

Abstract
We have studied 72 members belonging to a large kindred with a hearing disorder inherited in an autosomal dominant pattern. We used audiological, physiological, and psychoacoustic measures to characterize the hearing disorders. The initial phenotypic features of the hearing loss are of an auditory neuropathy (AN) with abnormal auditory nerve and brainstem responses (ABRs) and normal outer hair cell functions [otoacoustic emissions (OAEs) and cochlear microphonics (CMs)]. Psychoacoustic studies revealed profound abnormalities of auditory temporal processes (gap detection, amplitude modulation detection, speech discrimination) and frequency processes (difference limens) beyond that seen in hearing impairment accompanying cochlear sensory disorders. The hearing loss progresses over 10–20 years to also involve outer hair cells, producing a profound sensorineural hearing loss with absent ABRs and OAEs. Affected family members do not have evidence of other cranial or peripheral neuropathies. There was a marked improvement of auditory functions in three affected family members studied after cochlear implantation with return of electrically evoked auditory brainstem responses (EABRs), auditory temporal processes, and speech recognition. These findings are compatible with a distal auditory nerve disorder affecting one or all of the components in the auditory periphery including terminal auditory nerve dendrites, inner hair cells, and the synapses between inner hair cells and auditory nerve. There is relative sparing of auditory ganglion cells and their axons.
Keywords: hereditary deafness, auditory neuropathy, sensorineural, cochlear implant
INTRODUCTION
Auditory neuropathy (AN) is a hearing disorder affecting auditory nerve function in the presence of preserved cochlear outer hair cell activity (Starr et al. 1996). The hearing loss is characterized by disproportionate effects on auditory temporal processes relative to pure tone thresholds with speech perception and binaural hearing being profoundly impaired (Starr et al. 1991; Zeng et al. 1999). The use of hearing aids to amplify acoustic signals is usually without benefit. The disorder can affect individuals of all ages and the etiologies are diverse (see Starr et al. 2001 for review). Detailed studies of the time course for clinical and auditory test abnormalities in individual patients with this clinical syndrome are still incomplete. The magnitude and distribution of the pure tone threshold loss when patients are first identified can vary widely (Sininger and Oba 2001), consistent with the diverse etiological factors associated with AN and the length of time the disorder may have been present before testing was performed. The investigation of affected family members with inherited forms of AN can provide a longitudinal view of the natural history of symptoms and findings in this disorder. For instance, several inherited forms of AN have now been described in association with peripheral neuropathies (Butinar et al. 1999; Kalaydjieva et al. 2000; Kovach et al. 2002; Starr et al. 2003), Refsum’s disease (Oysu et al. 2001), Friedreich’s ataxia (Satya–Murti et al. 1980) and also in families without peripheral neuropathies accompanying mutations of the otoferlin (OTOF) gene (Yasunaga et al. 1999; Varga et al. 2003) and mitochondria (Merchant et al. 2001; Zwirner et al. 2001). Both the age of onset and the rate of progression vary in these different genetic forms of the disorder. In hereditary sensory motor neuropathy (HSMN) identified in Roma families in Lom, Romania, with deafness (Kalaydjieva et al. 1998; Butinar et al. 1999), the hearing disorder begins in the second decade, producing profound deafness within a few years. In contrast, deafness accompanying HSMN due to a mutation in the myelin protein zero (MPZ) gene begins in the third and fourth decades and progresses slowly (Starr et al. 2003).
The cochlear pathology in an autopsied patient with AN and hereditary peripheral neuropathy associated with the MPZ gene showed extensive loss (>90%) of auditory nerve and ganglion cells, while both inner and outer hair cells were normal in number and morphology, a pattern of loss consistent with a primary disorder of auditory nerve (Starr et al. 2003). A similar pattern of auditory nerve atrophy with normal hair cells had been noted previously in temporal bone studies of individual patients with deafness and hereditary neuropathy (Spoendlin 1974; Hallpike et al. 1980) before the availability of clinical test procedures [otoacoustic emissions (OAEs), auditory brainstem responses (ABRs)] now used to distinguish between disorders of hair cells and auditory nerve (Starr et al. 1991).
A disorder of auditory nerve function in the presence of preserved cochlear outer hair cell activity could also be possible if there were a loss or functional, impairment in the auditory periphery, affecting inner hair cells, their synapses with auditory nerve, or terminal dendrites sparing proximal auditory nerve and ganglion cells (Starr et al. 2000; Rapin and Gravel 2003). A distal auditory lesion was identified in premature infants who failed an infant hearing screening test (ABRs) and died shortly thereafter. The temporal bones in some of these infants showed loss of inner hair cells with preservation of ganglion cells (Amatuzzi et al. 2001). Animal models of distal auditory nerve and inner hair cell lesions use neurotoxic agents, such as kainic acid applied to the cochlea (Zheng et al. 1996; Salvi et al. 2000) or systemic anoxia (Harrison 1998). The pathology in these experimental models first affects inner hair cells and nerve terminals and can also lead to degeneration of auditory ganglion cells (Ding et al. 1999). A progressive loss of components in the auditory periphery (hair cells, terminal dendrites, and ganglion cells) is also seen in a genetic model of deafness in the Bronx Waltzer mouse (Sobkowicz et al. 1999).
We have had the opportunity to characterize the phenotype of hearing loss in a large kindred spanning seven generations with deafness inherited as an autosomal dominant disorder mapping to a novel locus called AUNA1 (auditory neuropathy, dominant, 1) on chromosome 13q14–21 (Kim et al. 2004). The hearing impairment in the initial stage is expressed as a disorder of auditory nerve function in the presence of preserved outer hair cell activity, a pattern of abnormality consistent with auditory neuropathy. As the hearing loss progresses, outer hair cell activity from high- and mid-frequency cochlear regions becomes impaired, consistent with a combined partial sensory and neural hearing loss. Finally, affected family members in the fifth and sixth decades of life are profoundly deaf and lose all outer hair cell functions, consistent with sensorineural deafness. There was no evidence of other cranial or peripheral neuropathies in affected family members. Results from physiological, psychophysical, and electrical stimulation of auditory nerve localized the site of disorder to distal auditory structures (hair cells, synapses, terminal dendrites) with sparing of auditory ganglion cells and their axons.
METHODS
Subjects
Family description
Family members recognize the prevalence of deafness in their kindred and both the hearing-impaired and unaffected family members are able to communicate with one another by reading lips without making sounds, a method of communication they call “silent talking.” We examined 72 individuals in the F4–F7 generations (see Table 1) and their lineage is plotted in Figure 1. Summary tables of the data are presented at appropriate points in the text. Family members were tested at various sites including community audiology offices, the University California Irvine research laboratories, their homes, and community centers. We were constrained by time and the availability of equipment at community sites and homes to perform all of the tests on each individual. A list of the tests performed is shown in Table 1. There were 72 individuals who had audiograms. Additional tests [e.g., ABRs, distortion product otoacoustic emissions (DPOAEs), muscle reflexes] were done on approximately one third of these family members to characterize both the type of hearing loss and the presence of other phenotypic features. Thus, 34 had DPOAEs, 22 had ABRs, 26 had psychoacoustic measures of gap detection, 30 had neurological examination, and 39 had otological examination. A family member was considered “affected” for genetic purposes if he/she had pure tone thresholds worse than the 90% percentile of thresholds for an age- and sex-matched unselected population (Robinson 1988). Deceased subjects were judged affected based on the recollections of family members.
Table 1.
Number of subjects tested for each procedure in each generation
| Generation | n | Audiogram | DPOAEs | ABRs | Reflexes | Gap | Speech | Neurological | Otological | DNA |
|---|---|---|---|---|---|---|---|---|---|---|
| F4 | 9 | 9 | 4 | 2 | 0 | 3 | 0 | 5 | 4 | 9 |
| F5 | 23 | 23 | 16 | 12 | 3 | 13 | 5 | 17 | 6 | 23 |
| F6 | 18 | 18 | 8 | 7 | 4 | 7 | 3 | 8 | 9 | 18 |
| F7 | 22 | 22 | 6 | 1 | 1 | 3 | 1 | 0 | 20 | 22 |
| Total | 72 | 72 | 34 | 22 | 8 | 26 | 9 | 30 | 39 | 72 |
n = number examined; DPOAEs = distortion product otoacoustic emissions; ABRs = auditory brainstem responses; Reflexes = acoustic middle ear muscle responses; Gap = threshold for detecting brief silent period in noise; Speech = word recognition; Neurological = clinical examination; Otological = clinical examination; DNA = blood or cheek swab for genetic analysis.
Figure 1.

Pedigree for hereditary dominant auditory neuropathy. Seven generations are shown. Affected members are indicated by filled symbols. The numbers 1–5 are placed below those subjects studied in detail with psychophysical and physiological methods. Nineteen unaffected members studied in generation VII are not included. The hearing status of the members of generation I was unknown. Consanguineous marriages are indicated by double lines.
Control subjects
Affected family members were compared with unaffected members and with normal controls. Affected family members with cochlear implants were compared with implanted subjects with deafness due to cochlear sensory disorders.
Institutional Review Board approvals for testing human subjects were obtained from the University California Irvine, University of Michigan, and Louisiana State University. A written informed consent was obtained from all subjects.
