface. As a result of these structural changes, this channel had a wider radius of 1.2 ± 0.2 Å on average, and frequently opened to a radius greater than 1.6 Å (Figure 6A). More than 1% of all configurations analyzed were open sufficiently wide to permit water or molecular oxygen passage, but there was less room for oxygen inside the active site and access to AA atom C11 was more hindered.
In the COX-2 average structure, M1 of monomer A is essentially closed (average radius of 0.8 ± 0.1 Å), with the gate residues Arg-120 and Tyr-355, along with Val-116, closely packed, completely blocking the active site opening (Figure 7C). However, we did observe transient openings in the gate region to radii of 1.3-1.6 Å (∼0.01% of configurations sampled). The average COX-2 monomer B structure (Figure 7D) has a partially opened gate region (average radius of 1.1 ± 0.2 Å), with 0.01% of the configurations open wide enough for passage of molecular oxygen. Unlike COX-1 where structural differences in the M1 channel arose from movement of helix D, in COX-2 the differences between monomers were primarily due to the orientation of side chains in the gate region, especially Tyr-355, relative to the AA carboxylate, suggesting that smaller local movements should be sufficient to allow molecular oxygen to enter the active site.
Two narrow channels extend from the upper, distal end of the cyclooxygenase active site into the dimer interface (Figure 8). These channels often contain water molecules in X-ray crystal structures and have been proposed to act as “release valves” for water upon substrate binding (39), as has been observed in simulation studies for other proteins (40). The first of these channels, D1, is bounded by two conserved glycine residues, Gly-227 and Gly-533, near the distal end of the active site (Figures 7 and 8). These two glycine residues do not make van der Waals contact in crystal structures or the current simulations, but instead form a narrow constriction site just at the junction with the dimer interface. The AA hydrocarbon chain packs tightly in the terminus of the D1 channel, completely filling the available volume. To probe the intrinsic channel radius with our channel_finder utility, we removed AA atoms C14-C20 from each trajectory prior to analysis. In general, the average width and structural fluctuations of the D1 channel were remarkably consistent for both A and B monomers in both enzyme isoforms, and there was no evidence of significant channel opening on a nanosecond timescale. Both COX-1 and COX-2 monomer A D1 channels displayed nearly identical fluctuations over the trajectories, and both have an average radius of 1.1 ± 0.1 Å (Figure 6B). The D1 channel in COX-2 monomer B exhibited slightly larger fluctuations during the simulation, with an average radius of 1.1 ± 0.2 Å, while the D1 channel in COX-1 monomer B was slightly wider, with an average radius of 1.2 ± 0.2 Å, due mainly to the conformational changes triggered by penetration of a water molecule into the M1 channel.
Figure 8.

Solid surface top views (rotated 90° towards the viewer along a horizontal axis relative to Figure 7) of the channels leading from the cyclooxygenase active site into the dimer interface for COX-1 (A, B) and COX-2 (C, D) average structures calculated from the MD trajectories. The D2 channel is closed to the 1.0 Å probe sphere in COX-1 monomers A and B, and the D1 channel is closed in COX-2 monomer B. Panel E shows a ribbon rendering of the view used in panels A-D along with the global orientation of the dimer. The coloring scheme is as described for Figure 7.
A second small channel, D2, extends at a right angle from the distal end of the cyclooxygenase active site and away from the D1 channel (Figure 8). In the X-ray crystal structures of both COX-1 and COX-2, the D2 channel splits into two separate arms, with one arm leading to the dimer interface and the other leading to the protein surface. This channel became constricted in the COX-1 simulation, due to the movement of residues Ser-126 and Pro-127 in the loop extending from the end of helix D. This constriction remained tightly closed in both monomers throughout the COX-1 simulation, with an average radius of 0.8 ± 0.1 Å. In the COX-2 simulation, monomer A maintained the more open configuration observed in the X-ray crystal structures, with an average radius of 1.2 ± 0.1 Å, while monomer B sampled both open and closed states leading to a slightly broader, nearly bimodal distribution, averaging 1.1 ± 0.2 Å. These simulation results suggest that the D2 channel is capable of interconverting between two states on a nanosecond timescale, however both are still quite narrow, with the more open state roughly equivalent to the D1 channel described above.
COX-2 has an active site side pocket (S1) that is insignificant in COX-1, and this pocket can open to form a channel to the protein surface (Figure 7). There was little structural fluctuation in this pocket during the COX-1 simulation, the average pocket radius was small (∼0.9 ± 0.1 Å), and there was no evidence of transient channel opening (Figure 6D). In the COX-2 simulation, however, both monomer A and B side pockets are larger, and this channel radius frequently increased to >1.6 Å in both monomers (0.06% of configurations). The existence of the COX-2 side pocket is due to three conserved mutations relative to COX-1, Ile-523-Val, Arg-513-His and Ile-434-Val. This is a region of key isoform specific differences, as the COX-2 selectivity of diarylheterocyclic NSAIDs, as well as the difference in activity for COX-1 versus COX-2 isoforms after acetylation of Ser-530 by aspirin, have been attributed to the larger side pocket volume in COX-2 (41, 42).