Audiology
Pure tone air conduction thresholds in subjects tested in their community used circumaural headphones in a quiet room. In subjects tested in audiology offices, pure tone audiograms, middle ear muscle reflexes, and speech recognition were defined using insert earphones in a sound-attenuating chamber. Middle ear muscle reflexes were tested using a tympanometer. DPOAEs reflecting activity of outer hair cells responsive to 1, 1.5, 2.1, 3, 4.2, 6, and 8 kHz were measured in 28 subjects in community centers and audiologist offices using presentation levels for f1 of 65 dB SPL and f2 of 55 dB sound pressure level (SPL). Six additional subjects were tested for DPOAEs with an AuDX handheld OAE screener in their home; DPOAEs were measured at 2, 3, 4, and 5 kHz. Abnormality of DPOAEs in those subjects tested in controlled sound environments (audiology or research offices) was defined by amplitudes at two or more adjacent frequencies falling at or below the test noise levels. In subjects tested in community centers or at home, results at 1 and 1.5 kHz were sometimes unsatisfactory due to the presence of environmental noise. If the tests were satisfactory, we classified the results as above. However, if the tests were unsatisfactory at the low frequencies, we used results beginning at 2 kHz.
Psychoacoustic and psychoelectric studies
Detailed quantitative measures of acoustic temporal, frequency, masking, and speech recognition functions were made in five hearing-impaired family members and two unaffected members. The tests were performed in a double-walled sound-attenuating chamber in a laboratory setting. A three-interval forced-choice (2-down, 1-up) adaptive procedure was used to quantify performance in these psychophysical tasks. The results for affected family members were compared to those of normal-hearing subjects and with hearing-impaired subjects due to cochlear sensory disorders.
Psychoelectric studies were performed in three of the five affected family members who had cochlear implants, one of whom had been previously studied psychoacoustically. Electric stimuli were delivered to the subjects via a customized research interface (Shannon et al. 1990). Procedures identical to those used in psychoacoustic experiments were used to collect the psychoelectrical data (Zeng et al. 1999). For gap detection, psychoelectrical results from the three implanted family members were compared individually to their psychoacoustical data from the same tasks that were obtained either presurgically in one case or postsurgically in the nonimplanted ear in all three cases. The postsurgical psychoelectrical results for gap detection and rate discrimination were also compared to data obtained from three additional cochlear implanted subjects whose deafness was presumably due to cochlear sensory impairment. The latter comparisons were analyzed to determine statistical differences from the obtained data.
Temporal process
Gap detection (acoustic). We tested five family members in a controlled-sound environment. The stimulus was a broadband (20 Hz–14 kHz) white noise, 500 ms in duration and 2.5-ms cosine-squared ramps, containing a silent interval or gap in the middle of the noise. It was presented monaurally through Sennheiser HDA200 headphones (for details, see Zeng et al. 1999). In addition, we tested 26 family members in a quiet office in their community center using a web-based version of gap detection ( http://www.ucihs.uci.edu/hesp/Onlinetest/gap/detection ). The web-based version used identical stimuli, headphones, and psychophysical procedures as the standard psychophysical tests employed in the laboratory controlled by a laptop computer in a quiet, but not sound-attenuated, room in the community center. Normative data for the web-based protocol were obtained from seven adult normal-hearing listeners, ages 18–36, naïve for psychoacoustic testing. Abnormality in family members was based on individual scores differing by two or more standard deviations (SDs) from the normal values obtained in this procedure.
Gap detection (electric). Gap detection thresholds to electric stimuli were measured at several loudness levels in the three family members with cochlear implants (Nucleus 24), one of whom was also studied with acoustic stimulation before implantation. Gap detection thresholds from three cochlear implanted subjects with deafness due to cochlear sensory disorder served as “controls” (two Nucleus 24 and one Clarion CI). Stimuli were 200-ms, 1-kHz biphasic pulse trains with pulse duration of 100 μs/phase (Nucleus 24) and 75 μs/phase (Clarion CI). The interval between the two pulse trains was varied to produce silent gaps.
Temporal integration (acoustic). Detection threshold was measured in two subjects as a function of noise duration.
Amplitude modulation detection (acoustic). A 500-ms-duration noise was used to define threshold for detecting amplitude modulation as a function of modulation frequency. Stimuli were presented at a “most comfortable” loudness.
Rate discrimination (electric). Pulse trains of different rates were delivered in a bipolar mode to apical electrodes. The duration of the pulse train was 200 ms and the pulse amplitude was presented at a “comfortable” loudness.
Frequency processes (acoustic)
Frequency discrimination was measured to tonal stimuli, 200 ms in duration and 2.5-ms cosine-squared ramps. Stimuli were presented at a subject’s most comfortable loudness level. Frequency difference limens were measured from 125 to 4 kHz in octave steps.
Simultaneous masking (acoustic)
Thresholds for pure tones (1 or 2 kHz), 200 ms in duration, presented at the temporal center of a 425-ms white noise burst were measured as a function of noise levels.
Speech recognition (acoustic and electric)
Sentence materials (Bench and Bamford 1979) were presented through a speaker in a sound-attenuating chamber at the most comfortable loudness level. A total of 10 sentences were used for each condition and each sentence contained 3–5 key words. Percent correct scores were calculated based on the number of words identified correctly.
Neurophysiology
Auditory brainstem responses (ABRs) in subjects tested in a sound-controlled environment were averaged separately to condensation and rarefaction clicks at 100 dB SPL presented monaurally at 21/s. A Neuro-Scan acquisition (with SynAmps) and stimulation (Stim) system was used to collect the data. Recordings were made between electrodes at vertex (Cz) referenced to ipsilateral and contralateral mastoids (Pratt et al. 1999). Five subjects tested in the research laboratory at Irvine also had measures of cochlear microphonic potentials to clicks and 1-kHz tone bursts. The stimuli were presented by electrically shielded earphones (Etymotic Type ER-3A) through a 10-cm-long plastic tube coupled to the earcanal by a foam insert. The acoustic stimuli used were 0.1-ms square wave pulses for “clicks” and 1-kHz tone bursts of 6-ms duration and 2-ms ramps. Separate tone burst averages were made to condensation and rarefaction signals to define cochlear microphonics by the appearance of phase-reversed potentials in the superimposed averages. Cochlear microphonics can be distinguished from recordings of electrical artifacts of the voltage applied to the earphone if clamping of the tube conducting the sound to the ear is accompanied by a marked attenuation of the potentials (see Starr et al. 1991 for details). Amplifier bandpass for clicks and tone bursts was set between Direct Current (DC) and 3 kHz. Sound intensities were calibrated for the different methods of stimulus coupling used between laboratories. Portable recording equipment (Navigator Pro, Bio-logic Systems) was used for ABRs in community center testing.
Auditory evoked cortical potentials (AEPs) were recorded from Fz, Cz, and Pz referenced to linked mastoids to 1-kHz tones (100-ms duration, 2-ms ramps) presented at a 960-ms interval and intensities over a 40-dB range above threshold. Amplifier bandpass for the cortical potentials was set between DC and 100 Hz (for additional details, see Starr et al. 2003). Peripheral nerve conduction (sural sensory, peroneal motor, and median sensory/motor) was tested in five affected family members using standard clinical procedures (Kimura 1989) and test equipment (Neuro Diagnostics, LBM-4).
Neurological examination
A clinical neurological examination testing function of cranial nerve, motor activity, reflexes, and sensation was performed on 30 individuals (26 affected and 4 unaffected family members).
Cochlear implant studies
Three family members with cochlear implants, one of whom had been studied both physiologically and psychoacoustically prior to implantation, were evaluated using electrical stimulation of the implant. We recorded electrically evoked auditory brainstem responses (EABRs) and electrically evoked auditory cortical evoked potentials (EAEPs) using the same recording amplifier bandpass as for the acoustic tests. The stimulus for EABRs and EAEPs was a biphasic pulse, 150-μs duration, applied to the implant in a bipolar mode between closely spaced electrodes at the apical region of the cochlea. We employed a bipolar mode of stimulation to reduce the spread of current to adjacent nonauditory structures such as the facial nerve. For EABRs, a single biphasic pulse was used; for EAEPs, four biphasic pulses with an interstimulus interval of 10 ms were used.
Phenotype of hearing function
The classification of the phenotype of hearing used the results of three hearing tests, audiogram. DPOAEs (or CMs), and ABRs, for identifying affected subjects. The classifications are described below.
“AN” (abnormal audiogram, normal DPOAEs from 1 to 4 kHz or higher, and abnormal ABRs).
“AN + outer hair cell” (abnormal audiogram, abnormal DPOAEs, and abnormal ABRs if tested). The distribution of DPOAE loss across the frequencies tested defined the outer hair cell disorder as “partial” (DPOAEs present only at low frequencies) or “total” (DPOAEs lost at all test frequencies).
“Hearing loss unspecified” (subjects with an abnormal audiogram but no DPOAEs and/or ABRs tests).
“Normal/unaffected” [subjects with an audiogram within normal limits for age and gender (Robinson 1998)]. Some of these subjects also had tests for DPOAEs and/or ABRs and their results were normal.
There were three male adults (ages 29, 37, 43) with moderate asymptomatic high-frequency hearing loss at 4, 6, and 8 kHz. The hearing loss was unilateral in one man and bilateral in the other two, DPOAEs were absent at these frequencies, and ABRs tested in two showed that Wave V latencies were normal. These three individuals had a history of noise exposure and were classified as having a hearing loss unrelated to the familial deafness. Their DNA (deoxyribonucleic acid) analyses were negative for the AUAN1 locus.
Data analysis
Individual test results were identified as abnormal if scores (psychoacoustic, neurophysiological) differed by more than two standard deviations (>2 ± SDs) from normal values. Repeated-measures analysis of variance procedures (ANOVA) were used to separately evaluate age and phenotype differences in the audiograms. Post hoc tests of differences among the means used the Tukey–Kramer procedure; the significance level was set at 5%. Group differences were also evaluated with independent t-tests for unequal samples in psychoacoustic (gap detection) and psychoelectric tests (gap detection, rate discrimination).
RESULTS
Inheritance
The presence of male-to-male transmission of the hearing loss trait excludes both X-linked and mitochondrial inheritance in the family (Fig. 1). The inheritance pattern is consistent with autosomal dominance as individuals in all generations were affected. There appears to be 100% penetrance since, among the individuals examined, there was no affected child born to unaffected parents. A marriage between affected first cousins in generation III produced six affected offspring, and another consanguineous marriage in generation V between affected second cousins had two affected children. There appeared to be no increased severity of the phenotype in any of the children of the consanguineous marriages.
Symptoms
Sixty-seven family members tested responded to a questionnaire regarding associated symptoms (tinnitus, dizziness, motor and sensory functions, etc.), age of onset, hearing-aid use, and risk indicators for acquired causes of hearing loss (ototoxicity, infections, trauma). We also included the age of onset from the two children who were affected to help clarify the age domain when hearing impairment can first develop. Age of onset of symptoms as determined by recollection in adults may be extremely inaccurate. For affected subjects their memory for the onset of hearing impairment ranged between 7 and 45 years of age (average = 18.6 years), and the hearing loss typically progressed to a profound deafness within 20 years. Diminished speech understanding was the most common initial and disabling symptom (88%). Background noise was noted to further impair speech understanding in 72%; an inability to understand when using a phone was noted in 61%; tinnitus was present in 59%; hearing aids were reported as “helpful” in 38%, making sounds louder but not improving speech understanding. When we examined the affected subjects, none was using hearing aids. Dizziness had been experienced in 19%, altered sensation (“numbness”) in 11%, and weakness (not specified further) in 6%. Seven affected individuals have received cochlear implants.
Audiology
The averaged pure tone audiograms from the right ear are shown in Figure 2 (left panel) for 34 hearing-impaired (filled symbols) and 35 unaffected family members (open symbols) grouped by age. The loss was symmetrical and only data from the right ear was graphed. The three unaffected family members with isolated high-frequency hearing loss were not included in this analysis. The hearing loss was moderate in affected young family members (<35 years old), severe at 35–50 years old, and profound in the group over 50 years of age. Thresholds were elevated at high frequencies to a greater extent than at low frequencies, with the average loss increasing as subjects aged. In unaffected normal family members, a mild to moderate high-frequency loss developed in the fifth and sixth decades of life, a common finding in the general population (Robinson 1988). ANOVA results using the lower frequencies between 250 Hz and 3 kHz indicated a significant overall threshold difference between unaffected normal (grouped) and affected (grouped) members [F(1, 56) = 245, p < 0.001]. Separate ANOVAs for unaffected normals [F(2, 26) = 5.2, p < 0.01] and affected members [F(2, 26) = 6.6, p < 0.005] indicated significant age effects in hearing thresholds. (Post-test in unaffected normals and in affected members confirmed that thresholds were significantly more elevated for older (>50) than younger (<35) individuals. The progression of the hearing loss for one of the affected family members studied over 13 years in shown in the right panel of Figure 2.
Figure 2.
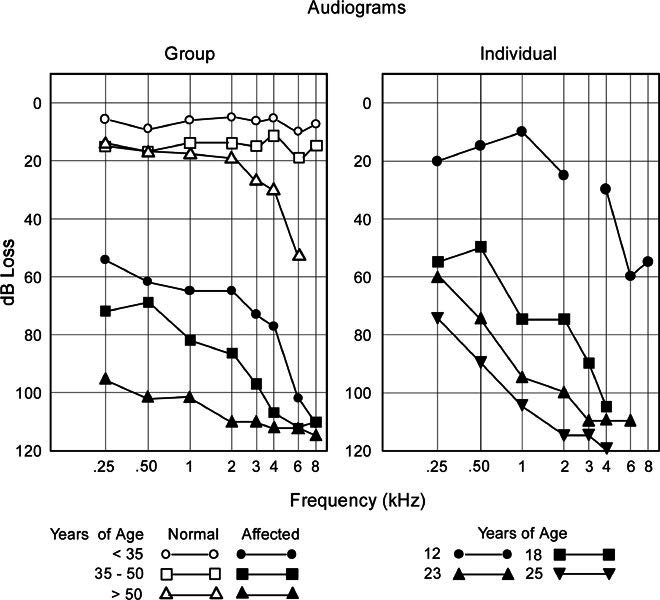
Left panel: Averaged pure tone audiogram as a function of age (< 35, 35–50, > 50 years of age) for unaffected normal-hearing family members (unfilled symbols) and affected hearing-impaired members (filled symbols). Affected family members have a progressive hearing loss, with high frequencies impaired to a greater extent than low frequencies. In unaffected family members a mild high-frequency loss develops after 35 years of age. Right panel: Serial audiograms from the left ear of a single individual showing the rate of progression over 13 years. Testing at age 12 (●—●) indicated speech discrimination of 92% for both ears (normal values > 90%) and absent acoustic reflexes; at age 18 (■—■) speech discrimination was 0% (right ear) and 35% (left ear) again with absent acoustic reflexes. ABRs from the left ear at age 12 were reported as “abnormal,” containing only short latency components (1.5 and 2.7 ms).
DPOAEs were present in 15 of the 23 affected subjects and in all 11 of the unaffected normal subjects tested. Eight affected subjects without DPOAEs had profound hearing loss and were, on average, 57 years of age. In contrast, the 15 affected family members with DPOAEs were considerably younger, averaging 37 years of age. The distribution of preserved DPOAEs in affected family members with DPOAEs showed low frequencies (2 kHz or less) to be present in all (15/15), whereas high frequencies (4 kHz or higher) were present in only 6 of 15. In the 11 unaffected normal family members, DPOAEs were present at 4 kHz or higher in eight members. The three unaffected family members without high-frequency DPOAEs had asymptomatic mild to moderate high-frequency loss thought to be secondary to noise exposure and unrelated to the AUAN1 locus.
ABRs were tested in 22 family members (18 affected and. 4 unaffected), ABRs were abnormal in all 18 affected subjects; ABRs were absent in 16 and showed a prolonged Wave V in two subjects. Cochlear microphonics (CMs) were identified in the ABR of nine of the affected subjects; all but one also had DPOAEs. The ABRs in unaffected family members were normal for Wave V latency.
Speech recognition measures were available in nine subjects, seven of whom had a profound hearing loss and were unable to understand speech without the use of visual cues. There were two young subjects with moderate pure tone hearing loss with speech recognition scores (67% and 26%) poorer than would have been expected for a moderate cochlear sensory hearing loss.
Acoustic middle ear muscle reflexes were absent in all eight subjects tested.
In summary, the pattern of audiological tests in younger subjects (<35 years old) is consistent with a disorder of auditory nerve in the presence of preserved outer hair cell function: ABRs were absent or abnormal while DPOAEs and/or CMs were present. In middle-aged subjects (35–50 years old), the hearing loss was severe; ABRs were absent and DPOAEs were lost to high frequencies, while DPOAEs to low frequencies remained preserved, a pattern of findings consistent with a mixed partial sensory disorder and neural dysfunction. Finally, subjects in their 50 s and 60 s were profoundly deaf and did not have evidence of both outer hair cell functions (absent DPOAEs, CMs) and auditory nerve and brainstem neural responses (absent ABRs).
Neurological examination and nerve conduction studies
The clinical neurological examination was normal except for the finding of sustained nystagmus on lateral gaze in 11 of the 26 affected family members beyond that typically found in normal subjects. We did not measure vestibular functions in the laboratory to resolve whether the clinical findings were due to a vestibular disorder. All but two affected family members had preserved ankle jerks and all had normal vibration sense in the toes. Motor and sensory conduction nerve velocities were normal in the five hearing-impaired family members we tested (Kimura 1989). These findings do not show evidence of a concomitant peripheral neuropathy.
Otological examination
The examination was normal in all 39 subjects without abnormality of the external auditory canal or tympanic membranes. Tympanometry was normal in the six affected family members examined. Temporal bone computerized tomography scans were obtained on seven patients and were normal.