Dimer Interface
Several architectural features observed in X-ray crystal structures for both COX-1 and COX-2 homodimers (12, 25) suggest possible mechanisms for direct transfer of the intermediate PGG2 from cyclooxygenase to peroxidase active sites. We conducted electrostatic potential calculations and mapped them to solvent accessible surfaces of the apo-enzymes to evaluate the dimer interface as a transit route between active sites. Using a top view of the dimer, it is possible to trace a direct path from the cyclooxygenase active site through the D1 channel and across the dimer interface to the peroxidase active site of the opposing monomer (Figure 9A, C). The dimer side view (Figures 9B and 10B) illustrates another possible PGG2 pathway from the lobby of the cyclooxygenase active site up through the dimer interface region and on to the peroxidase sites (25). The results of the electrostatic potential calculations indicate a strong, positive potential in the dimer interface region that results from a cluster of positive charges present in both COX-1 and COX-2. In the COX-1/AA crystal structure, four arginine residues (Arg-374 and Arg-376 from each monomer) flank the central reservoir, while two lysine residues (Lys-532) and two glutamate residues (Glu-543) are positioned just below the reservoir region, and exposed to the underside of the dimer (Figure 10). These are the only charged residues in the COX-1 dimer interface. The COX-2 crystal structure displays a similar pattern of charged residues, although two arginine residues (Arg-374) are replaced by glutamine, and electrostatic potential calculations indicate a slight decrease of the positive potential in the central reservoir.
Figure 9.

Electrostatic potential surface views for COX-1 (A, B) and COX-2 (C, D) average structures calculated from the MD trajectories, along with a crystal structure of human myeloperoxidase, MPO (E, F). Panels A, C, and E show top views looking down on the dimers from above the plane of the membrane (as in Figure 1B). Panels B, D, and F show side views of the dimers (as in Figure 1A), which are related to the top views by a 90° rotation about the horizontal axis indicated in panel A. Surfaces were generated using a standard 1.5 Å probe sphere radius, and color-coded (red for negative, blue for positive) according to an electrostatic potential map generated using GRASP (34). Each hollow surface has been sliced with a vertical clipping plane (cut surface appears black) to show the solvent accessible interior cavity surfaces (gray/red/blue), as well as the position of the substrates located in the interior of the protein inaccessible to the 1.5 Å probe sphere.
Figure 10.

Close-up views of Figure 9 panels A, B, C and D, with acidic and basic residues lining the solvent-filled cavity in the dimer interface explicitly shown. The views and surface are as described for Figure 9 (panels B and D are related to A and C by 90° rotation about a horizontal axis). For COX-1 (panels A and B), the displayed residues are: Arg-374, Arg-376, Lys-532 and Glu-543 (Gln in COX-2) from each monomer. For COX-2 (panels C and D), the displayed residues are: Gln-374, Arg-376, Lys-532 and Asp-125 (Pro in COX-1) from each monomer.
Two more water-filled channels are visible near the dimer interface in the COX crystal structures, although there is little previous discussion of them in the literature. These channels extend from the lobby, up through the interface region between the EGF-like domain and the catalytic domain of each monomer, and end at the peroxidase active sites (Figure 11). The EGF-like domain forms the amino terminus in each monomer, and is tethered to the catalytic domain by a disulfide bond near the top surface of the enzyme. There is no well-defined function for the EGF-like domain in COX enzymes, although EGF domains frequently mediate protein-protein interactions in other systems. This channel also appears to provide a direct, logical transit pathway for PGG2 from the cyclooxygenase site to the peroxidase site. The largest probe spheres able to traverse this channel in the COX-1 and COX-2 crystal structures have radii of 1.6 Å and 1.7 Å, respectively. We observed noticeable structural fluctuations in this channel during our simulations, with transient constrictions as small as a 1.1 Å radius in COX-1, and expansion to a 2 Å radius in COX-2.
Figure 11.