Phenotype of hearing disorder
The hearing loss affected high frequencies to a greater extent than low frequencies (Table 2, Fig. 3). For subjects categorized as AN (n = 6, average age = 28, range = 13–41), the pure tone loss ranged from mild to profound, click-evoked ABRs were abnormal (3) or absent (3), and DPOAEs were present from 1 to 4 kHz or higher. The audiograms, DPOAEs, and ABRs for three affected family members (Nos. 4, 5, and 3) with features of AN are shown in Figure 4. The audiogram for two of the subjects (Nos. 4 and 5) shows a mild loss at low frequencies, increasing to a moderate loss at 6 and 8 kHz. Their speech recognition scores were lower than would be expected for a cochlear hearing loss of this degree. DPOAEs were present at all test frequencies. A delayed Wave V is present superimposed on a slow potential shift in the click ABR for subject No. 4 and in the tone burst ABR for subject No. 5. Phase-reversed potentials representing CMs were evident in both the click and tone burst ABRs. Subject No. 3 had a profound hearing loss affecting all frequencies with a corresponding profound loss of speech recognition. DPOAEs were present from 1 kHz through 4 kHz but not at 6 kHz. Both click and tone burst ABRs were absent and CMs were present to the 1-kHz tones but not to clicks, For subjects with “AN + partial OHC (outer hair cell)” disorder (n = 10, average age = 45, range = 27–59), the hearing loss was severe, DPOAEs were present to low but absent to high frequencies, and ABRs tested in seven did not contain neural components, For subjects with “AN + total OHC (outer hair cell)” disorder (n = 7. average age = 55, range = 32–77), the hearing loss was profound and DPOAEs were absent at all test frequencies. One of the latter subjects (No. 1) is shown in Figure 4 with a profound hearing loss, absent DPOAEs at all tested frequencies, absent CMs, and absent neural components in the ABRs. There were 11 subjects with “hearing loss unspecified” since two of the auditory tests (OAEs, ABRs) used for the classifying phenotype were not performed. There were 35 family members (average age = 25) with normal audiograms, and, in the few we tested, normal DPOAEs and ABRs.
TABLE 2.
Audiological tests and phenotype
| Diagnosis | n | Mean age | Pure tone average | DPOAEs Present | ABRs |
|---|---|---|---|---|---|
| AN | 6 | 28 | Moderate loss | 1–4 kHz | Abnormal |
| AN + partial OHC | 10 | 43 | Severe loss | <2.1 kHz | Abnormal |
| AN + total OHC | 7 | 57 | Profound loss | Absent | Absent |
| Hearing loss unspecified | 11 | 49 | Profound loss | NT | NT |
| Normal | 38 | 25 | Normala | 1–4 kHza | Normalb |
Pure tone average = average threshold for 0.5, 1.0, and 2.0 kHz; NT = not tested; DPOAEs = distortion product otoacoustic emissions; OHC = outer hair cell; ABRs = auditory brainstem responses.
aThere were three subjects with high-frequency hearing loss unrelated to the AUAN1 abnormality; DPOAE tests in this group were normal except for these three subjects who showed absent DPOAEs at 4, 6, and 8 kHz.
bIncidence of normal tests in this group was 4 out of 4 tested.
Figure 3.
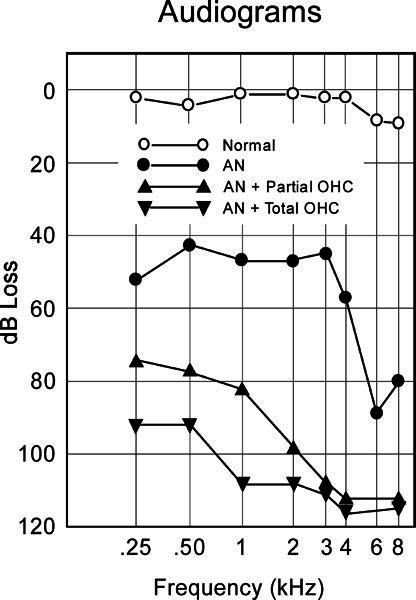
Averaged audiogram as a function of phenotype of hearing loss; “normal” = unaffected family members (n = 35); “AN” = auditory neuropathy (n = 6); “AN + partial outer hair cell” = auditory neuropathy and preserved low-frequency DPOAEs (n = 10); “AN + total outer hair cell” = auditory neuropathy and loss of DPOAEs to all tested frequencies (n = 7).
Figure 4.
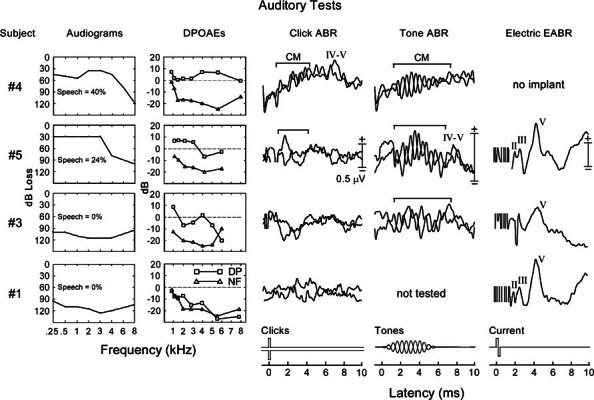
Cochlear and brainstem test measures are shown for four affected family members; audiograms and speech recognition (first column), distortion product otoacoustic emissions (DPOAEs, second column), reflecting amplitudes of both distortion products (DP) and noise floor (NF), auditory brainstem responses (ABRs) to clicks (third column), and to l–kHz tones (fourth column). Electrically evoked auditory brainstem responses (EABRs, fifth column) are shown. Acoustic ABRs are superimposed averages to separately presented condensation and rarefaction stimuli shown in the bottom traces. Phase-reversed potentials marked by brackets are cochlear microphonics (CMs). Subjects 4 (age 18). 5 (age 35), and 3 (age 41) had test results consistent with AN. Audiograms showed a mild to moderate loss for Nos 4 and 5 affecting high frequencies to a greater extent than low frequencies. Subject No. 3 had a severe, flat loss. DPOAEs were present for all test frequencies for Nos. 4 and 5 and were absent only for 6 kHz for No. 3. ABR neural components to clicks were absent in Nos. 3 and 5; No. 4 displayed a delayed Wave V superimposed on a sustained slow potential shift to clicks; No. 5 showed a Wave V to tones. CMs (clicks and/or 1–kHz tones) were present in all. Subject No. 1 (age 39) had test results consistent with AN plus outer hair cell disorder. Audiograms showed a profound loss. ABRs were absent and both DPOAEs and CMs were not detected. Three of these subjects had cochlear implants (fifth column). EABRs in all showed a normal latency Wave V, with Waves II and III also present for subjects 5 and 1. The ABR and EABR in subject No. 5 were to stimulation of the same ear before and after cochlear implantation. There are electrical recording artifacts present at the beginning of the EABRs, and the origins of the negative spike at 2 ms in subject No. 3 are unclear.
ANOVA results on the audiograms from affected family members revealed a significant (F(2, 18) = 19.2, p < 0.001] threshold hearing loss effect related to phenotype classification. Post hoc tests indicated that thresholds for AN + partial OHC and AN + total OHC were significantly elevated compared with AN; no significant differences were found between AN + partial OHC and AN + total OHC.
Psychophysical and neurophysiological measures before and after cochlear implantation
Temporal processes
Gap detection (acoustic). Three affected family members (Nos. 2, 4, and 5) and two unaffected family members were compared to seven normal-hearing subjects who were naïve in performing the psychoacoustic tasks (Table 3, Fig. 5, top). The normative control data are presented as a range depicting the mean plus or minus two standard deviations. The two unaffected family members had gap detection thresholds within the normal range at all sensation levels. The three affected subjects had significantly elevated gap thresholds at 30-dB sensation levels compared to controls [t(8) = 9.1, p < 0.001].
TABLE 3.
Psychoacoustic and psychoelectric measures in family members before and after cochlear implantation
| Tests | Affected family members | Cochlear deafness | Normal | ||||
|---|---|---|---|---|---|---|---|
| #3 | #1 | #5 | #4 | #2 | |||
| Acoustic stimulation | |||||||
| Gap detection: 30 dBSL (ms)c | CNT | CNT | 23 | 32 | 21 | 3a | <7.6 |
| AM: Peak sensitivity of detection (dB) | CNT | CNT | 0 | −1 | NT | −22a | −20 |
| AM: Cut-off frequency (Hz) | CNT | CNT | <4 | 16 | NT | 235a | 238 |
| Temporal integration (dB/doubling duration) | CNT | CNT | 3 | 4 | NT | 3a | 3 |
| Frequency discrimination (1 kHz) | CNT | CNT | 70 | 72 | 204 | 8 | |
| Frequency discrimination (4 kHz) | CNT | CNT | 280 | 220 | 498 | 38 | |
| Slope of the masking function (dB/dB) | CNT | CNT | 1 | 1 | NT | 1a | 1 |
| Speech recognition (%) | 0 | 0 | 0 | 4 | 10 | 80a | >90 |
| Electric stimulation of cochlear implant | |||||||
| Gap detection: apical electrode (ms)d | 18 | 4 | 2 | 5 | |||
| Rate discrimination: 100 Hz (in Hz)d | 20 | 36 | 21 | <43 | |||
| Speech recognition (%) | 4 | 58 | 65 | 70–80b | |||
Bold values for individual subjects for acoustic stimulation tests are >2 SD from normal, and for electric stimulation tests are >2 SD from implanted subjects with cochlear deafness.
CNT = could not test, threshold too elevated to perform; NT = not tested.
afrom Zeng et al. (1999).
bfrom averaged Nucleus 24 users (Skinner et al. 2002).
cStatistical tests for group differences (family members vs normals) were significant (p < 0.01) for acoustic gap detection.
dStatistical tests for group differences (affected family members vs. cochlear deafness) were not significant, p> 0.05 for either gap or rate discrimination.
Figure 5.
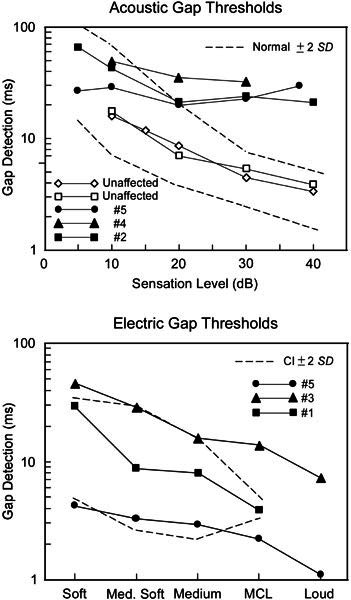
Gap detection to acoustic (top) and electric (bottom) stimuli as a function of sound intensity (acoustic) or “loudness” (electric); MCL in the abscissa of electric gap refers “most comfortable loudness.” The broken lines outline the ± 2 (SD) range for normal-hearing subjects (acoustic) and for three cochlear implant subjects (CI) without auditory neuropathy (electric). Acoustic gap detection (top) was abnormal at suprathreshold levels in three affected family members and normal in unaffected family members. Gap detection to electrical stimulation of the cochlear implant (subjects 1 and 5) shows values comparable to implanted subjects with cochlear sensory deafness. Subject No. 3 (▲—▲) was tested two months after implantation. The right ear of subject No. 5 (●—●) was tested before and after implantation and showed reduction in thresholds from 28 ms at 30 dB to 1.8 ms at MCL.
Twenty-six family members were tested on a web-based protocol in their community center. Nine had a profound hearing loss and were unable to perform the task. Eleven unaffected family members had normal gap detection thresholds. Six family members had elevated gap detection thresholds (20–192 ms), five of whom were “affected,” and one had a normal audiogram. Acoustic gap detection is typically unaffected in cochlear hearing loss after adjusting for loudness (Moore et al. 1989). This elevated gap detection threshold at high sensation levels has been reported as a signature of auditory neuropathy in a large set of psychophysical studies (Zeng et al. 1999).
Gap detection (electric). Three subjects with cochlear implants were studied (Nos. 1, 3, and 5), one of whom (No. 5) had also been studied with acoustic gap measures prior to implantation. In the latter subject tested six months after implantation, gap detection at “most comfortable loudness” shortened from 28 ms before implantation to 1.8 ms after implantation (Fig. 5, bottom). In the two other subjects with implants, acoustic gap detection thresholds could not be measured in the unimplanted ears because of the profound hearing loss. However, when the implanted ears were tested using electrical stimulation, gap detection was 3.9 ms in subject No. 1 (similar to a “control” group of implanted subjects without auditory neuropathy) and 18 ms in subject No. 3 tested only a few months after implantation. Compared with the gap detection data collected in three cochlear implant subjects whose deafness was presumably due to cochlear impairment, no significant difference was found for the gap detection data in the three affected family members who had received a cochlear implant [t(4) = 0.75, p = 0.49].
Amplitude modulation detection (acoustic). Results in two affected family members showed them to be profoundly impaired. Subject No. 5 was not able to detect 100% modulation rates even as low as 4 Hz. Subject No. 4 was unable to detect modulation higher than 16 Hz and again had elevated thresholds (−6 to −1 dB or 50%–90% modulation). Normal subjects can detect a 20-dB or 10% modulation up to 128 Hz. In contrast, cochlear hearing loss has little influence on amplitude modulation detection when intensities are adjusted to correct for the threshold loss (Moore and Glasbery 2001; Zeng et al. 1999).
Temporal integration (acoustic). In subjects 4 and 5, thresholds decreased at a relatively normal rate of 3–4 dB per doubling of the signal duration. This result was similar to what was reported in a large set of auditory neuropathy subjects (Zeng et al. 1999).
Rate discrimination (electric). The three affected family members produced rate difference limen of 20, 36, and 21 Hz, respectively, at the 100-Hz standard rate (Table 3). These values were not significantly different from that obtained in typical cochlear implant subjects whose deafness was presumably due to cochlear impairment [t(4) = 1.79, p = 0.15].
Frequency processes (acoustic). Frequency discrimination was abnormal in the three subjects tested, with difference limens from 70–280 Hz at low frequencies (1 kHz) to 220–480 Hz at 4 kHz. These values are greatly in excess of those found in cochlear sensory hearing loss (Thai-Van et al. 2002).
Simultaneous masking (acoustic). Masking functions in the two AN subjects showed abnormal threshold elevations but normal slopes of the growth for a noise level greater than 20 dB SPL with a 1-dB increase in noise level resulting in a 1-dB increase in the tone thresholds. The expected thresholds were about 20 dB higher than in normal-hearing subjects, suggesting that there were “dead regions” in the cochlea (Moore et al. 2000).
Speech recognition (acoustic and electric). The three implanted subjects were unable to recognize sentences prior to implantation. After implantation, two of the subjects (Nos. 1 and 5) achieved a 65% and 58% correct performance, respectively, for speech recognition in quiet and maintained a similar level of performance in the presence of noise (15-dB signal-to-noise ratio). Subject No. 3 achieved only a 4% correct performance in quiet when tested two months after the implant had been activated. One year after these tests, subject No. 3 engaged in phone conversations using the implanted ear.
In summary, psychophysical measures in affected family members revealed abnormal acoustic temporal discriminations (gap detection, amplitude modulation detection), abnormal frequency discrimination, but normal temporal integration and growth of masking with noise. Electrical activation of the cochlear implant restored gap detection to normal. Moreover, speech comprehension, absent to acoustic signals, was remarkably improved after the implant. Thus electrical stimulation through the implant was effective in normalizing auditory temporal processes that were impaired in affected family members.
Taken together, the significant difference observed in acoustic data and the nonsignificant difference in electric data suggest that, at least in these three affected family members, the pathological abnormality of the auditory nerve lies distal to the region of the ganglion cell bodies, the area activated by electrical stimulation from the cochlear implant. The distal structures that could be affected include inner hair cells, synapses between the inner hair cells and auditory nerve, and terminal dendrites of the auditory nerve. These conclusions, however, should be viewed as suggestive since the number of family members with implants tested was small.
Neurophysiological measures
ABRs and electrically evoked ABRs (EABRs) following cochlear implantation
Figure 4 (column 3) contains superimposed, separately averaged, click-evoked ABRs to condensation and rarefaction clicks from four affected subjects (Nos. 1, 3, 4, and 5). ABRs from three of the affected family members (Nos. 1, 3, and 5) do not contain Waves I–V, while the ABR from subject No. 4 showed a delayed Wave V at 6.95 ms superimposed on a slow potential shift.
Three of these subjects (Nos. 1, 3, and 5) had cochlear implants and electrical stimulation of the implants evoked EABRs (Fig. 4, column 5), with a Wave V of large amplitude (ca. 0.5 μV) at 4-ms latency, similar to patients with cochlear implants without auditory neuropathy (Firszt et al. 2002). Waves II and III were also evident in subjects 1 and 5. The ABRs and EABRs in subject No. 5 were tested by stimulating the same ear prior to and after implantation, while the non implanted ear was tested to acoustic stimulation for subjects 1 and 3.
Auditory long-latency evoked potentials
Auditory long-latency evoked potentials (acoustic AEPs) were recorded to a 1-kHz tone from five subjects (1–5). That for subject No. 5 is shown in Figure 6 as a function of signal intensity (left panel). The subject’s behavioral threshold was 75 dB SPL. To stimuli 5 and 15 dB above threshold, the N100 component had a peak latency of 180 ms, a value much delayed compared with normal N100 peak latency of approximately 110 ms evoked by intensities close to threshold. The latency of N100 in subject No. 5 shortened to 110 ms to intensities 40 dB above threshold but remained prolonged compared to normal latency values of 70–100 ms at similar intensity levels. N100 latency in the other affected family members were also delayed to acoustic stimulation.
Figure 6.
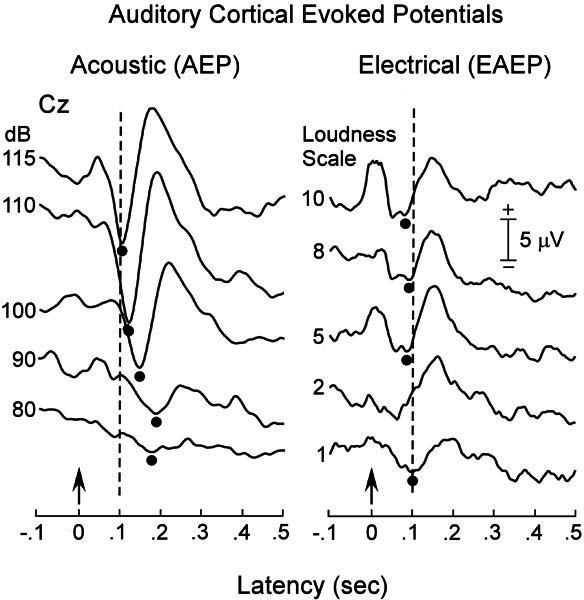
Auditory evoked potentials (AEPs, left panel) in subject No. 5 from stimulation of the right ear to acoustic signal (1–kHz tone, 100-ms duration) before, and to electric stimulation six months after cochlear implantation. Note the reduction in the latency of the N100 component in the electric (EAEP) compared to the acoustic (AEP) stimuli. The positive deflection in the initial 40 ms of the EAEPs is a recording artifact of the 40-ms train of stimulus pulses. Arrows indicate time of stimulus presentation. The broken vertical lines are at 100 ms, the typical latency of the acoustically evoked N100 in normal-hearing subjects.