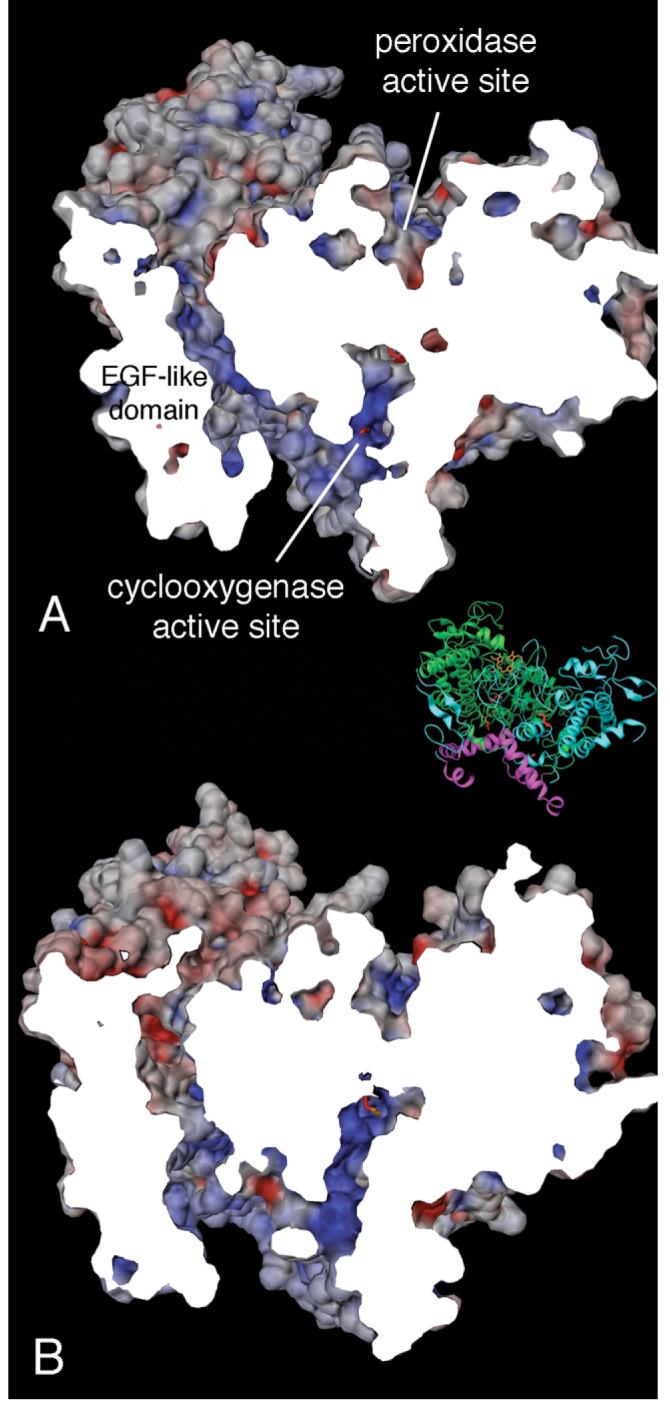
Solid electrostatic potential surface views of COX-1 (A) and COX-2 (B) crystal structure dimers showing the tunnel between the EGF-like domain and the catalytic domain, which connects the cyclooxygenase and peroxidase active sites of a single monomer. An analogous tunnel exists in the opposite monomer (not shown). The view is rotated 90° along the vertical C2 axis of the dimer relative to the side views shown in Figure 9 (panels B and D). The inset shows a ribbon rendering of the view and clipping plane used in this figure to clarify the global orientation of the dimer. Surfaces were generated using a small, 0.7 Å probe sphere radius in order to show the entire tunnel with a single vertical clipping plane (cut surface appears white).
Discussion
Experimental studies have shown that while Arg-120 is important for high-affinity AA binding to COX-1 (43), COX-2 can tolerate loss of the ionic component of the interaction (e.g., Arg-120-Gln), but not loss of both ionic and hydrogen bonding components (e.g., Arg-120-Leu) (44). Our simulation results provide a plausible explanation for this observation. In COX-1, the His-513 side chain will likely be only partially protonated at intermediate pH values, and is too small in any case to form an optimal hydrogen bond with AA unless the substrate molecule shifts significantly in the active site. While the COX-2 Arg-513 residue was also thought to be too distant to interact directly with the AA carboxyl group, our simulations show that the carboxyl group can make contact with both Arg-120 and Arg-513, with only minor changes in the active site side chain conformations. An AA binding configuration that involves simultaneous interactions with both COX-2 Arg-120 and Arg-513 has been suggested for several AA derivatives, namely 2-arachidonylglycerol (45), arachidonylethanolamide (46), and N-arachidonylglycine (47). The authors suggest that direct interaction of these derivatives with Arg-513 is critical for proper positioning of the substrates in the COX-2 active site and subsequent efficient oxygenation, thus explaining the COX-2 selectivity for these compounds. All of these experimental results suggest that Arg-513 can form important stabilizing contacts with ligands, and our simulations indicate that such interactions are even plausible for AA in wild-type COX-2 without any significant enzyme conformational changes.
The simulations reported here reveal considerable variability in AA bound conformations between COX-1 and COX-2 isoforms and individual monomers. In the COX-1 simulation, AA in monomer A maintained a “catalytically productive” conformation throughout the trajectory, with the C13 pro-S hydrogen atom well positioned for abstraction by the Tyr-385 radical. These simulation results demonstrate clearly how COX-1, by controlling the AA C12-C13 and C13-C14 torsion angles, can effectively control the stereospecificity of hydrogen abstraction and facilitate radical intermediate formation in AA oxygenation. In COX-1 monomer B, however, this control mechanism appeared to break down, likely due to perturbation of the active site resulting from introduction of a water molecule. AA became trapped in what appears to be an unproductive conformation, with both C13 methylene hydrogen atoms shielded from residue Tyr-385. While this particular AA conformation is probably not catalytically relevant, these simulations do suggest that distinct AA conformations are possible in the COX-1 cyclooxygenase active site. Previous experimental results suggest that there are at least three distinct, catalytically competent AA conformations possible in the COX-1 active site, each leading to a different product: PGG2, 11-hydroperoxy-5Z,8Z,12E,14Z-eicosatetraenoic acid (11-HPETE), or 15-hydroperoxy-5Z,8Z,11Z,13E-eicosatetraenoic acid (15-HPETE) (48). PGG2 is the predominant product (∼95%) for native COX-1, but the product ratio can be shifted to produce greater amounts of one or more of the minor products through mutagenesis of various key active site residues. Thuresson et al. proposed that these mutations might affect product ratios by altering the preferred AA conformation in the active site (48), just as the perturbation we observed in monomer B altered the preferred AA conformation in our simulation.