Electric long latency AEPs
Electrically evoked auditory cortical potentials (EAEPs) were present in the three implanted subjects (Nos. 1, 4, and 5). Results from subject No. 5 are shown in Figure 6 (right panel) presented as a function of stimulus current. The electrically evoked N100 peak ranged from 70 ms when stimulus levels were high (a subjective “10” on a 1–10 loudness scale) to 100 ms when intensity was close to threshold, a subjective “1.” Thus, electrical stimulation evoked N100 components that were 40–80-ms shorter than the N100 component to comparably loud acoustic stimuli prior to implantation. There is a striking amplitude difference in the cortical potentials evoked by electrical and acoustic stimulation. The low amplitude of the electrically evoked potential may be related to the use of bipolar stimulation through adjacent electrodes that limited the number of auditory ganglion cells and nerve fibers activated. Acoustically evoked cortical potentials were of similar low amplitude when the intensity of the tone burst was reduced to 10 dB above threshold, a procedure that also restricts the number of auditory nerve fibers activated.
In summary, physiological studies of affected family members early in the course of the disorder showed ABRs to be absent or profoundly abnormal, whereas OAEs and CMs were preserved, findings consistent with a disorder of function of the auditory nerve in the presence of preserved outer hair cell activities. The ability of electrical stimulation of the cochlea to restore ABR neural components (EABRs) and normalize the latency of cortical potentials (EAEPs) indicates that ganglion cells and their axons in affected family members were capable of normal synchronized activation.
DISCUSSION
Phenotypes of deafness
The family of this report has a dominantly inherited deafness that expresses phenotypic features that change as deafness progresses. Auditory physiological tests in affected family members when hearing loss is moderate are consistent with AN, showing absence or profound abnormalities of auditory nerve and brainstem responses (ABRs) in the presence of preserved cochlear receptor outer hair cell activities (OAEs, cochlear microphonics). In subjects whose hearing loss had progressed to a severe impairment, DPOAEs from middle and high frequencies were lost, consistent with both AN and a partial sensory receptor disorder. Finally, when subjects were profoundly deaf, all DPOAEs were lost, a feature of a profound sensory and neural hearing loss. Affected family members do not have any evidence of peripheral and/or cranial nerve involvement and are distinguished from other forms of hereditary AN occurring with peripheral neuropathies (Butinar et al. 1999; Kalaydjieva et al. 2000; Kovach et al. 2002; Starr et al. 2003), Refsum’s disease (Oysu et al. 2001), and Friedreich’s ataxia (Satya–Murti et al. 1980) or other cranial neuropathies (Ceranic and Luxon 2004). In the peripheral myelin protein 22 (PMP22) form of AN (Kovach et al. 2002; Sambuughin et al. 2003), subjects may also have impaired outer hair cell functions (absent OAEs), resulting in a sensorineural phenotype similar to some affected family members in the present report. OAEs have been noted to be absent in approximately 30% of AN subjects followed serially for several years (Starr et al. 2001), suggesting that AN and sensorineural deafness may be expressed at different times in certain types of deafness.
Families with nonsyndromic auditory neuropathy have also been described with autosomal dominant (Bonfils et al. 1991), autosomal recessive (Varga et al. 2003; Wang et al. 2003), and X-linked recessive (Wang et al. 2003) patterns of inheritance. Mutations in the OTOF gene have been implicated in autosomal recessive AN (Rodriguez–Ballesteros et al. 2003; Varga et al. 2003). Many loci have been mapped and genes cloned for nonsyndromic hereditary hearing impairment (Hereditary Hearing Loss home page), but few of the families in these studies were examined with ABRs and/or OAEs; thus, how many might actually have auditory neuropathy with or without hair cell disorder is unknown.
The type of deafness in the family of the present study changed in an orderly manner from AN in younger subjects to sensorineural deafness in older subjects. The identification of the AN phenotype in the family was recognized only when we tested young individuals with mild to moderate hearing loss. The phenotype of affected family members with severe or profound hearing loss was consistent with a sensorineural deafness because of the absence of OAEs. The identification of the responsible gene in this family would allow experimental studies of mechanisms underlying both the nerve and hair cell dysfunctions and the temporal sequence of expression of these disorders.
Psychophysics of the auditory deficit
Affected family members tested in the initial stages of the disorder show profound impairment of auditory temporal processes similar to those found in other patients with AN (Zeng et al. 1999). In contrast, once the loudness cue is controlled, temporal processes are usually normal in hearing loss due to cochlear sensory disorders (Bacon and Gleitman 1992). Affected family members as well as other subjects with AN are proficient lip readers, a skill that compensates for their auditory temporal processing deficits (Ramirez and Mann, ipersonal communication). Affected family members also have a profound disorder of frequency discrimination across a wide frequency range, whereas frequency discrimination disorders in cochlear sensory deafness are relatively mild and may even be better than normal at frequencies located at the edge of the hearing loss (Thai-Van et al. 2002). The extent of impaired frequency discrimination in affected family members is relatively uniform across frequencies; in contrast to the selective low-frequency discrimination impairment found in a family with AN due to a primary degeneration of the auditory nerve in the presence of normal hair cells (Starr et al. 2003). The distinction between the two families was also evident in measures of the slope of masking growth that was normal in the present family but abnormal in hereditary degeneration of auditory nerve (Starr et al. 2003). The results of psychoacoustic studies show that the underlying auditory processing abnormalities in affected family members are distinct from those found in a cochlear sensory deficit and differ from AN due to primary degeneration of the auditory nerve.
Cochlear implantation and locus of auditory dysfunction
Electrical stimulation of the auditory nerve through cochlear implant (see Fig. 7) is thought to activate ganglion cells and their immediately adjacent processes (Shepherd and Javel 1997) to restore synchronous discharges in auditory nerve fibers, and not by activating structures within the cochlear partition such as the inner hair cells, the adjacent nerve terminals, and their intervening synapses. Experimental animal studies suggest that there is a need for both a minimum number of functioning auditory nerve fibers and an ability to discharge synchronously to obtain averaged EABRs to electrical stimulation (Shepherd and Javel 1997). Diseases of auditory nerve that diminish the numbers of fibers will attenuate the level of excitation reaching the brainstem, while diseases of axons and myelin of the remaining fibers will disrupt the speed of conduction and the rate and temporal consistency of discharge. Both pathological processes would compromise the recording of normal averaged EABRs and also affect auditory perceptions dependent on neural synchrony.
Figure 7.
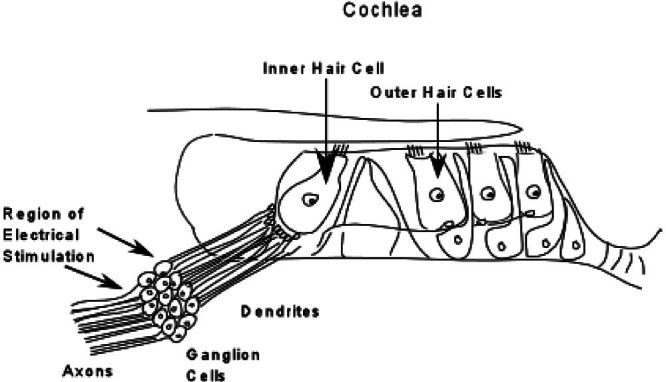
Schematic representation of the cochlea showing auditory nerve bipolar ganglion cells, their axons directed to the cochlear nucleus, and their dendrites directed to the hair cells. Approximately 95% of the dendrites project to inner hair cells with each inner hair cell making synaptic connections with up to 25 dendrites. Electrical stimulation through a cochlear implant bypasses the terminal portions of the auditory nerve and activates the nerve in the region of the ganglion cells.
Psychophysical and electrophysiological studies of electrical stimulation of cochlear implants in affected family members of this study suggest that the site of auditory nerve dysfunction is localized to distal regions of the auditory nerve (dendrites, inner hair cells, or their synapses) rather than at the proximal auditory nerve (ganglion cells or axons). It also appears that in the three subjects with implants, both the numbers and the integrity of the auditory nerve fibers are sufficiently preserved to allow normalization of objective measures of auditory brainstem (EABRs) and cortical (EAEPs) activities as well as behavioral measures of auditory temporal processes (gap detection). Speech recognition in two subjects was improved while a third subject, studied only four months after the implant had been activated, showed little benefit. However, this latter subject has subsequently improved and now engages in telephone communication using the implant. Other studies (Shallop et al. 2001; Buss et al. 2002, Madden et al. 2002) have also documented the appearance of normal EABRs and speech comprehension in children with AN secondary to anoxia or hyperbilirubinemia. In contrast, cochlear implantation in a child with deafness and Friedreich’s ataxia, a condition that is accompanied by degeneration of auditory nerve, did not result in improvement in word discrimination (Miyamoto et al. 1999).
We have observed EABRs to be absent in several AN patients, with an associated peripheral neuropathy consistent with involvement of the auditory nerve. The use of the cochlear implant did not benefit speech recognition or psychophysical temporal processes in these patients, but they were grateful for their improved ability to lip read and to detect sounds. It would be premature, therefore, to conclude that cochlear implantation is without benefit for deafness due to primary degeneration of the auditory nerve (Type I neuropathy). There are also a number of patients with presumed cochlear deafness with cochlear implants who also do not have EABRs and have little speech perception benefit (Firszt et al. 2002). However, our knowledge is incomplete as to the relative role of afferent versus central mechanisms in predicting effective use of cochlear implants. For instance, speech discrimination performance may not depend solely on the number of remaining ganglion cells in cochlear implant subjects (Nadol et al. 2001). Objective measures of auditory pathway activities such as EABRs and EAEPs may help clarify sites of auditory nerve damage, numbers of fibers affected, and central auditory compensatory processes accompanying cochlear implant use.
We propose that the locus of the auditory abnormality in the family of this report involves one or several of the constituents of the auditory periphery including inner hair cells, the synapses, and terminal dendrites, sparing to a great extent the ganglion cells and their axons. There are no methods available now that can distinguish between disorders of these closely adjacent sites in the auditory periphery.
Pathology of auditory neuropathy
The cochlear pathology in several hereditary forms of AN with peripheral neuropathy (Spoendlin 1974; Hallpike et al. 1980; Starr et al. 2003) shows a selective loss of ganglion cells with preservation of both inner and outer hair cells, findings consistent with a primary auditory nerve degeneration. We suggest that AN with involvement of the ganglion cells, axons, and proximal dendrites be designated as a proximal AN or type I AN. A depletion of afferent auditory nerve fibers with hair cell damage has also been identified in temporal bones from individuals with “sensorineural deafness,” reflecting a late or secondary consequence of the inner hair cell loss (Zimmerman et al. 1995). Experimental animal studies with progressive hearing loss show that distal dendrites are affected in keeping with a retrograde degeneration occurring as a late consequence of damage to the auditory periphery (White et al. 2000). We have data to suggest that the auditory nerve can also atrophy in the family of the present report. One of the affected family members in her 50 s developed unilateral electric-like facial pain, diagnosed as tic doloreux. The neurosurgeon defined the trigeminal nerve to be in proximity to a small arterial blood vessel. During a posterior fossa operation to correct the problem, the surgeon examined the auditory nerve because of the deafness and noted “atrophy of the statoacoustic nerve.” The statoacoustic nerve was not compromised by the blood vessel. The atrophy of the statoacoustic nerve in this affected family member may be a delayed consequence of a distal site of pathology in the auditory periphery.
We suggest that the clinical picture of AN accompanying disorders of distal components of the auditory periphery (terminal dendrites, inner hair cells, synapses) be designated as a distal AN or type II AN to be distinguished from proximal or type I AN due to a primary degeneration of the auditory nerve; the latter may or may not be accompanied by the presence of a peripheral neuropathy. Temporal bone studies of distal auditory nerve disorders will identify which of these components in the auditory periphery are affected. Moreover, when the gene at the AUNA1 locus is identified, the use of experimental animal models may allow the identification of both the gene’s normal functions and the pathophysiological consequences accompanying alterations of the gene. As these data become available, we anticipate that the categorization of hearing disorders as sensory, neural, and sensorineural will also include the affected domains within neural and sensory structures to influence the development of therapies directed at specific mechanisms underlying these different disorders.
Acknowledgments
We thank the family members who were cooperative and a major force in making the study occur. They provided encouragement, time, and their organization in the data collection. Bio-logic Equipment Company (Mundelein, IL) provided portable ABR recording equipment for the field studies. Drs. Hillel Pratt and Sharon Fujikawa provided valuable discussion of issues related to auditory function (Pratt) and hearing (Fujikawa). Ms. Anna Howell assisted in the data analysis. Theresa Kim assisted in preparation of figures. Sheng Liu assisted in speech data collection. This work was supported in part by the National Institutes of Health with Grants DC-02618 (A. Starr), DC-00161 (M. Lesperance), and DC-02267 (F.G. Zeng).
References
- Amatuzzi MG, Northrop C, Liberman MC, Thornton A, Halpin C, Herrmann B, et al. Selective inner hair cell loss in premature infants and cochlea pathological patterns from neonatal intensive care unit autopsies. Arch Otolaryngol Head Neck Surg. 2001;127:629–636. doi: 10.1001/archotol.127.6.629. [DOI] [PubMed] [Google Scholar]
- Bacon SP, Gleitman RM. Modulation detection in subjects with relatively flat hearing losses. J. Speech Hear. Res. 1992;35:642–653. doi: 10.1044/jshr.3503.642. [DOI] [PubMed] [Google Scholar]
- Bench, J, Benford, J (eds) (1979) Speech-hearing tests and the spoken language of hearing-impaired children, Academic Press, London
- Bonfils P, Avan P, Londero A, Narcy P, Trotoux J. Progressive hereditary deafness with predominant inner hair cell loss. Am. J. Otol. 1991;12:203–206. [PubMed] [Google Scholar]
- Buss E, Labadie RF, Brown CJ, Gross AJ, Gross JH, Pillsbury HC. Outcome of cochlear implantation, in pediatric auditory neuropathy. Otol. Neurotol. 2002;23:328–332. doi: 10.1097/00129492-200205000-00017. [DOI] [PubMed] [Google Scholar]
- Butinar D, Zidar J, Leonardis L, Popovic M, Kalaydjieva L, Angelicheva D, et al. Hereditary auditory, vestibular, motor, sensory neuropathy in a Slovenian Roma (Gypsy) kindred. Ann. Neurol. 1999;46:36–44. doi: 10.1002/1531-8249(199907)46:1<36::AID-ANA7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Ceranic B, Luxon LM. Progressive auditory neuropathy in patients with Leber’s hereditary optic neuropathy, J. Neurol. Neurosurg. Psychiatry. 2004;75:626–630. doi: 10.1136/jnnp.2003.017673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding DL, Wang J, Salvi R, Henderson D, Hu BH, Mcfadden SL, et al. Selective loss of inner hair cells, type-I ganglion neurons in carboplatin-treated chinchillas. Mechanisms of damage and protection. Ann. N. Y. Acad. Sci. 1999;884:152–170. doi: 10.1111/j.1749-6632.1999.tb08640.x. [DOI] [PubMed] [Google Scholar]
- Firszt JB, Chambers RD, Kraus N, Reeder RM. Neurophysiology of cochlear implant users. I: effects of stimulus current level and electrode site on the electrical ABR, MLR, and N1-P2 response. Ear. Hear. 2002;23:502–515. doi: 10.1097/00003446-200212000-00002. [DOI] [PubMed] [Google Scholar]
- Hallpik CS, Harriman DG, Wells CE. A case of afferent neuropathy and deafness. J. Laryngol. Otol. 1980;94:945–964. doi: 10.1017/s0022215100089696. [DOI] [PubMed] [Google Scholar]
- Harrison RV. An animal model of auditory neuropathy. Ear. Hear. 1998;19:355–361. doi: 10.1097/00003446-199810000-00002. [DOI] [PubMed] [Google Scholar]
- Kalaydjieva L, Nikolova A, Turnev I, Petrova J, Hristova A, Ishpekova B, et al Hereditary motor and sensory neuropathy—Lom, a novel demyelinating neuropathy associated with deafness in gypsies. Clinical, electrophysiological and nerve biopsy findings. Brain. 1998;121:399–408. doi: 10.1093/brain/121.3.399. [DOI] [PubMed] [Google Scholar]
- Kalaydjieva L, Gresham D, Gooding R, Heather L, Baas F, De Jonge R, et al. N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom. Am. J. Hum. Genet. 2000;67:47–58. doi: 10.1086/302978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TB, Issacson B, Sivakumaran TA, Starr A, Keats BJ, Lesperance M. A gene responsible for autosomal dominant auditory neuropathy maps to a novel locus (AUNA1) J. Assoc. Res. Otolaryngol. Abstr. 242, Winter Meeting. 2004;.:.. doi: 10.1136/jmg.2004.020628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura J. Electrodiagnosis in diseases of nerve and muscle: Principles andractice. 2. Philadelphia: F.A. Davis; 1989. [Google Scholar]
- Kovach MJ, Campbell KC, Herman K, Waggoner B, Gelber D, Hughes LF, et al. Anticipation in a unique family with Charcot–Marie–Tooth syndrome and deafness: delineation of the clinical features and review of the literature. Am. J. Med. Genet. 2002;108:295–303. doi: 10.1002/ajmg.10223. [DOI] [PubMed] [Google Scholar]
- Madden C, Hilbert L, Rutter M, Greinwald J, Choo D. Pediatric cochlear implantation in auditory neuropathy. Otol. Neurotol. 2002;23:163–168. doi: 10.1097/00129492-200203000-00011. [DOI] [PubMed] [Google Scholar]
- Merchant SN, McKenna MJ, Nadol JB, Jr, Kristiansen AG, Tropitzsch A, Lindal S, Tranebjaerg L. Temporal bone histopathologic and genetic studies in Mohr–Tranebjaerg syndrome (DFN-1) Otol. Neurotol. 2001;22:506–511. doi: 10.1097/00129492-200107000-00017. [DOI] [PubMed] [Google Scholar]