In COX-1, acetylation of Ser-530 by aspirin irreversibly inhibits AA oxygenation, but acetylated COX-2 can still produce 15R-HPETE, a product that exhibits inverted stereochemistry at C15 compared to the products of the unmodified enzyme (49). Experimental studies have also shown that occlusion of the COX-2 side pocket via substitution of the corresponding COX-1 residues (V434I/R513H/V523I) yields a mutant COX-2 enzyme that is inactivated by Ser-530 acetylation, suggesting that the ability of wild-type COX-2 to tolerate acetylation is intimately coupled to the larger COX-2 active site volume and the presence of the side pocket (42). Site-directed mutagenesis studies have shown that Ser-530 substitution with valine, threonine or methionine also inactivates COX-1, but the analogous substitutions in COX-2 yield an active enzyme that produces predominantly 15R-prostaglandins, i.e., products with inverted stereochemistry at C15 (50). In our simulations, the AA carboxylate occupies multiple different positions in the COX-2 gate region and exhibits variable degrees of interaction with side pocket residue Arg-513, while in COX-1, the AA carboxylate position is remarkably consistent, even despite non-trivial structural perturbation in monomer B. While AA displays greater conformational fluctuations when bound to COX-2 relative to COX-1, the COX-2 homodimer deviates only modestly from the reference crystal structure, and the structural fluctuations are generally smaller than those observed for the COX-1 homodimer. Therefore, enzyme structural fluctuations cannot explain the greater AA conformational flexibility in COX-2. However, it is clear that the COX-2 active site is larger than the COX-1 active site; in addition to the larger COX-2 side pocket, the COX-2 central channel region volume is 17% larger than the corresponding region in COX-1 (51). These simulations clearly demonstrate the inherent flexibility of AA within the larger COX-2 active site. These larger substrate conformational fluctuations observed in COX-2, along with the addition of Arg-513 to the gate hydrogen bond network, appear to explain why COX-2 can better tolerate some active site mutations (e.g., R120Q or S530M) or acetylation of Ser-530, as well as accept a wider variety of substrates than COX-1.
The analysis of protein channel structures and structural fluctuations in these simulations suggest interesting, and potentially mechanistically important, possibilities. Our simulations show that movement of lobby helix D opens the gate region sufficiently to permit molecular oxygen access to the cyclooxygenase active site, without disruption of the structurally important gate region hydrogen bond network. The X-ray crystal structure of a COX-2 complex with a zomepirac-pyridazine inhibitor displays a unique helix D orientation that results in a larger gate region opening, demonstrating the potential for conformational diversity in the gate region without significant conformational change elsewhere in the binding site (51). While helix D residues 118-122 are disordered in an unliganded COX-2 crystal structure, these residues display much smaller B factors in COX-2/ligand complexes, demonstrating that ligand binding reduces helix D disorder (41). These crystal structures and our COX-1 simulation results suggest that gate opening or closure appears to be determined primarily by the relative shift of lobby helix D towards the dimer interface. However, the COX-2 simulation demonstrated that smaller scale channel opening, sufficient for passage of molecular oxygen, could also be achieved through more local side chain movement in the gate region. The latter observation might be isoform specific, related to the increased number of possible configurations for the hydrogen bond network provided by the addition of Arg-513 to the gate region in COX-2.
The transit pathway for intermediate products from the cyclooxygenase active site to the peroxidase active site remains an intriguing question (39). There is growing evidence that substrate channeling, the direct or facilitated trafficking of a reaction intermediate from one active site to another within or between enzymes, may be a common phenomenon, providing enhanced reaction control and catalytic efficiency for multifunctional enzymes and multi-enzyme complexes (52). In the COX enzymes, the primary cyclooxygenase site product, PGG2, must travel to the peroxidase site where it is reduced to the hydroxide product, PGH2. PGG2 is a relatively unstable hydroperoxide, and since cells work to control the spread of lipid oxidation products, it seems logical that the COX enzymes would evolve some mechanism to efficiently channel the PGG2 intermediate to the peroxidase active site. However, the requirement for peroxidase-dependent activation of the cyclooxygenase active site has prompted the suggestion that initial activation of some COX enzymes leads to the production of PGG2, which can then go on to activate neighboring COX enzymes (5). This dispersal theory would argue against perfect, 1:1 channeling of the intermediate between active sites. In 1991, Eling et al. reported that exogenous PGG2 could compete with endogenously generated PGG2 for reduction at the peroxidase site in purified, detergent-solubilized COX preparations, but that endogenous PGG2 was reduced preferentially in microsomal membrane preparations (53). Their results suggest that if PGG2 is indeed being channeled between active sites in the COX enzymes, the mechanism is dependent on the interaction with the membrane, and channeling would not be observed in detergent-solubilized systems.
The juxtaposition of the cyclooxygenase and peroxidase active sites of opposing monomers across the dimer interface is compelling, suggesting an efficient route for trafficking of the intermediate through the D1 channel. However, previous COX mutagenesis studies involving Gly-533 argue against this proposed ligand-trafficking mechanism (54). In COX-2, Rowlinson, et al. found that G533V or G533L mutations completely abolished arachidonate oxygenation, but only partially affected oxygenation for shorter fatty acids such as linolenic or stearidonic acid, suggesting that the mutations caused a steric occlusion at the end of the substrate binding pocket that could be overcome by shortening the fatty acid chain (54). The valine and leucine mutations at Gly-533 would not only reduce the volume of the cyclooxygenase active site, but would also presumably occlude the D1 channel at the dimer interface junction. Rowlinson et al. concluded that the D1 channel does not function as a product exit route, since the G533V and G533L mutations did not inhibit oxygenation of the shorter fatty acids. Additionally, our simulations showed the D1 channel to have the least variability in structural fluctuations, suggesting it would rarely open wide enough to permit PGG2 to exit into the dimer interface reservoir, even in the absence of additional steric occlusion. It will be important to analyze the D1 channel fluctuations in the COX/PGG2 simulations currently in progress, to see if larger channel openings are observed for that complex.
In 2000, Kiefer et al. proposed that PGG2 could exit the cyclooxygenase active site through the main channel past the gate region, and enter the dimer interface from the lipid bilayer, before migrating up through the dimer interface to either peroxidase active site (25). Protein side chains occlude this pathway in the COX-1 and COX-2 crystal structures, but the average structure computed from our COX-1 trajectory shows an open channel beginning to form (Figure 9B, 10B). Electrostatic potential calculations indicate that the surface along this nascent channel is strongly positive, providing a favorable environment for the more hydrophilic, negatively charged PGG2. This is conceptually quite similar to the situation observed for acetylcholinesterase, where a strong negative electrostatic potential “steers” the positively charged choline product out through a “backdoor” channel in the enzyme, facilitating rapid exit of products and contributing to the rapid turnover of the enzyme (55). There appears to be sufficient room in the reservoir for the arginine side chains (Arg-374 from each monomer) that block the pathway to rotate, enabling passage of PGG2 up to one of the peroxidase active sites.
A protein dimer interface dominated by positively charged residues may seem somewhat counterintuitive, but it is unlikely that the cluster of basic residues in the COX dimer interface is coincidental. These basic residues are highly conserved among all vertebrate COX enzymes. Additionally, a related homodimeric heme peroxidase, human myeloperoxidase or MPO (pdb entry 1CXP, 56), also exhibits a dimer interface dominated by positive residues, with a strong positive electrostatic potential over the dimer interface surface (Figure 9E, F). MPO exhibits 22% sequence identity with COX and shares a similar catalytic domain tertiary fold, but unlike COX does not have a cyclooxygenase active site, EGF domain or membrane-binding domain. This homology has prompted the suggestion that the COX enzymes and other peroxidases like MPO arose from a common ancestor, and the unique features of the COX enzymes evolved over time (57). Along these lines, the accumulation of positive charge in the dimer interface may serve a similar function in MPO, directing substrates to the peroxidase site, or it could relate to the enzymes’ shared peroxidase ancestry in a more general way.
Finally, we note that it may be tempting to try and interpret the loss of homodimer symmetry during the simulations in the context of important mechanistic details (e.g., loss of homodimer symmetry might be correlated with the experimental observation that only one monomer of the homodimer is catalytically active at any given time), but we feel it is inappropriate to do so at this time. As discussed above, the loss of symmetry is due primarily to penetration of a water molecule into the monomer B substrate binding pocket during the COX-1 simulation. While this might simply be a computational artifact, we have shown previously in long protein simulations that we can reproduce equilibrium behavior of hydration shell waters extremely well (58), so we expect that the simulation protocol we have used in this study will yield reasonable water behavior. However, the observed water penetration might reflect a chance rare event, and much more extensive simulation is needed to correlate any observed symmetry disruptions with mechanistic details.
Despite the wealth of COX crystal structures now available, questions remain regarding COX function, mechanistic details, and isoform-specific behaviors. The equilibrium MD simulation results presented here for COX-1 and COX-2 with bound substrate yield insight into some of these questions, and also provide useful reference points for future COX simulations with bound reaction intermediates and products. This complete set of simulations will provide a detailed, dynamic picture for COX-ligand complexes, and should help us to address many lingering questions for the COX enzymes.
Supplementary Material
Acknowledgment
We thank Dr. Claus Schneider, Prof. Alan Brash and Prof. Larry Marnett for helpful discussions, and Dr. Jarrod Smith for invaluable assistance with the channel_finder utility.
1. Abbreviations
- AA
arachidonic acid
- BOG
β-octyl glucoside
- COX
cyclooxygenase
- MD
molecular dynamics
- NSAIDs
non-steroidal anti-inflammatory drugs
- PGG2
prostaglandin G2
- PGH2
prostaglandin H2
- HETE
hydroxyeicosatetraenoic acid
- HPETE
hydroperoxyeicosatetraenoic acid
- RMSD
root mean square deviation
Footnotes
This work was supported in part by NIH grant NS33290 (T.P.L.) and HL17921 (N.A.P.). K.E.F. was supported by training grant T32 GM08320 from the National Institutes of Health and by the Warren Fellowship. D.A.P was supported by NSERC Canada and the Warren Fellowship.