- Miyamoto RT, Kirk KI, Renshaw J, Hussain D. Cochlear implantation in auditory neuropathy. Laryngoscope. 1999;109:181–185. doi: 10.1097/00005537-199902000-00002. [DOI] [PubMed] [Google Scholar]
- Moore BC, Glassberg BR. Temporal modulation transfer functions obtained using sinusoidal carriers with normally hearing, hearing-impaired listeners. J. Acoust. Soc. Am. 2001;110:1067–1073. doi: 10.1121/1.1385177. [DOI] [PubMed] [Google Scholar]
- Moore BC, Glasbergb BR, Donaldson E, McPherson T, Plack CJ. Detection of temporal gaps in sinusoids by normally hearing and hearing-impaired subjects. J. Acoust. Soc. Am. 1989;85:1266–1275. doi: 10.1121/1.397457. [DOI] [PubMed] [Google Scholar]
- Moore BC, Huss M, Vickers DA, Glasberg Br, Alcantara JI. A test for the diagnosis of dead regions in the cochlea. Br. J. Audiol. 2000;34:205–224. doi: 10.3109/03005364000000131. [DOI] [PubMed] [Google Scholar]
- Nadol JB, Jr, Shiao JY, Burgess BJ, Ketten DR, Eddingtqn DK, Gantz BJ, et al. Histopathology of cochlear implants in humans. Ann. Otol. Rhinol. Laryngol. 2001;110:883–891. doi: 10.1177/000348940111000914. [DOI] [PubMed] [Google Scholar]
- Oysu C, Aslan I, Basaran B, Baserer N. The site of the hearing loss in Refsum’s disease. Int. J. Pediatr Otorhinolaryngol. 2001;61:129–134. doi: 10.1016/s0165-5876(01)00559-6. [DOI] [PubMed] [Google Scholar]
- Pratt H, Aminoff M, Nuwer MR, Starr A. Short latency auditory evoked potentials. Electroencephalogr Clin. Neurophysiol. Suppl. 1999;52:69–77. [PubMed] [Google Scholar]
- Ramirez, JA, Mann, V () “Using auditory–visual integration to probe the basis of noise-impaired speech perception in reading disability and auditory neuropathy” Personal Communication [DOI] [PubMed]
- Rapin I, Gravel J. “Auditory neuropathy”: physiologic and pathologic evidence calls for more diagnostic specificity. Int. J. Pediatr Otorhinolaryngol. 2003;67:707–728. doi: 10.1016/s0165-5876(03)00103-4. [DOI] [PubMed] [Google Scholar]
- Robinson DW. Threshold of hearing as a function of age and sex for the typical unscreened population. Br. J. Audiol. 1988;22:5–20. doi: 10.3109/03005368809077793. [DOI] [PubMed] [Google Scholar]
- Rodriguez–Ballesteros M, Del Castillo FJ, Martin Y, Moreno–Pelayo MA, Morera C, Prieto F, et al. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF) Hum. Mutat. 2003;22:451–456. doi: 10.1002/humu.10274. [DOI] [PubMed] [Google Scholar]
- Salvi RJ, Ding D, Wang J, Jiang HY. A review of the effects of selective inner hair cell lesions on distortion product otoacoustic emissions, cochlear function and auditory evoked potentials. Noise Health. 2000;2:9–26. [PubMed] [Google Scholar]
- Sambuughin N, De Bantel A, Mc Williams S, Sivakumar K. Deafness and CMT disease associated with a novel four amino acid deletion in the PMP22 gene. Neurology. 2003;60:506–508. doi: 10.1212/01.wnl.0000044048.27971.fc. [DOI] [PubMed] [Google Scholar]
- Satya–Murti S, Cacace A, Hanson P. Auditory dysfunction in Friedreich ataxia: result of spiral ganglion degeneration. Neurology. 1980;30:1047–1053. doi: 10.1212/wnl.30.10.1047. [DOI] [PubMed] [Google Scholar]
- Shallop JK, Peterson A, Facer GW, Fabry LB, Driscoll CL. Cochlear implants in five cases of auditory neuropathy: postoperative findings and progress. Laryngoscope. 2001;111:555–62. doi: 10.1097/00005537-200104000-00001. [DOI] [PubMed] [Google Scholar]
- Shannon RV, Adams DD, Ferrel RL, Palumbo RL, Grandgenett M. A computer interface for psychophysical and speech research with the Nucleus cochlear implant. J. Acoust. Soc. Am. 1990;87:905–907. doi: 10.1121/1.398902. [DOI] [PubMed] [Google Scholar]
- Shepherd RK, Javel E. Electrical stimulation of the auditory nerve. I. Correlation of physiological responses with cochlear status . Hear. Res. 1997;108:112–144. doi: 10.1016/S0378-5955(97)00046-4. [DOI] [PubMed] [Google Scholar]
- Sininger Y, Oba S. Patients with auditory neuropathy: who are they and what can they hear? In: Sininger Y, Starr A, editors. Auditory neuropathy. San Diego: Singular Press; 2001. pp. 15–35. [Google Scholar]
- Skinner MW, Arndt PL, Staller SJ. Nucleus 24 advanced encoder conversion study: performance versus performance. Ear. Hear. 23. 2002;23(1 Suppl):2S–17S. doi: 10.1097/00003446-200202001-00002. [DOI] [PubMed] [Google Scholar]
- Sobkowicz HM, Inagaki M, August BK, Slapnick SM. Abortive synaptogenesis as a factor in the inner hair cell degeneration in the Bronx Waltzer (bv) mutant mouse. J. Neurocytol. 1999;28:17–38. doi: 10.1023/A:1007059616607. [DOI] [PubMed] [Google Scholar]
- Spoendlin H. Optic cochleovestibular degenerations in hereditary ataxias. II. Temporal bone pathology in two cases of Friedreich’s ataxia with vestibulo-cochlear disorders. Brain. 1974;97:41–48. doi: 10.1093/brain/97.1.41. [DOI] [PubMed] [Google Scholar]
- Starr A. Neurology of auditory neuropathy. In: Sininger Y, Starr A, editors. Auditory neuropathy. San Diego: Singular Press; 200l. pp. 37–50. [Google Scholar]
- Starr A, Mcpherson D, Patterson Jv, Don M, Luxford W, Shannon R, et al. Absence of both auditory evoked potentials and auditory percepts dependent on timing cues. Brain. 1991;114:1157–1180. doi: 10.1093/brain/114.3.1157. [DOI] [PubMed] [Google Scholar]
- Starr A, Picton TW, Sininger Y, Hood LJ, Berlin CI. Auditoiy neuropathy. Brain. 1996;119:741–753. doi: 10.1093/brain/119.3.741. [DOI] [PubMed] [Google Scholar]
- Starr A, Sininger YS, Pratt H. The varieties of auditory neuropathy. J. Basic Clin. Physiol. Pharmacol. 2000;11:215–230. doi: 10.1515/jbcpp.2000.11.3.215. [DOI] [PubMed] [Google Scholar]
- Starr A, Sininger Y, Nguyen T, Michalewski HJ, Oba S, Abdala C. Cochlear receptor (microphonic and summating potentials, otoacoustic emissions) and auditory pathway (auditory brain stem responses) activity in auditory neuropathy. Ear. Hear. 2001;22:91–99. doi: 10.1097/00003446-200104000-00002. [DOI] [PubMed] [Google Scholar]
- Starr A, Michalewski HJ, Zeng FG, Fujikawa–Brooks S, Linthicum F, Kim CS, Winnier D, Keats BJ. Pathology and physiology of auditory neuropathy with a novel mutation in the MPZ gene Tyr145→Ser) Brain. 2003;126:1604–1619. doi: 10.1093/brain/awg156. [DOI] [PubMed] [Google Scholar]
- Thai–Van H, Micheyl C, Norena A, Collet L. Local improvement in auditory frequency discrimination is associated with hearing-loss slope in subjects with cochlear damage. Brain. 2002;125:524–537. doi: 10.1093/brain/awf044. [DOI] [PubMed] [Google Scholar]
- Varga R, Kelley PM, Keats BJ, Starr A, Leal Sm, Cohn E, Kimberling WJ. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J. Med. Genet. 2003;40:45–50. doi: 10.1136/jmg.40.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Gu R, Han D, Yang W. Familial auditory neuropathy. Laryngoscope. 2003;113:1623–1629. doi: 10.1097/00005537-200309000-00041. [DOI] [PubMed] [Google Scholar]
- White JA, Burgess BJ, Hall RD, Nadol JB., Jr. Pattern of degeneration of the spiral ganglion cell and its processes in the C57BL/6 J mouse. Hear. Res. 2000;141:12–18. doi: 10.1016/S0378-5955(99)00204-X. [DOI] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Cohen–Salmon M, El-Amraoui A, Mustapha M, Salem N, et al. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat. Genet. 1999;21:363–369. doi: 10.1038/7693. [DOI] [PubMed] [Google Scholar]
- Zeng FG, Oba S, Garde S, Sininger. Y, Starr A. Temporal and speech processing deficits in auditory neuropathy. Neuroreport. 1999;103:429–435. doi: 10.1097/00001756-199911080-00031. [DOI] [PubMed] [Google Scholar]
- Zheng XY, Wang J, Salvi RJ, Henderson D. Effects of kainic acid on the cochlear potentials and distortion product otoacoustic emissions in chinchilla. Hear. Res. 1996;95:161–167. doi: 10.1016/0378-5955(96)00047-0. [DOI] [PubMed] [Google Scholar]
- Zimmermann CE, Burgess BJ, Nadol JB., Jr. Patterns of degeneration in the human cochlear nerve. Hear. Res. 1995;90:192–201. doi: 10.1016/0378-5955(95)00165-1. [DOI] [PubMed] [Google Scholar]
- Zwirner P, Wilichowski E. Progressive sensorineural hearing loss in children with mitochondrial encephalomyopathies. Laryngoscope. 2001;111:515–521. doi: 10.1097/00005537-200103000-00024. [DOI] [PubMed] [Google Scholar]