References
- 1.Funk CD. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 2.Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclooxygenase-2 in vivo is anti-inflammatory and non-ulcerogenic. Proc. Natl. Acad. Sci. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solomon DH, Glynn RJ, Levin R, Avorn J. Nonsteriodal anti-inflammatory drug use and acute myocardial infarction. Arch. Intern. Med. 2002;162:1099–1104. doi: 10.1001/archinte.162.10.1099. [DOI] [PubMed] [Google Scholar]
- 4.FitzGerald GA. Coxibs and cardiovascular disease. N. Engl. J. Med. 2004;351:1709–1711. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- 5.Rouzer CA, Marnett LJ. Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem. Rev. 2003;103:2239–2304. doi: 10.1021/cr000068x. [DOI] [PubMed] [Google Scholar]
- 6.Hamberg M, Samuelsson BJ. On the mechanism of the biosynthesis of prostaglandin E1 and F1α. J. Biol. Chem. 1967;242:5336–5343. [PubMed] [Google Scholar]
- 7.Marnett LJ, Maddipati KR. Prostaglandin H synthase. In: Everse J, Everse KE, Grisham MB, editors. Peroxidases in Chemistry and Biology. Vol. 1. CRC Press; Boca Raton, FL: 1991. pp. 293–334. [Google Scholar]
- 8.Karthein R, Dietz R, Nastainczyk W, Ruf HH. Higher oxidation states of prostaglandin H synthase. EPR study of a transient tyrosyl radical in the enzyme during the peroxidase reaction. Eur. J. Biochem. 1988;171:313–320. doi: 10.1111/j.1432-1033.1988.tb13792.x. [DOI] [PubMed] [Google Scholar]
- 9.Shimokawa T, Kulmacz RJ, DeWitt DL, Smith WL. Tyrosine 385 of prostaglandin endoperoxide synthase is required for cyclooxygenase catalysis. J. Biol. Chem. 1990;265:20073–20076. [PubMed] [Google Scholar]
- 10.Peng S, Okeley NM, Tsai AL, Wu G, Kulmacz RJ, van der Donk WA. Structural characterization of a pentadienyl radical intermediate formed during catalysis by prostaglandin H synthase-2. J. Am. Chem. Soc. 2001;123:3609–3610. doi: 10.1021/ja015599x. [DOI] [PubMed] [Google Scholar]
- 11.Peng S, Okeley NM, Tsai AL, Wu G, Kulmacz RJ, van der Donk WA. Synthesis of isotopically labeled arachidonic acids to probe reaction mechanism of prostaglandin H synthase. J. Am. Chem. Soc. 2002;124:10785–10796. doi: 10.1021/ja026880u. [DOI] [PubMed] [Google Scholar]
- 12.Malkowski MG, Ginell SL, Smith WL, Garavito RM. The productive conformation of arachidonic acid bound to prostaglandin synthase. Science. 2000;289:1933–1937. doi: 10.1126/science.289.5486.1933. [DOI] [PubMed] [Google Scholar]
- 13.Filizola M, Perez JJ, Palomer A, Mauleon D. Comparative molecular modeling study of the three-dimensional structures of prostaglandin endoperoxide H2 synthase 1 and 2 (COX-1 and COX-2) J. Mol. Graph. 1997;15:290–300. doi: 10.1016/s1093-3263(97)00107-1. [DOI] [PubMed] [Google Scholar]
- 14.Molnar F, Norris LS, Schulten K. Simulated (un)binding of arachidonic acid in the cyclooxygenase active site of prostaglandin H2 synthase-1. Prog. React. Kin. Mech. 2000;25:263–298. [Google Scholar]
- 15.Cukier RI, Siebold SA. Molecular dynamics simulations of prostaglandin endoperoxide H synthase-1: Role of water and the mechanism of compound I formation from hydrogen peroxide. J. Phys. Chem. B. 2002;106:12031–12044. [Google Scholar]
- 16.Nina M, Berneche S, Roux B. Anchoring of a monotopic membrane protein: the binding of prostaglandin H2 synthase-1 to the surface of a phospholipids bilayer. Eur. Biophys. J. 2000;29:439–454. doi: 10.1007/pl00006649. [DOI] [PubMed] [Google Scholar]
- 17.Garcia-Nieto R, Perez C, Gago F. Automated docking and molecular dynamics simulations of nimesulide in the cyclooxygenase active site of human prostaglandin-endoperoxide synthase-2 (COX-2) J. Comput.-Aided Mol. Des. 2000;14:147–160. doi: 10.1023/a:1008110924479. [DOI] [PubMed] [Google Scholar]
- 18.Pouplana R, Lozano JJ, Ruiz J. Molecular modeling of the differential interaction between several non-steroidal anti-inflammatory drugs and human prostaglandin endoperoxide H synthase-2 (h-PGHS-2) J. Mol. Graph. 2002;20:329–343. doi: 10.1016/s1093-3263(01)00133-4. [DOI] [PubMed] [Google Scholar]
- 19.Sahi S, Srinivasan M, Kothekar V. 530 ps molecular dynamics simulation of indoprofen and NS398 with COX-1 and COX-2. Study of perturbative changes in the complexes. J. Molec. Struct. 1999;498:133–148. [Google Scholar]
- 20.Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comp. Chem. 2000;21:1049–1074. [Google Scholar]
- 21.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Zakrzewski VG, Montgomery JA, Jr., Stratmann RE, Burant JC, Dapprich S, Millam JM, Daniels AD, Kudin KN, Strain MC, Farkas O, Tomasi J, Barone V, Cossi M, Cammi R, Mennucci B, Pomelli C, Adamo C, Clifford S, Ochterski J, Petersson GA, Ayala PY, Cui Q, Morokuma K, Malic DK, Rabuck AD, Raghavachari K, Foresman JB, Cioslowski J, Ortiz JV, Baboul AG, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Gomperts R, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Andres JL, Gonzalez C, Head-Gordon M, Replogle ES, Pople JA. Gaussian, Inc.; Pittsburgh, PA: 1998. [Google Scholar]
- 22.Becke AD. Density-functional thermochemistry. III. the role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 23.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr., Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 24.Case DA, Pearlman DA, Caldwell JW, Cheatham TE, III, Wang J, Ross WS, Simmerling CL, Darden TA, Merz KM, Stanton RV, Cheng AL, Vincent JJ, Crowley M, Tsui V, Gohlke H, Radmer RJ, Duan Y, Pitera J, Massova I, Seibel GL, Singh UC, Weiner PK, Kollman PA. AMBER 7. University of California; San Francisco: 2002. [Google Scholar]
- 25.Kiefer JR, Pawlitz JL, Moreland KT, Stegeman RA, Hood WF, Gierse JK, Stevens AM, Goodwin DC, Rowlinson SW, Marnett LJ, Stallings WC, Kurumbail RG. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature. 2000;405:97–101. doi: 10.1038/35011103. [DOI] [PubMed] [Google Scholar]
- 26.Swanson E. PSSHOW Version 2.4. Seattle, WA: 1995. [Google Scholar]
- 27.Berendsen HJC, Grigera JR, Straatsma TP. The missing term in effective pair potentials. J. Phys. Chem. 1987;91:6269–6271. [Google Scholar]
- 28.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-integration of cartesian equations of motion of a system with constraints - molecular-dynamics of N-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 29.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 30.Darden T, York D, Pedersen L. Patical mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 31.Callahan TJ, Swanson E, Lybrand TP. MD display: An interactive graphics program for visualization of molecular dynamics trajectories. J. Mol. Graph. 1996;14:39–41. doi: 10.1016/0263-7855(95)00088-7. [DOI] [PubMed] [Google Scholar]
- 32.Tai K, Shen T, Borjesson U, Philippopoulos M, McCammon JA. Analysis of a 10-ns molecular dynamics simulation of mouse acetylcholinesterase. Biophys. J. 2001;81:715–724. doi: 10.1016/S0006-3495(01)75736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanner MF, Spehner J-C, Olson AJ. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 34.Nicholls A, Bharadwaj R, Honig B. GRASP - Graphical representation and analysis of surface-properties. Biophys. J. 1993;64:A166. [Google Scholar]
- 35.Philippsen A. DINO: Visualizing Structural Biology. 2003 http://www.dino3d.org.
- 36.Malkowski MG, Thuresson ED, Lakkides KM, Rieke CJ, Micielli R, Smith WL, Garavito RM. Structure of eicosapentaenoic and linoleic acids in the cylcooxygenase site of prostaglandin endoperoxide H synthase-1. J. Biol. Chem. 2001;276:37547–37555. doi: 10.1074/jbc.M105982200. [DOI] [PubMed] [Google Scholar]
- 37.Thuresson ED, Malkowski MG, Lakkides KM, Rieke CJ, Mulichak AM, Ginell SL, Garavito RM, Smith WL. Mutational and x-ray crystallographic analysis of the interaction of dihomo-γ-linolenic acid with prostaglandin endoperoxide H synthases. J. Biol. Chem. 2001;276:10358–10365. doi: 10.1074/jbc.M009378200. [DOI] [PubMed] [Google Scholar]
- 38.Gupta K, Selinsky BS, Kaub CJ, Katz AK, Loll PJ. The 2.0 Å resolution crystal structure of prostaglandin H2 synthase-1: Structural insights into an unusual peroxidase. J. Mol. Biol. 2004;335:503–518. doi: 10.1016/j.jmb.2003.10.073. [DOI] [PubMed] [Google Scholar]
- 39.Picot D. The Three-Dimensional Structure of Cyclooxygenases. Lung Biol. Health Dis. 1998;114:161–186. Eicosanoids, Aspirin and Asthma. [Google Scholar]
- 40.Hyre DE, Amon LK, LeTrong I, Stenkamp RE, Lybrand TP, Stayton PS. Early mechanistic events in biotin dissociation from streptavidin. Nature Struct. Biol. 2002;9:582–585. doi: 10.1038/nsb825. [DOI] [PubMed] [Google Scholar]
- 41.Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stalllings WC. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644–648. doi: 10.1038/384644a0. [DOI] [PubMed] [Google Scholar]
- 42.Rowlinson SW, Crews BC, Goodwin DC, Schneider C, Gierse JK, Marnett LJ. Spatial requirements for 15-(R)-hydroxy-5Z,8Z,11Z,13E-eicosatetraenoic acid synthesis within the cyclooxygenase active site of murine COX-2. Why acetylated COX-1 does not synthesize 15-(R)-HETE. J. Biol. Chem. 2000;275:6586–6591. doi: 10.1074/jbc.275.9.6586. [DOI] [PubMed] [Google Scholar]
- 43.Bhattacharyya DK, Lecomte M, Rieke CJ, Garavito RM, Smith WL. Involvement of arginine 120, glutamate 524, and tyrosine 355 in the binding of arachidonate and 2-phenylpropionic acid inhibitors to the cyclooxygenase active site of ovine prostaglandin endoperoxide H synthase-1. J. Biol. Chem. 1996;271:2179–2184. doi: 10.1074/jbc.271.4.2179. [DOI] [PubMed] [Google Scholar]
- 44.Rieke CJ, Mulichak AM, Garavito RM, Smith WL. The role of arginine 120 of human prostaglandin endoperoxide H synthase-2 in the interaction with fatty acid substrates and inhibitors. J. Biol. Chem. 1999;274:17109–17114. doi: 10.1074/jbc.274.24.17109. [DOI] [PubMed] [Google Scholar]
- 45.Kozak KR, Prusakiewicz JJ, Rowlinson SW, Schneider C, Marnett LJ. Amino acid determinants in cyclooxygenase-2 oxygenation of the endocannabinoid 2-arachidonylglycerol. J. Biol. Chem. 2001;276:30072–30077. doi: 10.1074/jbc.M104467200. [DOI] [PubMed] [Google Scholar]
- 46.Kozak KR, Prusakiewicz JJ, Rowlinson SW, Prudhomme DR, Marnett LJ. Amino acid determinants in cyclooxygenase-2 oxygenation of the endocannabinoid anandamide. Biochem. 2003;42:9041–9049. doi: 10.1021/bi034471k. [DOI] [PubMed] [Google Scholar]
- 47.Prusakiewicz JJ, Kingsley PJ, Kozak KR, Marnett LJ. Selective oxygenation of N-arachidonylglycine by cyclooxygenase-2. Biochem. Biophys. Res. Commun. 2002;296:612–617. doi: 10.1016/s0006-291x(02)00915-4. [DOI] [PubMed] [Google Scholar]
- 48.Thuresson ED, Lakkides KM, Smith WL. Different catalytically competent arrangements of arachidonic acid within the cyclooxygenase active site of prostaglandin endoperoxide H synthase-1 lead to the formation of different oxygenated products. J. Biol. Chem. 2001;275:8501–8507. doi: 10.1074/jbc.275.12.8501. [DOI] [PubMed] [Google Scholar]
- 49.Lecomte M, Laneuville O, Ji C, DeWitt DL, Smith WL. Acetylation of human prostaglandin endoperoxide synthase-2 (cyclooxygenase-2) by aspirin. J. Biol. Chem. 1994;269:13207–13215. [PubMed] [Google Scholar]
- 50.Schneider C, Boeglin WE, Prusakiewicz JJ, Rowlinson SW, Marnett LJ, Samel N, Brash AR. Control of prostaglandin stereochemistry at the 15-carbon by cyclooxygenases-1 and -2: A critical role for serine 530 and valine 349. J. Biol. Chem. 2002;277:478–485. doi: 10.1074/jbc.M107471200. [DOI] [PubMed] [Google Scholar]
- 51.Luong C, Miller A, Barnett J, Chow J, Ramesha C, Browner MF. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nature Struct. Biol. 1996;3:927–933. doi: 10.1038/nsb1196-927. [DOI] [PubMed] [Google Scholar]
- 52.Huang X, Holden HM, Raushel FM. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu. Rev. Biochem. 2001;70:149–180. doi: 10.1146/annurev.biochem.70.1.149. [DOI] [PubMed] [Google Scholar]
- 53.Eling TE, Glasgow WC, Curtis JF, Hubbard WC, Handler JA. Studies on the reduction of endogenously generated prostaglandin G2 by prostaglandin H synthase. J. Biol. Chem. 1991;266:12348–12355. [PubMed] [Google Scholar]
- 54.Rowlinson SW, Crews BC, Lanzo CA, Marnett LJ. The binding of arachidonic acid in the cyclooxygenase active site of mouse prostaglandin endoperoxide synthase-2 (COX-2). A putative L-shaped binding conformation utilizing the top channel region. J. Biol. Chem. 1999;274:23305–23310. doi: 10.1074/jbc.274.33.23305. [DOI] [PubMed] [Google Scholar]
- 55.Gilson MK, Straatsma TJ, McCammon JA, Ripoll JR, Faerman CH, Axelsen PH, Silman I, Sussman JL. Open “back door” in a molecular dynamics simulation of acetylcholinesterase. Science. 1994;263:1276–1278. doi: 10.1126/science.8122110. [DOI] [PubMed] [Google Scholar]
- 56.Fiedler TJ, Davey CA, Fenna RE. X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 Å resolution. J. Biol. Chem. 2000;275:11964–11971. doi: 10.1074/jbc.275.16.11964. [DOI] [PubMed] [Google Scholar]
- 57.Garavito RM, Picot D, Loll PJ. Prostaglandin H synthase. Curr. Opin. Struct. Biol. 1994;4:529–535. [Google Scholar]
- 58.Hyre DE, Amon LK, LeTrong I, Stenkamp RE, Lybrand TP, Stayton PS. The role of specific water molecules in the biotin-streptavidin dissociation reaction. Nature Struct. Biol. 2002;9:582–585. doi: 10.1038/nsb825. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.